

Abstract
In response to a DNA double-strand break (DSB), cells undergo a transient cell cycle arrest prior to mitosis until the break is repaired. In budding yeast (Saccharomyces cerevisiae), the DNA damage checkpoint is regulated by a signaling cascade of protein kinases, including Mec1 and Rad53. When DSB repair is complete, cells resume cell cycle progression (a process called “recovery”) by turning off the checkpoint. Recovery involves two members of the protein phosphatase 2C (PP2C) family, Ptc2 and Ptc3, as well as the protein phosphatase 4 (PP4) enzyme, Pph3. Here, we demonstrate a new function of these three phosphatases in DSB repair. Cells lacking all three phosphatases Pph3, Ptc2, and Ptc3 exhibit synergistic sensitivities to the DNA-damaging agents camptothecin and methyl methanesulfonate, as well as hydroxyurea but not to UV light. Moreover, the simultaneous absence of Pph3, Ptc2, and Ptc3 results in defects in completing DSB repair, whereas neither single nor double deletion of the phosphatases causes a repair defect. Specifically, cells lacking all three phosphatases are defective in the repair-mediated DNA synthesis. Interestingly, the repair defect caused by the triple deletion of Pph3, Ptc2, and Ptc3 is most prominent when a DSB is slowly repaired and the DNA damage checkpoint is fully activated.
DNA double-strand breaks (DSBs) trigger a protein kinase cascade to activate the DNA damage checkpoint. By arresting the cell cycle at the G2/M phase, the checkpoint provides enough time for cells to repair DSBs before the cells enter mitosis. In response to DNA damage in budding yeast (Saccharomyces cerevisiae), Mec1 and Tel1 (yeast orthologues of mammalian ATR and ATM, respectively) are initially activated. Mec1 is more critical for the activation of the DNA damage checkpoint (5). Mec1 phosphorylates the checkpoint signal transducer kinases, Chk1 and Rad53 (the yeast homologue of mammalian Chk2), as well as the checkpoint mediator protein, Rad9 (2, 23). Following the induction of DSBs, Rad9 accumulates in the region surrounding the breaks through its interactions with phosphorylated histone H2A on serine 129 (hereafter termed γ-H2AX) as well as methylated histone H3 on lysine 79 (H3-K79me) (4, 34). The Mec1-dependent phosphorylation of Rad53 leads to autophosphorylation of Rad53 (21, 27). The hyperphosphorylation of Rad53 that modulates downstream effectors for cell cycle regulation is maintained during the checkpoint (21).
As DNA damage is repaired, cells deactivate the checkpoint to reenter the cell cycle. This process is called “recovery.” In addition, cells can resume the cell cycle even in the presence of an unrepaired DNA break, a process referred to as “adaptation” (13, 29). Both adaptation and recovery are accompanied by the disappearance of hyperphosphorylated Rad53 (14, 21, 31). Turning off the DNA damage checkpoint requires dephosphorylation of Rad53 and probably other substrates. Ptc2 and Ptc3 were the first identified protein phosphatases that are required for deactivation of the DNA damage checkpoint (15). Ptc2 and Ptc3 are protein phosphatase 2C (PP2C) family members (17). In the absence of Ptc2 and Ptc3, the hyperphosphorylated Rad53 persists even after a DSB is repaired (3, 15). Direct dephosphorylation of Rad53 by Ptc2 was demonstrated with an immunopurified Ptc2 in vitro (15). The FHA (forkhead-associated) domain in Ptc2 can be phosphorylated by casein kinase II (CKII), which is involved in DNA damage adaptation (3, 29). When the CKII phosphorylation site of Ptc2 is mutated, Rad53 dephosphorylation is impaired (3). These results suggest a model in which Ptc2 is phosphorylated by CKII and thereby dephosphorylates Rad53.
Another protein phosphatase, Pph3, has been shown to play a role in the checkpoint recovery subsequent to DSB repair (10, 18). Pph3 and its two interacting proteins, Psy2 and Psy4, form a complex that resembles the mammalian phosphoprotein phosphatase 4 (PP4) family (6). In the absence of Pph3, recovery of the cells from the checkpoint-mediated arrest is delayed (10). Pph3 regulates the dephosphorylation of γ-H2AX in vivo and in vitro (10). Thus, it has been proposed that Pph3 mediates the checkpoint inactivation by dephosphorylating γ-H2AX. A recent study by O'Neill et al. showed that, after activation of the DNA replication checkpoint (different from that induced by a DSB), Psy2 directly binds to Rad53 and the complex comprising Pph3 and Psy2, but not Psy4, dephosphorylates Rad53 (18). They argued the complex containing Pph3 can modulate its substrate specificity by changing the composition of the complex. This study implies the possibility that there could be multiple substrates of Pph3.
To repair a DSB by homologous recombination (HR), cells can utilize different HR-mediated repair pathways, which include gene conversion (GC) and single-strand annealing (SSA). When both ends of broken DNA share homology with template sequences either within the same chromosome or within a different chromosome, the DSB is repaired by GC (Fig. 1A), which is short-patch repair transferring genetic information from the homologous template to the damaged site, usually by a mechanism termed “synthesis-dependent strand annealing” (19). On the other hand, SSA occurs when a DSB occurs between two flanking homologous sequences (Fig. 1B). In the SSA pathway, the 5′-to-3′ resection activity beginning at the broken end continues until the complementary sequences are exposed, leading to annealing of the homologous sequences. After nonhomologous (NH) tails have been removed from the annealed DNA, the resulting gaps are filled in by DNA synthesis and ligation. Consequently, intervening sequences between two repeats and a single copy of repeated sequences are deleted in SSA-mediated DSB (19). By utilizing budding yeast mating-type systems with an inducible HO endonuclease for the formation of a DSB, the pathways have been extensively studied (25). Different HR processes display different kinetics of repair, with intrachromosomal MAT switching (Fig. 1A, panel a) requiring about 1.5 to 2 h to complete, whereas interchromosomal ectopic gene conversion involving the same MAT locus (Fig. 1A, panel b) is first seen in 3 h after the induction of a DSB (10). On the other hand, in the specific SSA event used in these studies, where one of the flanking homologies is about 30 kb distal to a homologous sequence (Fig. 1B), repair is completed only in 6 h (9, 31).
FIG. 1.

Strains used to study the roles of Pph3, Ptc2, and Ptc3 in HR-mediated DSB repair. (A, panel a) Strain YJK139 contains the endogenous HMLα-inc locus but lacks the HMRa locus. An HO-induced DSB at MAT on chromsome 3 (Chr3) is repaired by intrachromosomal gene conversion (Intra-GC) using HMLα-inc on the same chromosome as a donor. The centromere is marked as a black dot. (b) Both endogenous HML and HMR loci are deleted in strain YJK17. An HO-induced DSB at MAT on Chr3 is repaired by interchromosomal gene conversion by copying the MATa-inc sequence on chromosome 5 (Chr5). (B) Strain YMV80 contains an HO cleavage site within the LEU2 sequence (leu2-cs) on Chr3. The 3′ partial sequence of LEU2 (U2) is located 30 kb apart from the leu2-cs. An HO-induced DSB on the leu2-cs is repaired by single-strand annealing (SSA) using the U2 sequence as a homologous donor. The centromere is marked as a black dot.
In keeping with these kinetic differences between the three assays, there are differences in the activation of the DNA damage checkpoint, as monitored by cell cycle arrest and by the phosphorylation of Rad53. Notably, intrachromosomal MAT switching provokes neither cell cycle delay nor Rad53 phosphorylation (21). Cells with these different systems to repair a DSB also exhibit significant differences in the failure to resume cell cycle progression after repair is complete. Cells lacking both Ptc2 and Ptc3 are severely impaired in recovery, using the SSA system, but they are not blocked in either ectopic or the intrachromsomal GC events (15). Similarly, deletion of SRS2, which encodes a helicase involved in the HR-mediated DSB repair, prevents recovery in both the SSA and ectopic repair processes but has a smaller effect on intrachromosomal MAT switching (31; W. M. Hicks., L. Kapulsky, and J. E. Haber, unpublished). Finally, cells lacking histone H3 chaperones Asf1 and CAF-1 show a severe recovery defect in SSA and a significant decrease in resuming mitosis in the ectopic system, but there is no effect on intrachromosomal GC (1, 11).
Here, we have determined a new role for three different phosphatases, Pph3, Ptc2, and Ptc3, in the DNA damage response by utilizing the different HR-mediated DSB repair systems described above. We show that the triple-deletion pph3Δ ptc2Δ ptc3Δ cells have striking defects in repairing an HO endonuclease-induced DSB, particularly in DSB repair-mediated DNA synthesis, while neither cells with the pph3Δ deletion alone nor cells with the ptc2Δ ptc3Δ double deletion have a defect in DSB repair itself. Furthermore, the defects are especially evident in situations where DSB repair is slow and the DNA damage checkpoint is strongly activated.
MATERIALS AND METHODS
Yeast strains.
All strains are listed in Table 1. YMV80 and its derivatives lack HO cut sites within MAT, HMLα, and HMRa on chromosome 3 (Chr3) but have a cut site within a centromere-proximal LEU2 gene (leu2-cs) on Chr3 (31). The homologous LEU2 sequences providing a donor region to repair an HO-induced DSB at leu2-cs lie 30 kb distal to the centromere. Cells isogenic to the YJK17 strain lack endogenous HMLα or HMRa on Chr3. Rather, the donor sequence to repair an HO-induced DSB at the MAT locus is an ectopic MAT locus containing an uncuttable HO site (MATa-inc) on chromosome 5 (10). YJK139 lacks endogenous HMRa, but contains HMLα-inc, which is the only homologous sequence to MAT.
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source or reference | Representative figure(s) in this study |
|---|---|---|---|
| YJK139 | MATα hoΔ HMLα-inc hmrΔ::ADE1 ade1-100 leu2,3-112 lys5 trp1::hisG ura3-52 ade3::GAL::HO | This study | 2D and 6B, C, and D |
| WH115 | YJK139 isogenic; pph3Δ::KanMX ptc2Δ::URA3MX ptc3Δ::NatMX | This study | 6B, C, and D |
| YJK17 | MATα hoΔ hmlΔ::ADE1 hmrΔ::ADE1 arg5,6Δ::HPH::MATa-inc ade1-100 leu2,3-112 lys5 trp::hisG ura3-52 ade3::GAL::HO | 10 | 2A, B, and C; 3B; and 5B, C, E, F, and G |
| YJK26 | YJK17 isogenic; pph3Δ::KanMX | 10 | 2A, B, C, and D; 3B; and 5B and C |
| YJK24 | YJK17 isogenic; ptc2Δ::KanMX ptc3Δ::NatMX | This study | 2A, B, and C; 3B; and 5B and C |
| YJK70 | YJK17 isogenic; pph3Δ::KanMX ptc2Δ::KanMX ptc3Δ::NatMX | This study | 2A, B, and C; 3B; and 5B, C, E, F, and G |
| YMV80 | hmlΔ::ADE mataΔ::hisG hmrΔ::ADE1 leu2-cs ade3::GAL::HO ade1-100 lys5 ura3-52 | 31 | 3A and 4B, C, D, and E |
| JY544 | YMV80 isogenic; pph3Δ::KanMX ptc2Δ::KanMX ptc3Δ::NatMX | This study | 3A and 4B, C, D, and E |
| tNS2368 | YMV80 isogenic; rad1Δ::KanMX | This study | 3C |
| R763 | MATabar1Δ::ADE3 hoΔ hmlΔ::ADE1 hmrΔ::ADE1 ade1-100 leu2,3-112 lys5 trp1::hisG ura3-52 ade3::GAL::HO hta1-S129A hta2-S129A | 10 | 2D |
| YJK35 | R763 isogenic; hta1-S129A hta2-S129A pph3Δ::KanMX | This study | 2D |
Western analysis.
Cell extracts were prepared by trichloroacetic acid (TCA) and then subjected to Western blot analysis. γ-H2AX was detected by polyclonal anti yeast γ-H2AX antibody (1:10,000) (a gift from C. Redon and W. Bonner, NIH). As a loading control, carboxypeptidase Y (CPY) was visualized with the monoclosnal anti-CPY antibody (1:10,000; Invitrogen).
ChIP assay.
Chromatin immunoprecipitation (ChIP) of Rad51 was carried out as previously described (24), using an antibody against yeast Rad51 (a gift from P. Sung, Yale University). The Rad51 ChIP signal was measured by quantitative PCR with multiple primer pairs specific to the chromatin regions surrounding the HO-induced DSB at the MAT locus on Chr3 as well as the region specific to the donor on Chr5. (Sequences of the oligonucleotides used in this study will be released upon request.) PCR was performed with a real-time PCR machine (MJ Research). With each primer pair, the number of amplification cycles that were required for the sample's response curve to reach a particular threshold fluorescence signal level was measured. The amount of chromatin-immunoprecipitated template DNA for the reaction was then estimated from a standard curve based on serial dilution of a standard PCR product. The ChIP signal at each locus was normalized to that at CEN8 in chromosome 8, where DSB was not induced. The fold increase of Rad51 was calculated by dividing the ChIP signal at different time points after HO induction (TX) by that without HO induction (T0).
RESULTS
In the absence of Pph3, Ptc2, and Ptc3 together, cells display enhanced sensitivity to DNA-damaging agents.
To investigate involvement of the protein phosphatases Pph3, Ptc2, and Ptc3 in the DNA damage response, we constructed mutant cells lacking the phosphatase genes in different combinations: cells with the pph3Δ mutation alone, as well as those with the ptc2Δ ptc3Δ double and pph3Δ ptc2Δ ptc3Δ triple mutations. None of the mutant cells showed a growth defect in the absence of DNA damage (Fig. 2A); however, treatment with DNA-damaging agents affected the viability of mutant cells. Cells lacking Pph3 alone were hypersensitive to methyl methanesulfonate (MMS), and ptc2Δ ptc3Δ cells showed ∼10-fold reduced viability in the presence of MMS, compared to wild-type (WT) cells. Triple deletion of PPH3, PTC2, and PTC3 synergistically reduced cell viability (Fig. 2A). A striking defect in the viability of pph3Δ ptc2Δ ptc3Δ cells was also observed in the presence of hydroxyurea (HU) or camptothecin (CPT), while neither pph3Δ nor ptc2Δ ptc3Δ cells showed sensitivity to HU and CPT (Fig. 2A). These results suggest that Pph3, Ptc2, and Ptc3 function redundantly in the pathway that is activated in response to DNA damage or stalled replication forks. On the other hand, the sensitivity of pph3Δ ptc2Δ ptc3Δ cells to UV treatment was moderate (Fig. 2A). This result implies that the redundant function of Pph3, Ptc2, and Ptc3 is more important for the DSB-inducible response than for the pathways activated by different kinds of DNA damage. Similar results were obtained when cells were treated with more acute doses of MMS for 1 h and then plated on MMS-free medium; here too, the triple mutant was hypersensitive to MMS (Fig. 2B).
FIG. 2.

pph3Δ ptc2Δ ptc3Δ cells are synergistically sensitive to DNA-damaging agents. (A) Wild-type and phosphatase deletion mutant cells were grown to the saturated phase in liquid YEP-dextrose medium. Ten-fold serial dilutions of the cultures were spotted on YEP-dextrose plates with or without DNA-damaging agents (0.02% methyl methanesulfonate [MMS], 50 mM hydroxyurea [HU], 1 μg/ml camptothecin [CPT]). To treat cells with UV, 10-fold serially diluted cells were spotted on YEP-dextrose plates, followed by irradiation with 50 J/m2 UV. The photographs were taken 3 days after spotting. (B) After growth of wild-type or mutant cells to exponential phase in liquid culture, 0.05%, 0.1%, 0.15%, and 0.2% MMS were added into the culture. One hour after MMS treatment, the cells were washed with MMS-free medium three times and then plated on YEP-dextrose plates. Viable colonies were counted 3 days later. Percentage of viability was calculated by dividing the number of colonies from the MMS-treated culture by that from the untreated culture. Error bars were obtained from three different experiments. (C) Wild-type and phosphatase deletion mutant cells were grown in liquid YEP-dextrose medium to exponential phase and treated with or without 0.2% MMS for 1 h. Whole-cell extracts were subjected to Western analysis with either anti-γ-H2AX or anti-carboxypeptidase Y (CPY) antibody. (D) PPH3 is deleted either in wild-type or in h2a-S129A strains. The sensitivities to MMS were assessed by spotting 10-fold serially diluted cultures onto YEP-dextrose plates with or without 0.02% MMS. The photographs were taken 3 days after spotting.
γ-H2AX is a known target of the phosphatase activity of Pph3 (10). We examined whether deletion of both PTC2 and PTC3 would also affect the phosphorylation status of γ-H2AX by Western analysis using anti-γ-H2AX antibody. Consistent with a previous report (10), pph3Δ cells had an enhanced level of γ-H2AX compared to wild-type cells in the absence of DNA-damaging agents (Fig. 2C). In addition, MMS treatment induced more γ-H2AX in pph3Δ cells. On the other hand, ptc2Δ ptc3Δ cells did not show a high level of γ-H2AX without DNA-damaging agents. However, in response to MMS treatment, more γ-H2AX was induced in ptc2Δ ptc3Δ cells than in wild-type cells. These results suggest that Ptc2 and Ptc3 play roles in the regulation of γ-H2AX only when the damage checkpoint is activated by induced DNA damage. A triple deletion of PPH3, PTC2, and PTC3 did not increase the level of γ-H2AX more than that seen in the pph3Δ mutant either in the absence or in the presence of MMS. These results suggest that Pph3 is the primary phosphatase for γ-H2AX and that when the DNA damage checkpoint is turned on, Ptc2 and Ptc3 work in the same pathway with Pph3 to regulate γ-H2AX. However, these results do not explain why DSB repair is synergistically impaired by deletion of PPH3, PTC2, and PTC3 together because the level of γ-H2AX in the pph3Δ mutant is not enhanced by deleting PTC2 and PTC3. We note that the sensitivity of pph3Δ cells to MMS was more striking than that of h2a-S129A mutant cells, in which formation of γ-H2AX is absent. In addition, deletion of PPH3 in the h2a-S129A mutant cells still resulted in the sensitivity of the cells to MMS (Fig. 2D). These results imply that Pph3 is involved in something more than γ-H2AX dephosphorylation during the DNA damage response.
The sensitivity of pph3Δ ptc2Δ ptc3Δ cells to a single DSB depends on the extent of checkpoint-mediated arrest.
Cells containing a defect either in DSB repair or in the DNA damage checkpoint can fail to proliferate after the induction of DNA damage. Since Pph3, Ptc2, and Ptc3 are involved in the checkpoint recovery, the synergistically reduced viability of pph3Δ ptc2Δ ptc3Δ cells in response to DNA damage might be due to the complete failure of inactivation of the DNA damage checkpoint. Alternatively, it is also possible that the combined deletion of PPH3, PTC2, and PTC3 somehow impairs DSB repair, whereas the repair is still functional either in pph3Δ cells or in ptc2Δ ptc3Δ cells. To determine which specific process in the DSB response pathway was defective in pph3Δ ptc2Δ ptc3Δ cells, we examined DSB repair by utilizing the HO-induced DSB strains in three different situations: intrachromosomal gene conversion (GC) during MAT switching (YJK139) (Fig. 1A, panel a), ectopic GC-mediated gene switching (YJK17) (Fig. 1A, panel b), and an extended single-strand annealing (SSA) process (YMV80) (Fig. 1B). We first tested the viability of the pph3Δ ptc2Δ ptc3Δ mutant in all three systems and found that there was a difference in the severity of the defect that corresponded to the degree of the checkpoint responses. The most severe effect was seen in strain YMV80, in which a galactose-induced HO endonuclease cleavage on chromosome 3 (Chr3) is repaired by SSA only after a slow 5′-to-3′ resection of the DSB ends allows homologous sequences 30 kb apart to anneal (31). Approximately 72% of wild-type cells and 76% of pph3Δ cells formed colonies on yeast extract-peptone (YEP)-galactose plates, where HO is induced, compared to cells plated on YEP-dextrose (Fig. 3A). In contrast, deletion of both PTC2 and PTC3 reduced viability to only 15%, as previously reported (15). Strikingly, the triple-deletion pph3Δ ptc2Δ ptc3Δ mutant showed only 1% of viability on galactose plates, which is significantly different from the viability of ptc2Δ ptc3Δ cells (P = 0.0221 by Student's t test).
FIG. 3.
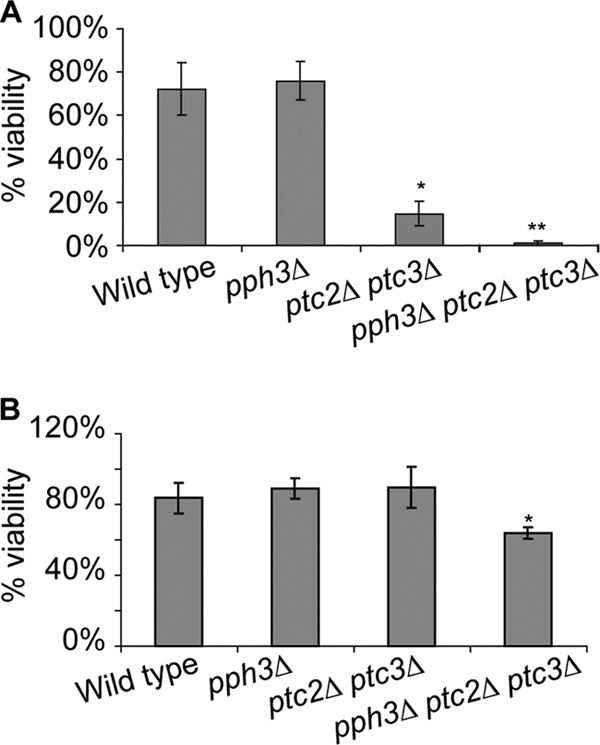
Triple deletion of PPH3, PTC2, and PTC3 reduces the viability of cells after the induction of a DSB by HO endonuclease. (A) Viabilities of the YMV80 wild-type strain and its derivatives lacking Pph3, Ptc2, or Pph3 on galactose plates. Wild-type and mutant cells were grown in liquid YEP-lactate culture to exponential phase, and then cells from each culture were plated either on YEP-dextrose or YEP-galactose plates. Three days after plating, the number of colonies on each plate was counted. The percentages of viability of the wild type and each mutant were calculated by dividing the number of colonies on the galactose plates by that on the dextrose plates. The graph shows an average of three different experiments. *, result significantly different from that for wild-type cells; **, significantly different viability compared to that of ptc2Δ ptc3Δ cells. (B) Percentages of viability of the YJK17 wild-type strain and its derivatives lacking Pph3, Ptc2, or Pph3 on galactose plates. Experiments were performed as described for panel A. *, result significantly different from that for wild-type cells.
Ectopic GC was studied in strain YJK17. A DSB was generated at the MATα locus on Chr3 by the galactose-inducible HO, and the break was repaired by gene conversion (GC)-mediated HR using an ectopic MATa-inc locus on chromosome 5 (Chr5) as a homologous donor (Fig. 1A, panel b) (10). The MATa-inc sequence cannot be cleaved by HO endonuclease. Wild-type cells repair the DSB efficiently, so that 84% of the wild-type cells survived when plated on YEP-galactose pates (where HO is induced) compared to those plated on YEP-dextrose. The viabilities of the pph3Δ single mutant and ptc2Δ ptc3Δ double mutant were 89% and 90%, respectively, which were not significantly different from that of wild-type cells (P = 0.33 and P = 0.41 by Student's t test) (Fig. 3B). On the other hand, 64% of pph3Δ ptc2Δ ptc3Δ cells were viable on galactose plates, which was lower than the other mutant combinations as well as wild-type cells. This reduction is statistically significant compared to wild-type cells as well as the other mutant cells (P = 0.0062 by Student's t test).
Intrachromosomal GC was monitored in strain YJK139 in which the HO-induced DSB at MATa on Chr3 is repaired by HMLα-inc, located 200 kb away on the same chromosome. Here, the viability after HO induction was greater than 90% and was not different in the pph3Δ ptc2Δ ptc3Δ mutant, which was derived from YJK139 (data not shown). These results indicate that the simultaneous absence of PPH3, PTC2, and PTC3 leads to a defect in cell viability after the induction of a DSB only when the DSB-mediated checkpoint is activated.
Triple-phosphatase-deletion pph3Δ ptc2Δ ptc3Δ cells are not just defective in recovery but are also defective in completing SSA-mediated DSB repair.
To determine which specific process in the DSB response pathway was defective in pph3Δ ptc2Δ ptc3Δ cells, we first monitored the SSA-mediated DSB repair in the YMV80 derivatives by Southern analysis using a probe specific to the region adjacent to the DSB site (Fig. 4A). The formation of an HO-induced DSB and the disappearance of the HO-cut band were efficient both in wild-type and in pph3Δ ptc2Δ ptc3Δ cells (Fig. 4B). The repair product appeared as early as 8 h after the induction of a DSB in wild-type cells. However, while more than 90% of wild-type cells completed the repair in 12 h, little repair product was detected in pph3Δ ptc2Δ ptc3Δ cells even 24 h after induction of the DSB. We note that our previous reports showed that neither the pph3Δ mutation alone nor the ptc2Δ ptc3Δ double mutation impairs the SSA-mediated DSB repair (10, 15). These results demonstrate the SSA-mediated DSB repair is defective only in the simultaneous absence of Pph3, Ptc2, and Ptc3.
FIG. 4.

SSA-mediated DSB repair is defective in pph3Δ ptc2Δ ptc3Δ cells. (A) Diagram showing the leu2-cs (centromere-proximal dark gray box), which contains an HO endonuclease cleavage site (pointed to with ▴) within the endogenous LEU2, and the U2 locus (centromere-distal dark gray box), which contains 3′ partial sequences of LEU2, on chromosome 3 in strain YMV80. The centromere is marked as a black dot. Vertical lines mark the recognitions sites of the Asp718 restriction enzyme, which was used for Southern analysis. Probe 1, marked as a black bar, is specific for the leu2-cs adjacent region that is centromere proximal. Probe 1 can detect three DNA fragments digested with Asp718, which are denoted as “uncut,” “cut,” and “repair.” (B) Either pph3Δ ptc2Δ ptc3Δ or wild-type cells were collected 1, 2, 4, 8, 12, and 24 h after as well as prior to HO induction. Genomic DNA purified from the cells was digested with an Asp718 restriction enzyme and subjected to Southern analysis using probe 1. (C) PCR-based nonhomologous (NH) tail removal assay. The diagram in the upper panels shows an intermediate step of SSA-mediated DSB repair in strain YMV80. The primers used for the NH tail cleavage assay are denoted as p1 and p2, which are specific to the leu2-cs adjacent region that is centromere distal and to the leu2-cs, respectively. The annealed homologous sequences are marked in gray boxes. The lower panel shows quantified PCR results using the p1/p2 primer pair. The PCR product by the p1/p2 primer was run on 1.5% agarose gel, and then the intensities of PCR product on the gel were measured by Quantity-One (Bio-Rad). Then the signal was normalized by the PCR signal with the CEN8 p1/p2 primer pair used as a loading control. The centromere 8 (CEN8)-specific region is amplified by the pair of CEN8 p1/p2 primers. The normalized PCR ratio before HO induction was set as 100%. (D) PCR-based primer extension assay. The upper panel shows a diagram demonstrating an intermediate step of SSA-mediated DSB repair in the strain YMV80. Newly synthesized donor-specific sequences are shown as waves, and the homologous regions are in gray. p3 and p4 denote primers specific to the donor and to the leu2-cs adjacent region that is centromere proximal, respectively. The lower panels show representatives of the PCR results obtained by using the p3/p4 pair as well as the CEN8 p1/p2 pair (loading control). (E) Quantification of PCR product shown in panel D. The band intensities on 1.5% agarose gel were measured by Quantity-One (Bio-Rad). The ratio of the p3/p4 PCR product to the loading control (PCR product by CEN8 p1/p2) at 24 h after HO induction in wild-type cells was referred to as 100%. Subsequently, the quantified PCR values at all time points were converted to percentages. The error bar was obtained from three different PCRs.
Next, we asked which particular step of the SSA-mediated DSB repair was defective in pph3Δ ptc2Δ ptc3Δ cells. A distinctive step of the SSA repair in YMV80 derivatives is removal of the long NH single-strand tail from annealed strands. Without cleavage of the tail, new DNA synthesis cannot be primed from the annealed strands (19). The defect in the SSA-mediated DSB repair might be accounted for by the roles played by the phosphatases in the regulation of NH single-strand tail removal. To examine this possibility, we examined cleavage of NH tail by PCR analysis (30). By 8 h after the induction of a DSB, more than ∼90% of the NH tail was removed from the 3′ exposed single-stranded DNA (ssDNA) in wild-type cells, while ∼50% of NH tail was still retained in cells lacking one of NH tail cleavage factors, Rad1 (Fig. 4C). At 8 h after the DSB induction, the amount of NH tail removed in pph3Δ ptc2Δ ptc3Δ cells was intermediate (∼65%) between wild-type cells and the NH tail cleavage-defective rad1Δ cells. These results suggest that the cleavage of NH tail is moderately defective in triple phosphatase deletion in pph3Δ ptc2Δ ptc3Δ cells.
In the pph3Δ ptc2Δ ptc3Δ cells derived from YMV80 strains, the degree of inviability was much more pronounced than the defect in the NH tail removal. In addition, the lack of viability was paralleled by Southern blot analysis, which exhibited a severe defect in the formation of DSB repair product. These results indicated that a step other than the NH tail cleavage was impaired in pph3Δ ptc2Δ ptc3Δ cells, leading to a prominent defect in the SSA-mediated repair. Therefore, we evaluated new DNA synthesis, the step following the removal of NH tail. New DNA synthesis from the annealed strand is required to fill in the adjacent single-strand region to complete repair. This step can be monitored by PCR using a primer specific to the U2 donor sequences and another primer specific to the region centromere proximal to the leu2-cs containing an HO-induced DSB (Fig. 4D). This analysis demonstrated that priming of new DNA synthesis occurred only in less than 20% of pph3Δ ptc2Δ ptc3Δ cells by 24 h after the induction of a break (Fig. 4D and E). We note that a similar DNA synthesis step is required on the other end of the annealed segment, so that the proportion of events that have completed both repair steps may be much smaller than 20%. This result suggests that the severe defect in the repair-mediated DNA synthesis subsequent to the NH tail removal is a main cause of the failure of DSB repair in the YMV80 derivative.
We conjectured that the defect in repair in the triple phosphatases was dependent on activation and maintenance of the DNA damage checkpoint. Consistent with this idea, we found that deletion of RAD9, which is required to activate the Mec1-dependent checkpoint (5), enabled pph3Δ ptc2Δ ptc3Δ cells to complete repair and to regain viability. As before, we plated cells on YEP-dextrose and YEP-galactose to assess the ability of YMV80 and its derivatives to complete repair. In comparison to an average viability of 64.7% for the wild-type strain and only 0.8% for the ptc2Δ ptc3Δ pph3Δ derivative, a rad9Δ ptc2Δ ptc3Δ pph3Δ strain exhibited 53.2% survival. These data strongly indicate that the failure of repair in the phosphatase mutant depends on activation of the DNA damage checkpoint.
GC-mediated DSB repair is also defective in pph3Δ ptc2Δ ptc3Δ cells.
Next, we asked whether the ectopic GC-mediated DSB repair in YJK17 derivatives was also affected by deletion of the three phosphatases. Using a probe specific to the MAT distal region, the formation of an HO-induced DSB and the repair of the break were detected (Fig. 5A). One hour after the induction of HO endonuclease, all of the tested cells, including pph3Δ ptc2Δ ptc3Δ cells, had an efficient HO cleavage at the MAT locus on Chr3 (Fig. 5B). In addition, the HO-cut band disappeared at almost the same rate in all cells tested, suggesting that the DNA resection was not defective in the mutant cells. Here, strand invasion occurs between perfectly matched sequences and there is no requirement for NH tail removal. In wild-type cells, the appearance of repair product was seen as early as 3 h after DSB induction, and by 9 h, more than 90% of the wild-type cells completed the repair (Fig. 5C). Both pph3Δ and ptc2Δ ptc3Δ cells had a slightly reduced formation of repair product compared to wild-type cells. About 76% of the mutant cells completed repair within 12 h after the induction of HO (Fig. 5C). The production of the repair product in triple-deletion pph3Δ ptc2Δ ptc3 cells appeared to be slower than those in the other mutant cells (Fig. 5B). Furthermore, only 54% of the pph3Δ ptc2Δ ptc3Δ cells completed the repair by 12 h after HO induction (Fig. 5C). The delayed and less efficient DSB repair in the triple-deletion mutant cells implies that the simultaneous absence of Pph3, Ptc2, and Ptc3 also impairs GC-mediated DSB repair, although to a degree that is less than what we observed with YMV80.
FIG. 5.

Interchromosomal ectopic GC-mediated DSB repair is defective in pph3Δ ptc2Δ ptc3Δ cells. (A) Diagram of MATα in Chr3 and MATa-inc in Chr5. The vertical lines indicate EcoRI cleavage sites. ▴ marks an HO-induced DSB. The black bar denotes probe 2, specific to the MAT region, which can detect a “MAT” fragment either prior to HO induction or when the DSB is repaired. Probe 2 can also detect the “HO cut” as well as “MATa-inc” on Chr5. (B) Exponentially growing wild-type and mutant cells were collected at different time points after as well as prior to the induction of HO. Genomic DNA from the cells at each time point was digested with EcoRI and then run on a 0.8% agarose gel for Southern analysis. (C) The percentage of value of the repair product at each time point was quantified by Quantity-One (Bio-Rad). The ratio of “MAT” to “MATa-inc” was calculated at each time point. The ratio was then was normalized with that prior to HO induction, which was referred to as 100%. (D) Diagram showing the formation of a synapsis between 3′-tailed single-strand DNA from Chr3 and the homologous duplex region in Chr5. Newly synthesized DNA specific to MATa-inc sequence in Chr5 is shown as waves. “a” denotes the region amplified by PCR following Rad51 ChIP, which is about 200 bp away form the HO-induced DSB. “b” indicates the Chr3-specific region, ∼500 bp distal to the HO-induced DSB. “c” is the Chr5-specific region. (E) Rad51 ChIP values at single-stranded regions (a and b) on Chr3 as well as a homologous donor region on Chr5 (c). The fold increase was measured by normalizing the Rad51 ChIP signal at each time point after HO induction with that prior to HO induction. (F) Representative results from PCR assays to show primer extension from the invading strand. Genomic DNA either from wild-type cells or from pph3Δ ptc2Δ ptc3Δ cells, which was purified for Southern analysis in Fig. 5B, was subjected to PCR using the p5 and p6 primers in panel D. As a loading control, the CEN8-specific sequences were amplified by CEN8 p1/p2 primers. (G) Percent value of PCR-based primer extension assay. The calculation was done as described in the legend to Fig. 4E.
The GC pathway is divided into multiple steps. (i) In step 1, single-strand DNA (ssDNA) is exposed from broken DNA by 5′-to-3′ DNA resection. (ii) In step 2, Rad51 is accumulated on 3′-tailed ssDNA. (iii) In step 3, the Rad51-ssDNA nucleofilament searches for and invades the homologous donor region. (iv) Finally, in step 4, new DNA is synthesized from the invading 3′-tailed strand. We determined which step was defective in the GC-mediated DSB repair in the triple-phosphatase-deletion pph3Δ ptc2Δ ptc3Δ cells. The Southern analysis in Fig. 5B showed that the formation of a DSB, as well as the DNA resection, was not defective in the mutant cells (steps 1 and 2). The subsequent steps following DNA resection were examined by chromatin immunoprecipitation (ChIP) analysis using anti-Rad51 antibody either in the wild-type or in the pph3Δ ptc2Δ ptc3Δ cells. By examining two different regions on the ssDNA (Fig. 5D), we checked the association of the Rad51 with MAT DNA, which we presume to be the nucleofilament formation. Both in wild-type and in pph3Δ ptc2Δ ptc3Δ cells, the accumulation of Rad51 on the ssDNA was detected as early as 3 h after the induction of a break (Fig. 5E, panels a and b). These results indicate that the assembly of Rad51 on ssDNA was not defective in the absence of Pph3, Ptc2, and Ptc3. We also examined the formation of a synapsis between the Rad51-ssDNA filament and the homologous donor region (26). By the Rad51 ChIP analysis, the interaction with the donor-specific region was detected in pph3Δ ptc2Δ ptc3Δ cells as well as in wild-type cells (Fig. 5E, panel c). This result indicates that the Rad51-coated ssDNA succeeds in searching for a homologous donor and in forming a synapsis with a donor without Pph3, Ptc2, and Ptc3 (step 3). However, the disappearance of Rad51 from all of the tested regions in the mutant cells was delayed ∼2 h compared to that in wild-type cells (Fig. 5E), implying that displacement of Rad51 from the ssDNA annealed to the homologous sequence is defective in pph3Δ ptc2Δ ptc3Δ cells.
New DNA synthesis from the invading strand, which follows synapsis between a donor and the invading strand, can be monitored by PCR using a primer specific to donor sequences and the other primer specific to the invading strand (7). Using the pair of primers shown in Fig. 5D, we checked the extension of the invading strand either in wild-type or in pph3Δ ptc2Δ ptc3Δ cells. In the mutant cells, the appearance of the PCR product was seen distinctively later than that in wild-type cells (Fig. 5F and G). This result indicates that the DSB repair-mediated DNA synthesis is delayed when three phosphatases, Pph3, Ptc2, and Ptc3, are simultaneously absent during GC-mediated DSB repair.
Intrachromosomal GC-mediated MAT switching is moderately defective in pph3Δ ptc2Δ ptc3Δ cells.
Both the cell viability assays after the induction of a DSB (Fig. 3) and the DSB repair assays (Fig. 4 and 5) demonstrate that the severity of the redundant effect of three phosphatases, Pph3, Ptc2, and Ptc3, is proportional to the extent of the damage-activated cell cycle checkpoint. To further determine the involvement of checkpoint activation in the effect of the phosphatases on DSB repair, we examined the intrachromosomal GC repair in YJK139, in which the damage checkpoint kinases Mec1 and Tel are not activated. The Southern analysis using a probe specific to MAT (Fig. 6A) demonstrated that the triple-deletion pph3Δ ptc2Δ ptc3Δ cells have a 30-min delay in product formation (Fig. 6B and C), as well as decreased levels of primer extension, compared to WT cells (Fig. 6D). These results again suggest that the DSB repair-mediated DNA synthesis is less efficient when three phosphatases are absent. However, the much less severe defects in YJK139 derivatives than those either in YMV80 or YJK17 derivatives indicate that the extent of the damage-activated checkpoint is important for the requirement of the phosphatases in repair-mediated DNA synthesis.
FIG. 6.

Intrachromosomal GC-mediated DSB repair is moderately defective in pph3Δ ptc2Δ ptc3Δ cells. (A, panel a) Diagram of MATa and HMLα-inc on Chr3. The vertical lines indicate StyI cleavage sites. ▴ marks an HO-induced DSB. The black bar denotes probe 3, specific to the MAT region, which can detect “MATa” fragment prior to HO induction. Probe 3 can detect the “HO cut,” “repair (MATα-inc),” and “MAT distal” fragments. Because of the sequence homology to HMLα-inc, probe 3 also detects “donor (HMLα-inc).” The centromere is marked as a black dot. (b) Diagram showing an intermediate step of intrachromosomal MAT switching. Newly synthesized DNA specific to HMLα-inc sequences is shown as waves. p7 and p8 mark the primers used for the PCR-based primer extension assay in panel D. (B) Southern analysis showing intrachromosomal MAT switching at 30, 45, 60, 90, 120, 150, 300 min after as well as prior to HO induction. *, nonspecific bands detected by probe 3. (C) Percentage values of repair products measured in panel B. The calculation was done as described in the legend to Fig. 5C. (D) Percentage values of the PCR-based primer extension assay during intrachromosomal MAT switching with the p7/p8 primer pair. The calculation was done as described in the legend to Fig. 4E.
DISCUSSION
Cell proliferation after completion of DSB repair requires not only activation of the DNA damage checkpoint but also its subsequent inactivation. Pph3, Ptc2, and Ptc3 appear to be the main phosphatases involved in the inactivation of the DNA damage checkpoint. In this study, we have discovered a redundant role for Pph3, Ptc2, and Ptc3 not just in regulating the damage checkpoint but also in DSB repair itself. More specifically, we have shown that simultaneous deletion of PPH3, PTC2, and PTC3 leads to a defect in the DSB repair-mediated DNA synthesis but not in DNA replication during the unperturbed S phase. A particularly interesting point is that the effect of the three phosphatases in DSB repair appears to be dependent on the extent of the damage checkpoint mediated by the kinetics of the repair, so that intrachromosomal GC shows the weakest phenotype and the long-delayed SSA assay shows a very strong defect. This raises an important aspect of the checkpoint activation, which can be inhibitory to efficient DSB repair when the balance between phosphorylation and dephosphosphorylation is impaired.
Given the results above, we suggest a model in which a redundant function of Pph3, Ptc2, and Ptc3 antagonizes a negative effect caused by the checkpoint activation on DSB repair. The damage-induced checkpoint mediates phosphorylation of multiple proteins such as replication protein A (22, 32). One possibility is that the phosphorylated form of an unidentified factor(s) may be inhibitory for DSB repair and may need to be dephosphorylated by Pph3, Ptc2, and Ptc3 to gain its function. Recently, Lee et al. have shown that PP4-dependent dephosphorylation of RPA2 mediates efficient loading of Rad51 (12), supporting this possibility; however, we saw no obvious defect in Rad51-mediated synapsis of the DSB end with its donor (Fig. 5E). We speculate that a protein other than RPA2 is a common target of the three phosphatases required for the DSB repairs in our systems. Alternatively, the phosphatases may affect the stability of an unidentified factor. A recent study showed that CK2-dependent phosphorylation of mammalian XRCC1 is involved in efficient DNA repair by maintaining the stability of XRCC1 (20). It is also feasible that the strongly activated damage checkpoint either stabilizes an inhibitory factor for DSB repair or depletes a component of the DSB repair machinery. The major defect we observed in both the YJK17 and in YMV80 strains is inefficient priming of the DSB repair-mediated DNA synthesis. Repair-mediated DNA synthesis shares many factors with DNA replication machinery, but gene conversion—as monitored by MAT switching—does not require the Mcm helicase proteins or Cdc45, nor does it require the Cdc7-Dbf4 kinase, although Cdk1 (Cdc28) appears to be needed at several steps (8, 16, 33). To prevent unscheduled replication outside of S phase, cells tightly regulate the replication machinery (28, 35). Therefore, it is possible that a redundant function of Pph3, Ptc2, and Ptc3 is required for the loading of a DNA synthesis factor on DNA break sites in a replication-independent manner. Recently we have shown that MAT switching also requires the Dpb11protein involved in assembling the normal DNA replication fork (W. M. Hicks, M. Yamaguchi, and J. E. Haber, unpublished data). Dpb11 represents another possible phosphatase target. Further studies to identify the factor that is redundantly regulated by the three phosphatases Pph3, Ptc2, and Ptc3 will give insight into understanding the mechanism of how the phosphatases function in repairing DSB.
Acknowledgments
W. M. Hicks was supported by NIH Genetics Training grant GM007122. This research is funded by NIH grants GM20056 and GM61766.
We thank Jake Harrison for making strain JY544 (a derivative strain of YMV80 lacking PPH3, PTC2, and PTC3).
Footnotes
Published ahead of print on 6 December 2010.
REFERENCES
- 1.Chen, C. C., et al. 2008. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134:231-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grenon, M., C. Gilbert, and N. F. Lowndes. 2001. Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat. Cell Biol. 3:844-847. [DOI] [PubMed] [Google Scholar]
- 3.Guillemain, G., et al. 2007. Mechanisms of checkpoint kinase Rad53 inactivation after a double-strand break in Saccharomyces cerevisiae. Mol. Cell. Biol. 27:3378-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammet, A., C. Magill, J. Heierhorst, and S. P. Jackson. 2007. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 8:851-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison, J. C., and J. E. Haber. 2006. Surviving the breakup: the DNA damage checkpoint. Annu. Rev. Genet. 40:209-235. [DOI] [PubMed] [Google Scholar]
- 6.Hastie, C. J., C. Vazquez-Martin, A. Philp, M. J. Stark, and P. T. Cohen. 2006. The Saccharomyces cerevisiae orthologue of the human protein phosphatase 4 core regulatory subunit R2 confers resistance to the anticancer drug cisplatin. FEBS J. 273:3322-3334. [DOI] [PubMed] [Google Scholar]
- 7.Holmes, A., and J. E. Haber. 1999. Physical monitoring of HO-induced homologous recombination. Methods Mol. Biol. 113:403-415. [DOI] [PubMed] [Google Scholar]
- 8.Ira, G., et al. 2004. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431:1011-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain, S., et al. 2009. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev. 23:291-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keogh, M. C., et al. 2006. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature 439:497-501. [DOI] [PubMed] [Google Scholar]
- 11.Kim, J. A., and J. E. Haber. 2009. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc. Natl. Acad. Sci. U. S. A. 106:1151-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee, D. H., et al. 2010. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat. Struct. Mol. Biol. 17:365-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee, S. E., et al. 1998. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94:399-409. [DOI] [PubMed] [Google Scholar]
- 14.Lee, S. E., et al. 2000. Arrest, adaptation, and recovery following a chromosome double-strand break in Saccharomyces cerevisiae. Cold Spring Harb. Symp. Quant. Biol. 65:303-314. [DOI] [PubMed] [Google Scholar]
- 15.Leroy, C., et al. 2003. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 11:827-835. [DOI] [PubMed] [Google Scholar]
- 16.Lydeard, J. R., et al. 2010. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 24:1133-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moorhead, G. B., L. Trinkle-Mulcahy, and A. Ulke-Lemee. 2007. Emerging roles of nuclear protein phosphatases. Nat. Rev. Mol. Cell Biol. 8:234-244. [DOI] [PubMed] [Google Scholar]
- 18.O'Neill, B. M., et al. 2007. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc. Natl. Acad. Sci. U. S. A. 104:9290-9295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pâques, F., and J. E. Haber. 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63:349-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsons, J. L., et al. XRCC1 phosphorylation by CK2 is required for its stability and efficient DNA repair. DNA Repair (Amst.) 9:835-841. [DOI] [PubMed]
- 21.Pellicioli, A., S. E. Lee, C. Lucca, M. Foiani, and J. E. Haber. 2001. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell 7:293-300. [DOI] [PubMed] [Google Scholar]
- 22.Pellicioli, A., et al. 1999. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 18:6561-6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez, Y., et al. 1996. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271:357-360. [DOI] [PubMed] [Google Scholar]
- 24.Shroff, R., et al. 2004. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 14:1703-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugawara, N., and J. E. Haber. 2006. Repair of DNA double strand breaks: in vivo biochemistry. Methods Enzymol. 408:416-429. [DOI] [PubMed] [Google Scholar]
- 26.Sugawara, N., X. Wang, and J. E. Haber. 2003. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell 12:209-219. [DOI] [PubMed] [Google Scholar]
- 27.Sweeney, F. D., et al. 2005. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr. Biol. 15:1364-1375. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka, S., et al. 2007. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 445:328-332. [DOI] [PubMed] [Google Scholar]
- 29.Toczyski, D. P., D. J. Galgoczy, and L. H. Hartwell. 1997. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90:1097-1106. [DOI] [PubMed] [Google Scholar]
- 30.Toh, G. W., et al. Mec1/Tel1-dependent phosphorylation of Slx4 stimulates Rad1-Rad10-dependent cleavage of non-homologous DNA tails. DNA Repair (Amst.) 9:718-726. [DOI] [PMC free article] [PubMed]
- 31.Vaze, M. B., et al. 2002. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell 10:373-385. [DOI] [PubMed] [Google Scholar]
- 32.Wang, H., J. Guan, A. R. Perrault, Y. Wang, and G. Iliakis. 2001. Replication protein A2 phosphorylation after DNA damage by the coordinated action of ataxia telangiectasia-mutated and DNA-dependent protein kinase. Cancer Res. 61:8554-8563. [PubMed] [Google Scholar]
- 33.Wang, X., et al. 2004. Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 24:6891-6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wysocki, R., et al. 2005. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell. Biol. 25:8430-8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zegerman, P., and J. F. Diffley. 2007. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445:281-285. [DOI] [PubMed] [Google Scholar]