

Abstract
Promoter methylation-mediated silencing is a hallmark of many established tumor suppressor genes. This review focuses on the methylation and suppression of a receptor-type tyrosine phosphatase gene, PTPRO, in a variety of solid and liquid tumors. In addition, PTPRO exhibits many other characteristics of a bona fide tumor suppressor. Reactivation of genes silenced by methylation using inhibitors of DNA methyltransferases and histone deacetylases, and the potential application of PTPRO as a molecular target for cancer therapy have been discussed.
Keywords: protein tyrosine phosphatase, PTPRO, DNA methylation, gene reactivation, DNA hypomethylating agents, HDAC inhibitors
Methylation of DNA at C-5 of cytosine within a CpG dinucleotide is the most striking modification in the eukaryotic genome.1 Although CpG is usually under-represented in much of the genome, short (500–2000 bp long) CpG regions, designated CpG islands, are found in the proximal promoters of almost half of the mammalian genes. These regions are generally unmethylated in normal cells unless they are associated with imprinted genes or genes located on the inactive X chromosome. On the other hand, many cancer cells exhibit global hypomethylation and regional or localized hypermethylation of specific genes.2,3 Hypermethylation leads to recruitment of a class of proteins, designated methyl C-binding proteins or MBDs, and subsequently that of many corepressors. The large repressor complex thus formed on the promoter consists of histone deacetylases (HDAC), Sin3A, RbAp46, RbAp48, Mi2, MTA-2 and other unidentified proteins.4–7 This epigenetic event causes alterations in chromatin structure, prevents binding of transcription factors to the regulatory elements and ultimately results in transcriptional repression. Many tumor suppressor genes are silenced in different types of cancers due to promoter methylation.8,9 Promoter hypermethylation and gene silencing also fit with the Knudson’s two-hit model that calls for two successive genetic “hits” in order to inactivate both alleles of tumor suppressor genes, resulting in the transformation of a normal cell into cancer cell.10 Accordingly, it is conceivable that one “hit” represents methylation of one of the alleles. Indeed, reports from many laboratories indicate that a gene can be inactivated in the cancer cell by stably maintaining mutations (the first “hit”) in one allele and hypermethylation in the other (the second “hit”).10–13 Alternatively, methylation of both alleles can contribute to both first and second “hits”, as proposed by Jones and co-workers.2 It is remarkable that hypermethylation is absent in the mutated allele, but is always associated with the wild-type allele of a gene.14 Since numerous reviews on DNA methylation have appeared in the past few years,2,3,15–17 this review will focus on a novel class of candidate tumor suppressor genes that are silenced by promoter methylation in rodent and human tumors, and their functional, diagnostic and therapeutic implications.
Protein tyrosine phosphatases (PTPs): physiological functions relevant to their potential role in cancer
Protein phosphorylation is an important post-translational modification that can affect various cellular processes, including differentiation, contact inhibition, cell cycle and immune response (see Hunter18 for a review). This protein modification is coordinated by the actions of protein kinases and phosphatases that must be precisely regulated in normal physiological processes. Protein tyrosine kinases (PTKs) form a major class of oncoproteins representing 80% of all oncogenes.19 A PTP that catalyzes the reverse reaction could, therefore, function as a tumor suppressor. PTPs could themselves promote signaling (positive effect) by dephosphorylation and activation of PTKs. Consequently, PTPs could coordinate rather than antagonize the functions of PTKs or inhibit signaling (negative effect) depending on the downstream effectors.20–23 For instance, tyrosine phosphatase, PTPN2, activates p53 and induces apoptosis in human tumor cell line,24 whereas PTP1B negatively regulates insulin signalling via dephosphorylation of insulin receptor kinase.25 The known physiological functions of several PTPs suggest their primary roles in neuronal differentiation (e.g. PTPRK, PTPRD, PTPRR, PTPRB, PTPRS, PTPRA), T-cell receptor signaling (e.g. PTPN22, PTPRC, PTPN7, PTPRJ, PTPN2) and cellular adhesion (e.g. PTPRF, PTPN14, PTPRM, PTPRS, PTPRA, PTPRT) (see Table 1 for details). Alteration in the expression or function of these PTPs could, therefore, lead to deregulation of tightly regulated signaling process resulting in uncontrolled cell growth among other pathological conditions.26
Table 1.
Protein tyrosine phosphatases that exhibit characteristics of a tumor suppressor
| Gene symbol | CpG island | Known physiological functions | Characteristic suggestive of tumor suppressor function |
|---|---|---|---|
| PTPN22 | No | Suppresses T-cell activation | Deleted and rearranged |
| PTPRF | Yes | Cell–cell contact at adherens junction, β-catenin signaling | Deleted and mutated |
| PTPRU | Yes | Cell–cell recognition and adhesion | Deleted |
| PTPN7 | No | T-cell antigen receptor signaling, suppress activity of MAP kinases | Deleted and duplicated |
| PTPN14 | Yes | Cell–cell adhesion via β-catenin | Deleted, duplicated, mutated When overexpressed, HeLa cells grow slower and adhere less well |
| PTPRG | Yes | Deleted and mutated | |
| PTPN23 | Yes | Deleted | |
| PTPN13 | Yes | Fas-mediated programmed cell death, regulator of rho signaling pathway | Deleted and mutated |
| PTPRK | Yes | Stimulates neurite outgrowth | Deleted and downregulated |
| PTPN12 | Yes | Controls shape and motility | Deleted |
| PTPRZ1 | Yes | Oligodendrocyte survival | Deleted |
| PTPRN2 | Yes | Autoantigen associated with IDDM | Deleted and methylated |
| PTPRD | Intron 1 | Neurite outgrowth and neuron axon guidance | Reduced expression in hepatomas |
| PTPN3 | ? | Deleted | |
| PTPRJ | Yes | T-cell receptor signaling | Deleted and mutated |
| PTPRO | Yes | Axonogenesis and differentiation, podocyte structure and glomerular filtration | Deleted, methylated and suppressed |
| PTPN6 | Upstream | Signaling pathways in hematopoietic cells | Deleted, methylated and suppressed |
| PTPN11 | Yes | Mutations involved in leukemogenesis | Mutated |
| PTPN21 | Yes | Liver regeneration and spermatogenesis | Deleted and duplicated |
| PTPN2 | Yes | Inhibits T-cell proliferation | Induces apoptosis via activation of p53 |
| PTPRS | Upstream | Cell–cell interactions, primary axonogenesis, axon guidance during embryogenesis | Deleted |
| PTPRH | Upstream | Negative regulator of integrin mediated signaling | Downregulated during dedifferentiation of human hepatocellular carcinomas Induces apoptotic cell death upon over-expression Inhibits cell growth and motility |
| PTPRA | ? | Regulation of integrin signaling, cell adhesion and proliferation, neuronal migration | Expression relates to low tumor grade |
| PTPRT | Yes | Signal transduction and cellular adhesion in CNS | Deleted and mutated |
In addition to the functional consequences that would arise upon loss of a PTP, genetic alterations of several PTPs in different types of cancers also strengthen their position as growth/tumor suppressors (see Table 1). Several PTPs (e.g. PTPRG, PTPRK, PTPRJ) have been localized to regions marked by loss of heterozygosity (LOH), a hallmark of many tumor suppressor genes.27–29 Inactivating mutations in some PTPs (e.g. PTPRF, PTPN14, PTPRG, PTPN13, PTPN11, PTPRT, PTPN3) are also associated with development of cancers.30 A recent study based on computational analysis of the human genome identified 38 classical PTPs, 19 of which mapped to regions frequently deleted in human cancers and 30 of these protein phosphatases have been implicated in different types of cancers.26,31 Several studies have implicated PTPs in cancer and a few have demonstrated their role as tumor suppressors using in vitro and in vivo assays.32 To this date, however, DEP1 is the only protein tyrosine phosphatase that is classified as an authentic tumor suppressor.29,31
Methylation and suppression of PTP receptor-type O: role as tumor suppressor in human cancer
Suppression of gene expression by promoter methylation is considered an important mechanism for the loss of function of many tumor suppressors. Our laboratory has shown that the gene for protein tyrosine phosphatase receptor-type O, PTPRO (also called Glepp1 or PTP-U2) is progressively methylated in the preneoplastic liver and primary hepatomas produced in rats fed methyl-deficient diet. This gene was identified in a genome-wide scan for methylation changes during tumor progression.8,33 It is also heavily methylated and silenced in a transplanted rat hepatoma.33 Further, when the animals bearing the tumor were treated with the DNA hypomethylating agent 5-azacytidine (5-AzaC), demethylation of the PTPRO promoter resulted in gene re-expression and reduction in tumor size. All these observations pointed towards the potential role of PTPRO as a tumor suppressor. The observation that PTPTO is methylated in preneoplastic liver of rats fed methyl-deficient diet33 suggests that this modification could emerge as an early tumor marker in hepatocellular and probably other tumors. We have since extended these studies to human tumors and have identified tumor-specific methylation of the PTPRO CpG island, located within the promoter region, in primary human hepatocellular carcinoma relative to the matching normal liver tissue. Analysis of 43 primary lung tumors and their matching normal adjacent lung tissues also revealed extensive methylation of PTPRO promoter in a large number of lung tumors, whereas the promoter was essentially methylation-free in the matching normal lung tissue.34 In many cases of hepatic and lung tumors, the promoter methylation inversely correlated with PTPRO expression. Although normal liver and lung do not express PTPRO to the same level as brain or kidney, it is noteworthy that PTPRO expression is abrogated in the majority of primary liver and lung tumors. Further, ectopic expression of PTPRO in human lung cancer line, A549 (where PTPRO is suppressed due to methylation) resulted in inhibition of anchorage-independent growth, delayed entry of the cells into cell cycle and increased susceptibility to apoptosis.34 Recent study also showed that PTPRO overexpression reduced the tumor forming potential of cells upon injection into immunocompromised mice (Motiwala T, Rosol T, Jacob ST, unpublished data). The suppressed PTPRO gene was reactivated following treatment of the nonexpressing A549 cells with DNA hypomethylating agents.34 Further, the PTPRO gene is localized to the chromosomal region 12p12.3 that is characterized by LOH in different types of cancer,31,34 another established characteristic of many tumor suppressor genes.35 Global expression profiling of microsatellite instability (MSI-H) colon cancer using cDNA microarray identified PTPRO as one of the 81 genes that are selectively downregulated and methylated.36 These data, taken together, support the notion that PTPRO is a candidate tumor suppressor.
Methylation and suppression of the truncated form of PTPRO (PTPROt) in cancer cells of lymphoid origin
Several variants of PTPRO are generated due to transcription from distinct promoters and alternative splicing (see Figs 1 and 2). The cells of lymphoid origin exclusively express PTPROt whereas most epithelial tissues express predominantly the full-length form. To this date, there has been only one report demonstrating the potential role of promoter methylation in the suppression of a PTP in tumors of lymphoid origin.37 This study, however, deals with a nonreceptor type PTP (SHP-1). It was of interest to investigate whether the PTPRO gene is methylated and silenced in primary human leukemia. Indeed, we were able to show that it is methylated and silenced in the majority of the peripheral blood lymphocytes from 92 chronic lymphocytic leukemia (CLL) patients whereas the CD19+ selected B-lymphocytes from normal individuals did not show methylation of this gene (T Motiwala, H Kutay, J Byrd, M Grever, S Jacob, unpublished data). Further, it could be reactivated in a CLL-like cell line (where PTPROt is suppressed) following treatment with a DNA hypomethylating agent. It is evident that PTPRO/PTPROt methylation and suppression is a common characteristic of many different types of tumors.
Figure 1.
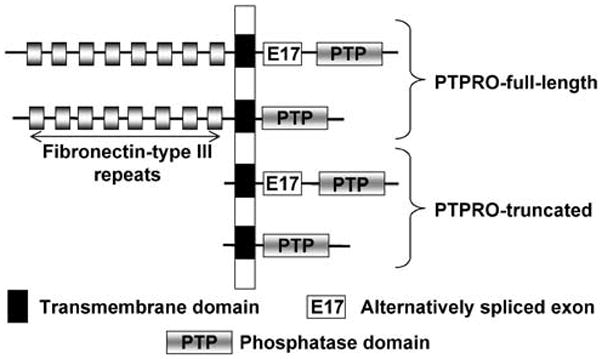
Protein isoforms of PTPRO. The full-length and truncated forms of PTPRO differ mainly with respect to their extracellular domains (fibronectin type III repeats). Each of these forms gives rise to two isoforms that are products of alternatively spliced transcripts, that is, by splicing of E17.
Figure 2.
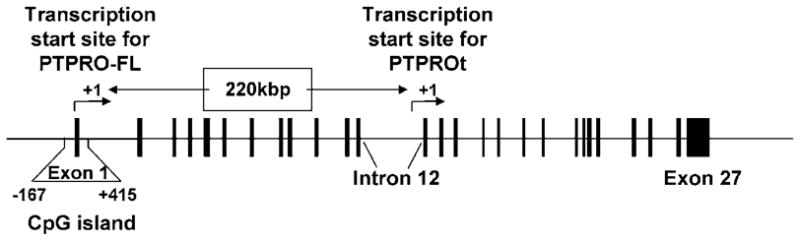
Schematic representation of the PTPRO gene.
Methylation of other receptor-type protein tyrosine phosphatases in leukemia
Are the methylation and subsequent suppression of tyrosine phosphatases confined to PTPRO or PTPROt? Recent analysis by Restriction Landmark Genomic Scanning of CLL genomic DNA has revealed that three other PTPs (PTPRN2, PTPRZ2, PTPN11) are preferentially methylated in CLL and acute myeloid leukemia (C Plass, S Jacob, unpublished data). It is, therefore, highly likely that other PTPs may also be targets for DNA methylation in human cancers. Analysis of the genomic sequences of PTPs shows that most PTPs (except PTPN22, PTPRC, PTPN7, PTPRR, PTPRQ, PTPRA) harbor a CpG island in proximity to their promoter regions (Table 1). The association of CpG islands with the vast majority of PTPs suggests that they have a potential for being regulated by methylation. While the effect of methylation on their expression has not been studied, it is logical to anticipate reduced expression of these PTP genes upon methylation. An important issue that needs to be explored is in regard to the molecular mechanisms for the methylation-mediated silencing of PTPRO or other PTPs in human cancer. It is of particular interest to investigate whether the mechanism for the altered expression of PTPRO is distinct from that of PTPROt (see below).
Proposed mechanisms of methylation-mediated suppression of PTPRO and PTPROt
The structural organization of PTPROt suggested the existence of an independent promoter downstream (220 kb) of the transcription start site of the full-length form of PTPRO (see Fig 2). Computational analysis provided a clue as to its probable location within intron 12 of the gene. To prove the existence of a distinct downstream promoter site, we selected a region ~1kb upstream of the potential +1 site of the PTPROt isoform that extends into the first exon (see Fig 2). Transient transfection studies using the promoter-luciferase reporter construct in several different cell lines demonstrated the presence of an active promoter in that region. This observation is consistent with the identification of an intronic promoter that drives the expression of PTP-oc (rabbit homolog of PTPROt).38 Interestingly, there is no CpG island within the PTPROt promoter. In fact, there are only five scattered CpG dinucleotides in this region. Can methylation of these specific CpGs alter expression of PTPROt in cells of lymphoid origin? Alternatively, can methylation of the CpG island located 220 kb upstream of the identified promoter regulate transcription of the truncated gene from the downstream +1 site? If methylation of the few CpGs in the intronic promoter is responsible for gene suppression, the molecular mechanism for the methylation-mediated alteration in PTPROt expression is likely to be different from that of PTPRO. In this context it is noteworthy that the methylation of a single CpG in NF1 and BRCA1 gene promoters can prevent interaction of a specific transcription factor, CREB, to the cognate element that contains the methylated CpG.39,40 Similarly, in mouse ribosomal RNA (rRNA) gene, methylation of cytosine in the only CpG located within the immediate promoter region was sufficient for inhibiting binding of the RNA polymerase- I-specific transcription factor UBF to its regulatory sequence (UCE) and subsequent inhibition of ribosomal gene transcription.41 It is, however, not known whether methylation of the few CpG residues in the PTPROt promoter also facilitates recruitment of methyl C-binding proteins (MBDs) that help recruit specific corepressors such as Sin3A, HDAC or HMT forming a large repressor complex on the promoter. Alternatively, methylation at the upstream CpG island results in positioning of inhibitory nucleosomes due to recruitment of MBD-corepressor complexes that deacetylate histone tails and methylate histone H3 at K9. K9 methyl histone H3 could then recruit HP1α protein to form heterochromatin structure that propagates through out the region to impede access of transactivators to cognate cis elements on both promoters.42,43 Interestingly, unlike the mouse and rat rRNA promoters that contain one and five CpGs, respectively, the corresponding human promoter exhibits a CpG island and an inverse correlation exists between methylation of the CpG island and human rRNA promoter activity.44 It would be of interest to determine whether multiple mechanisms operate in the methylation-mediated suppression of rRNA genes as well as PTPRO and PTPROt. Further study is needed to address these important issues.
Reactivation of genes silenced by promoter methylation: epigenetic therapy
A quarter century ago, Taylor and Jones45 demonstrated differentiation of mouse cells (3T3 and C3H/10T1/2/2CL8) into new mesenchymal phenotypes following treatment with the DNA hypomethylating agent, 5-AzaC or 5-AzadC. This observation set the stage for many studies in the ensuing decades that explored reactivation of suppressed genes in different cell types, particularly cancer cells (for reviews, see Baylin,46 Karpf and Jones,47 and Jain48). Reactivation of the suppressed genes can also occur in animals treated with 5-AzaC,33,49,50 which rules out the possibility of any artifacts produced in cell culture. Epigenetic modification alters the expression of a gene without affecting DNA sequence. Suppression of a gene by promoter methylation could, therefore, be alleviated either by removing the methyl groups from DNA or altering the histone tail modifications such as acetylation or methylation. It is, therefore, conceivable that demethylation and the consequent reactivation of these genes will be a rational approach to treat cancer. In preclinical studies, 5-AzadC appears to exhibit greater potency over 5-AzaC, with respect to inhibition of DNA methyltransferase and initiation of differentiation process51 making it a more ideal chemotherapeutic agent for epigenetic therapy.
In an effort to restore the activity of the epigenetically silenced genes, 5-AzaC or its congener 5-AzadC has been used for cancer therapy (for a review, see Christman52). Nearly 100 clinical trials with either 5-AzaC or 5-AzadC alone or in combination with a HDAC inhibitor have been reported in the National Cancer Institute database. While different types of malignancies have been treated with these agents, the most promising results were achieved in the therapy of hematopoietic neoplasms.53 The 5-Aza compounds probably demethylate the tumor suppressor genes leading to their reactivation and ultimately reduced tumor growth. In this context, the diminished expression of PTPROt, a potential tumor suppressor gene, in the peripheral blood lymphocytes from CLL patients suggests that PTPROt may indeed be a novel molecular target for CLL therapy.
How do the DNA hypomethylating agents function? The most straightforward mechanism involves conversion of the 5-Aza nucleosides to the nucleoside triphosphates, their incorporation into DNA (5-AzadC) or both RNA and DNA (5-AzaC) and subsequent alteration in protein synthesis. These drugs could also inhibit de novo thymidylate synthesis following their deamination into the respective uridines and conversion into uridine triphosphates. A noteworthy mechanism proposed more than two decades ago is based on their ability to inhibit DNA methylation following incorporation into DNA.54 Of the three functional DNA methyltransferases (DNMT 1, 3A, 3B) identified in mammals (for a review, see Jeltch55 Bestor56). DNMT3A and DNMT3B exhibit predominantly de novo methyltransferase activity whereas DNMT1 is exclusively involved in the methylation of hemimethylated DNA. Since DNMT1 requires hemimethylated DNA as the substrate, it is anticipated that the effect of 5-AzaC or 5-AzadC will not be fully exerted until the 5-AzadC-incorporated DNA undergoes replication. The stable and irreversible binding of DNMTs to 5-AzadC-incorporated DNA results in trapping and depletion of these DNMTs. Several recent observations suggest the covalent conjugate formation between DNMT and 5-AzaC-substituted DNA alone cannot explain many aspects of its action. First, DNMT activity decreases much faster than incorporation of 5-AzaC into DNA.51 Second, recent microarray analysis of human colon cancer cells (HCT116) revealed alterations in the gene expression profiles of 5-AzaC-treated cells irrespective of growth stage.57 It also appears that among three DNMTs only DNMT1 is the target of 5-AzaC. Since gene expression profile of the inhibitor-treated HCT116 cells is similar to that of DNMT1 null cells, DNMT1 is likely to be the preferential target of 5-azaC.57 This observation is consistent with our earlier report that DNMT1 is selectively and rapidly degraded in animal cells following exposure to 5-azaC.58 Recent study in our laboratory has shown that this selective degradation of DNMT1 is mediated by the proteasomal pathway (K Ghoshal and S Jacob, unpublished data), which offers an alternative mechanism of action for these potent anticancer drugs. The expression of less than 1% of the genes was elevated in response to 5-AzadC,59 which rules out the possibility that cellular transformation is initiated by increasing global hypomethylation by DNMT inhibitors.
Another promising therapeutic approach in the treatment of cancer involves agents that alter histone post-translational modifications. Histones are modified by acetylation, methylation, phosphorylation, ADP-ribosylation and ubiquitination. There is substantial evidence for a link between the characteristic modifications on histone tails and gene regulation through changes in chromatin configuration (“histone code”).42 Transcriptionally active chromatin is usually associated with hyperacetylated histones, while transcriptionally inactive heterochromatin is associated with hypoacetylated histones. 60 Unlike histone acetylation, histone methylation can either activate (K4 of histone H3) or repress (K9 of H3) gene transcription.61 Genetic evidence suggests a link between histone modification and DNA methylation, which has prompted epigenetic targeting to reexpress genes by increasing histone acetyl transferase enzyme activity and/or inhibiting histone deacetylase (HDAC) enzymes. Three major classes of HDAC enzymes exhibit specific function of modifying histone and nonhistone proteins.62
Many studies have shown that combined regimen of DNMT and HDAC inhibitors can regress or prevent tumor growth.58,63,64 A variety of clinically available agents that include depsipeptide, MS-275, SAHA and valproic acid inhibit HDAC, specifically Class I HDAC enzymes.65 The advantage of this regimen is that a relatively lower dose of DNMT inhibitors can be used, which may reduce their toxic effects. These agents can modify the expression of many cellular genes in a synergistic manner.58,66 We have elucidated the molecular mechanism for the activation of a specific promoter (metallothionein-I or MT-1 promoter) by combined treatment of mouse lymphosarcoma cells with 5-AzaC and a HDAC inhibitor such as trichostatin or clinically potent depsipeptide.58 In this study, the synergistic effects of the two classes of drugs in the MT-I re-expression was found to be due to promoter demethylation, altered association of different factors that leads to reorganization of the chromatin, and the resultant increase in accessibility of the promoter to the activated transcription factor MTF-1.58 It is noteworthy that the whole MT locus comprising of MT-IV, MT-III, MT-IIA and several isoforms of MT-I on human chromosome 16q13 are reactivated upon treatment with this drug,57 probably due to open chromatin structure.
Concluding remarks
We have presented evidence for the tumor suppressor, potential of a receptor-type protein tyrosine phosphatase, PTPRO. These include (a) its methylation-mediated silencing in primary human tumors; (b) inhibition of anchorage-independent growth and increased sensitivity of cells to apoptosis upon expression of PTPRO in nonexpressing cells; (c) inhibition of growth of PTPRO-expressing cells in nude mice; and (d) localization in the chromosomal region characterized by LOH. If PTPRO is an authentic tumor suppressor, knocking down PTPRO by stably transfecting the cells expressing this gene with RNAi is likely to induce cell transformation. Glepp1 (PTPRO) knockout mice do not exhibit developmental abnormalities in brain or hematopoietic system. The targeting approach used for knockout was, however, specific for the full-length isoform.67 Further, since these mice were not challenged with a carcinogen, it is difficult to assess the effect of the PTPRO deletion in tumorigenesis.
It is important to recognize that DNA hypomethylation also plays a significant role in tumor progression (for a recent review, see El-Osta17). In fact, DNA hypomethylation, an early event in tumorigeneisis, can predispose cells to chromosomal rearrangements and destabiliztion of chromosomal organization.17,68 In this context, it is noteworthy that methyl deficient diet can lead to both global DNA hypomethylation and localized or regional hypermethylation of genes such as tumor suppressor genes.33,69 The most accepted concept is to envisage early changes in the hypomethylation of genes such as oncogenes followed by hypermethylation of genes such as tumor suppressor genes during cell transformation or in tumorigenesis, both processes resulting in gene deregulation. It remains to be determined as to what fraction of tumor incidence is caused by alteration in genomic methylation.70
Identification of the key substrate(s) of PTPRO will be critical to the understanding of the role of this enzyme in cancer. As some of the PTKs, established oncogenes, are known to be the substrates of PTPs, dephosphorylation of these kinases by overexpression of PTPs could have important functional implications. Characterization of the substrates of PTPRO and its isoforms is, therefore, crucial to understand its role in cancer. Based on the methylation of PTPRO, and its suppression in the primary human tumors derived from three different tissues/cells (liver, lung and CLL), it is not unrealistic to expect similar downregulation of this gene in other tumors as well. Further study involving both liquid and solid tumors will resolve this issue. If the reduced expression of PTPRO occurs widely in many primary human tumors besides hepatocellular and lung tumors and CLL, methylation of PTPRO could be a prognostic marker of these diseases. Accordingly, one can anticipate significant methylation of PTPRO in tumors and negligible methylation as tumor regresses. Further, suppression of PTPRO expression due to methylation will provide an important molecular target for epigenetic therapy of cancer. In this context, the methylation of other PTPs in CLL is of interest. It is important to determine whether these PTPs are also downregulated in cancer, and whether such modification occurs in preneoplastic stage.
Finally, reactivation of the silenced genes deserves comment. Although considerable efforts have been expended on the identification of novel HDAC inhibitors, the DNA hypomethylating agents frequently used in therapy are those discovered more than two decades ago. It should also be recognized that HDAC inhibitors alone do not reactivate the genes suppressed by methylation. While combining DNMT inhibitors with an HDAC inhibitor can minimize the clinically effective dose, it is crucial to determine the right combination doses for therapy of each type of cancer. It is, therefore, desirable to explore other less toxic and more potent inhibitors of DNA methyltrasferases or drugs that can interfere with other factors in the DNA methylation machinery.
Acknowledgments
We thank Drs Kalpana Ghoshal and Sarmila Majumder for useful comments. We regret that many excellent papers relevant to this review could not be cited due to space limitation. The work in the authors’ laboratory was supported by grants (CA 81024 and CA 86978) from the National Cancer Institute and ES 10874 from the National Institute of Environmental Health Science.
References
- 1.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 4.Wade PA, Gegonne A, Jones PL, et al. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23:62–66. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 5.Wade PA, Jones PL, Vermaak D, et al. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr Biol. 1998;8:843–846. doi: 10.1016/s0960-9822(98)70328-8. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Ng HH, Erdjument-Bromage H, et al. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakai H, Urano T, Ookata K, et al. MBD3 and HDAC1, two components of the NuRD complex, are localized at Aurora-A-positive centrosomes in M phase. J Biol Chem. 2002;277:48714–48723. doi: 10.1074/jbc.M208461200. [DOI] [PubMed] [Google Scholar]
- 8.Costello JF, Fruhwald MC, Smiraglia DJ, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–138. doi: 10.1038/72785. [see comments] [DOI] [PubMed] [Google Scholar]
- 9.Baylin SB, Herman JG, Graff JR, et al. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- 10.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 11.Sakai T, Toguchida J, Ohtani N, et al. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am J Hum Genet. 1991;48:880–888. [PMC free article] [PubMed] [Google Scholar]
- 12.Myohanen SK, Baylin SB, Herman JG. Hypermethylation can selectively silence individual p16ink4A alleles in neoplasia. Cancer Res. 1998;58:591–593. [PubMed] [Google Scholar]
- 13.Batova A, Diccianni MB, Yu JC, et al. Frequent and selective methylation of p15 and deletion of both p15 and p16 in T-cell acute lymphoblastic leukemia. Cancer Res. 1997;57:832–836. [PubMed] [Google Scholar]
- 14.Esteller M, Fraga MF, Guo M, et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet. 2001;10:3001–3007. doi: 10.1093/hmg/10.26.3001. [DOI] [PubMed] [Google Scholar]
- 15.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [see comment] [DOI] [PubMed] [Google Scholar]
- 16.Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- 17.El-Osta A. The rise and fall of genomic methylation in cancer. Leukemia. 2004;18:233–237. doi: 10.1038/sj.leu.2403218. [DOI] [PubMed] [Google Scholar]
- 18.Hunter T. Signaling — 2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 19.Fischer EH. Cell signaling by protein tyrosine phosphorylation. Adv Enzyme Regul. 1999;39:359–369. doi: 10.1016/s0065-2571(98)00014-4. [DOI] [PubMed] [Google Scholar]
- 20.Maroun CR, Naujokas MA, Holgado-Madruga M, et al. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol. 2000;20:8513–8525. doi: 10.1128/mcb.20.22.8513-8525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 22.Palka HL, Park M, Tonks NK. Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J Biol Chem. 2003;278:5728–5735. doi: 10.1074/jbc.M210656200. [DOI] [PubMed] [Google Scholar]
- 23.Hermiston ML, Xu Z, Majeti R, et al. Reciprocal regulation of lymphocyte activation by tyrosine kinases and phosphatases. J Clin Invest. 2002;109:9–14. doi: 10.1172/JCI14794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S, Radha V, Sudhakar C, et al. A nuclear protein tyrosine phosphatase activates p53 and induces caspase-1-dependent apoptosis. FEBS Lett. 2002;532:61–66. doi: 10.1016/s0014-5793(02)03628-1. [DOI] [PubMed] [Google Scholar]
- 25.Salmeen A, Andersen JN, Myers MP, et al. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 26.Alonso A, Sasin J, Bottini N, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 27.Panagopoulos I, Pandis N, Thelin S, et al. The FHIT and PTPRG genes are deleted in benign proliferative breast disease associated with familial breast cancer and cytogenetic rearrangements of chromosome band 3p14. Cancer Res. 1996;56:4871–4875. [PubMed] [Google Scholar]
- 28.Zhang Y, Siebert R, Matthiesen P, et al. Cytogenetical assignment and physical mapping of the human R-PTP-kappa gene (PTPRK) to the putative tumor suppressor gene region 6q22.2–q22.3. Genomics. 1998;51:309–311. doi: 10.1006/geno.1998.5323. [DOI] [PubMed] [Google Scholar]
- 29.Ruivenkamp CA, van Wezel T, Zanon C, et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31:295–300. doi: 10.1038/ng903. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Shen D, Parsons DW, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 31.Andersen JN, Jansen PG, Echwald SM, et al. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- 32.Ardini E, Agresti R, Tagliabue E, et al. Expression of protein tyrosine phosphatase alpha (RPTPalpha) in human breast cancer correlates with low tumor grade, and inhibits tumor cell growth in vitro and in vivo. Oncogene. 2000;19:4979–4987. doi: 10.1038/sj.onc.1203869. [DOI] [PubMed] [Google Scholar]
- 33.Motiwala T, Ghoshal K, Das A, et al. Suppression of the protein tyrosine phosphatase receptor type O gene (PTPRO) by methylation in hepatocellular carcinomas. Oncogene. 2003;22:6319–6331. doi: 10.1038/sj.onc.1206750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Motiwala T, Kutay H, Ghoshal K, et al. Protein tyrosine phosphatase receptor-type O (PTPRO) exhibits characteristics of a candidate tumor suppressor in human lung cancer. Proc Natl Acad Sci USA. 2004;101:13844–13849. doi: 10.1073/pnas.0405451101. Epub 12004 Sep 13848. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Imreh S, Klein G, Zabarovsky ER. Search for unknown tumor-antagonizing genes. Genes, Chromosomes Cancer. 2003;38:307–321. doi: 10.1002/gcc.10271. [DOI] [PubMed] [Google Scholar]
- 36.Mori Y, Yin J, Sato F, et al. Identification of genes uniquely involved in frequent microsatellite instability colon carcinogenesis by expression profiling combined with epigenetic scanning. Cancer Res. 2004;64:2434–2438. doi: 10.1158/0008-5472.can-03-3508. [DOI] [PubMed] [Google Scholar]
- 37.Oka T, Ouchida M, Koyama M, et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002;62:6390–6394. [PubMed] [Google Scholar]
- 38.Amoui M, Baylink DJ, Tillman JB, et al. Expression of a structurally unique osteoclastic protein-tyrosine phosphatase is driven by an alternative intronic, cell type-specific promoter. J Biol Chem. 2003;278:44273–44280. doi: 10.1074/jbc.M303933200. [DOI] [PubMed] [Google Scholar]
- 39.Mancini DN, Singh SM, Archer TK, et al. Site-specific DNA methylation in the neurofibromatosis (NF1) promoter interferes with binding of CREB and SP1 transcription factors. Oncogene. 1999;18:4108–4119. doi: 10.1038/sj.onc.1202764. [DOI] [PubMed] [Google Scholar]
- 40.DiNardo DN, Butcher DT, Robinson DP, et al. Functional analysis of CpG methylation in the BRCA1 promoter region. Oncogene. 2001;20:5331–5340. doi: 10.1038/sj.onc.1204697. [DOI] [PubMed] [Google Scholar]
- 41.Santoro R, Grummt I. Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol Cell. 2001;8:719–725. doi: 10.1016/s1097-2765(01)00317-3. [DOI] [PubMed] [Google Scholar]
- 42.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 43.Kass SU, Wolffe AP. DNA methylation, nucleosomes and the inheritance of chromatin structure and function. Novartis Found Symp. 1998;214:22–35. doi: 10.1002/9780470515501.ch3. discussion 36–50. [DOI] [PubMed] [Google Scholar]
- 44.Ghoshal K, Majumder S, Datta J, et al. Role of human ribosomal RNA (rRNA) promoter methylation and of methyl-CpG-binding protein MBD2 in the suppression of rRNA gene expression. J Biol Chem. 2004;279:6783–6793. doi: 10.1074/jbc.M309393200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor SM, Jones PA. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell. 1979;17:771–779. doi: 10.1016/0092-8674(79)90317-9. [DOI] [PubMed] [Google Scholar]
- 46.Baylin SB. Reversal of gene silencing as a therapeutic target for cancer — roles for DNA methylation and its inter-digitation with chromatin. Novartis Found Symp. 2004;259:226–233. discussion 234–227. [PubMed] [Google Scholar]
- 47.Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene. 2002;21:5496–5503. doi: 10.1038/sj.onc.1205602. [DOI] [PubMed] [Google Scholar]
- 48.Jain PK. Epigenetics: the role of methylation in the mechanism of action of tumor suppressor genes. Ann NY Acad Sci. 2003;983:71–83. doi: 10.1111/j.1749-6632.2003.tb05963.x. [DOI] [PubMed] [Google Scholar]
- 49.Majumder S, Ghoshal K, Datta J, et al. Role of de novo DNA methyltransferases and methyl CpG-binding proteins in gene silencing in a rat hepatoma. J Biol Chem. 2002;277:16048–16058. doi: 10.1074/jbc.M111662200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Ghoshal K, Majumder S, Li Z, et al. Suppression of metallothionein gene expression in a rat hepatoma because of promoter-specific DNA methylation. J Biol Chem. 2000;275:539–547. doi: 10.1074/jbc.275.1.539. [DOI] [PubMed] [Google Scholar]
- 51.Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2′-deoxycytidine. J Biolog Chem. 1982;257:2041–2048. [PubMed] [Google Scholar]
- 52.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 53.Claus R, Lubbert M. Epigenetic targets in hematopoietic malignancies. Oncogene. 2003;22:6489–6496. doi: 10.1038/sj.onc.1206814. [DOI] [PubMed] [Google Scholar]
- 54.Jones PA, Taylor SM, Wilson VL. Inhibition of DNA methylation by 5-azacytidine. Rec Results Cancer Res. 1983;84:202–211. doi: 10.1007/978-3-642-81947-6_15. [DOI] [PubMed] [Google Scholar]
- 55.Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [erratum appears in Chembiochem 2002 May 3;3(5):382] [DOI] [PubMed] [Google Scholar]
- 56.Bestor TH. The DNA methyltransferases of mammals. Humn Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 57.Gius D, Cui H, Bradbury CM, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell. 2004;6:361–371. doi: 10.1016/j.ccr.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 58.Ghoshal K, Datta J, Majumder S, et al. Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure. Mol Cell Biol. 2002;22:8302–8319. doi: 10.1128/MCB.22.23.8302-8319.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki H, Gabrielson E, Chen W, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31:141–149. doi: 10.1038/ng892. [see comment] [DOI] [PubMed] [Google Scholar]
- 60.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 61.Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14:286–298. doi: 10.1016/s0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 62.de Ruijter AJ, van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Belinsky SA, Klinge DM, Stidley CA, et al. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 2003;63:7089–7093. [PubMed] [Google Scholar]
- 64.Shaker S, Bernstein M, Momparler RL. Antineoplastic action of 5-aza-2′-deoxycytidine (Dacogen) and depsipeptide on Raji lymphoma cells. Oncol Rep. 2004;11:1253–1256. [PubMed] [Google Scholar]
- 65.Kouraklis G, Theocharis S. Histone deacetylase inhibitors and anticancer therapy. Curr Med Chem — Anti-Cancer Agents. 2002;2:477–484. doi: 10.2174/1568011023353921. [DOI] [PubMed] [Google Scholar]
- 66.Cameron EE, Bachman KE, Myohanen S, et al. Synergy of demethylation and histone deacetylase inhibition in the reexpression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 67.Wharram BL, Goyal M, Gillespie PJ, et al. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J Clin Invest. 2000;106:1281–1290. doi: 10.1172/JCI7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu GL, Bestor TH, Bourc”his D, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 69.Pogribny IP, Miller BJ, James SJ. Alterations in hepatic p53 gene methylation patterns during tumor progression with folate/methyl deficiency in the rat. Cancer Lett. 1997;115:31–38. doi: 10.1016/s0304-3835(97)04708-3. [DOI] [PubMed] [Google Scholar]
- 70.Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell. 2002;1:299–305. doi: 10.1016/s1535-6108(02)00061-2. [DOI] [PubMed] [Google Scholar]