

Abstract
Cells experience multiple environmental stimuli simultaneously. To survive, they must respond accordingly. Unfortunately, the proper response to one stress easily could make the cell more susceptible to a second coexistent stress. To deal with such a problem, a cell must possess a mechanism that balances the need to respond simultaneously to both stresses. Our recent studies of ompR malT(Con) double mutants show that elevated expression of LamB, the outer membrane porin responsible for maltose uptake, causes cell death when the osmoregulator OmpR is disabled. To obtain insight into the nature of the death experienced by ompR malT(Con) mutants, we described the death process. On the basis of microscopic and biochemical approaches, we conclude that death results from a loss of membrane integrity. On the basis of an unbiased genome-wide search for suppressor mutations, we conclude that this loss of membrane integrity results from a LamB-induced envelope stress that the cells do not sufficiently perceive and thus do not adequately accommodate. Finally, we conclude that this envelope stress involves an imbalance in the lipopolysaccharide/porin composition of the outer membrane and an increased requirement for inorganic phosphate.
The ability to perceive environmental changes and modulate gene activity/metabolism accordingly is essential for bacterial survival. Dysfunctional proteins and/or regulatory mechanisms often result in improper gene expression and cell death. Such is the case with the ompR malT(Con) mutant of Escherichia coli, which dies due to elevated expression of LamB in the absence of a functional OmpR (58).
OmpR is a two-component response regulator that controls a set of genes associated with outer membrane biogenesis, envelope stress, and osmoregulation (49, 53). As osmolality increases, OmpR becomes activated by acceptance of a phosphoryl group from its cognate sensor kinase EnvZ (28, 43). MalT is the master regulator of a regulon that encodes proteins involved in transport and metabolism of maltose and maltodextrins (6, 7). LamB, a member of the MalT regulon, is an outer membrane porin (OMP) that facilitates the uptake of maltose and maltodextrins across the outer membrane (36). malT(Con) is an allele that encodes a constitutively active mutant MalT protein that is insensitive to both the inducer maltotriose and the primary inhibitor MalK (58, 59). The result is dysregulated expression of the MalT regulon, including LamB (15, 19).
Lethality of the ompR malT(Con) mutant is conditional. Death does not ensue under conditions that downregulate LamB expression. For example, the ompR malT(Con) mutant survives on minimal medium supplemented with glucose as the sole carbon source (58). Survival most likely occurs because glucose causes catabolite repression, which reduces malT transcription and thus LamB expression (11, 12).
To understand the basis for the ompR malT(Con) lethality, we first described the death process and then performed an unbiased genome-wide search for mutations that suppress death. On the basis of microscopic and biochemical approaches, we conclude that death results primarily from a loss of membrane integrity. Since many suppressor mutations disrupted malT(Con) and lamB, we conclude that elevated expression of LamB is the primary cause of death. Since other suppressor mutations disrupted rseA and rseB, which encode inhibitors of the extracytoplasmic stress-responsive sigma factor σE, we further conclude that LamB causes an envelope stress that the cells do not sufficiently perceive and thus do not adequately accommodate. Finally, several suppressor mutations constitutively activated the PhoB regulon. Since we traced this suppression to increased expression of the OMP PhoE, we conclude that ompR malT(Con) lethality results, at least in part, from an imbalance in the lipopolysaccharide (LPS)/porin composition of the outer membrane. Because inorganic phosphate rescued the ompR malT(Con) mutant in a PhoE-dependent manner, this imbalance likely involves some phosphorylated compound.
MATERIALS AND METHODS
Bacterial strains, bacteriophage, transcriptional fusions, and plasmids.
All bacterial strains used in this study are listed in Table 1. All strains evaluated were derivatives of E. coli AJW678 (37). Derivatives were constructed by generalized transduction with P1vir, as described previously (73).
TABLE 1.
Strains, plasmids, and reporter fusions used in this study
| Strain, plasmid, or fusion | Relevant genotype | Reference or source |
|---|---|---|
| Strains | ||
| AJW678 | thi-1thr-1(Am) leuB6metF159(Am) rpsL136lacX74 | 37 |
| AJW2050 | AJW678 ompR::Tn10 | 58 |
| AJW2051 | AJW678 ompR::Tn10malT(Con)(T949A) ackA::Km | 58 |
| AJW3098 | AJW678 ompR::Tn10malT(Con)(T949A) | 58 |
| AJW3499 | AJW678 malT(Con)(T949A) | 58 |
| AJW3780 | AJW678 ompR::Tn10malT(Con)(T949A) phoU::Km | This study |
| AJW3781 | AJW678 ompR::Tn10malT(Con)(T949A) pstA::Km | This study |
| AJW3782 | AJW678 ompR::Tn10malT(Con)(T949A) pstB::Km | This study |
| AJW3783 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Km | This study |
| AJW3785 | AJW678 ompR::Tn10malT(Con)(T949A) pstS::Km | This study |
| AJW3815 | AJW678 rseA::Km | This study |
| AJW3816 | AJW678 ompR::Tn10rseA::Km | This study |
| AJW3817 | AJW678 malT(Con)(T949A) rseA::Km | This study |
| AJW3818 | AJW678 ompR::Tn10malT(Con)(T949A) rseA::Km | This study |
| AJW3855 | AJW678 ompR::Tn10malT(Con)(T949A) rseA::Tn5 | This study |
| AJW3945 | AJW678 ompR::Tn10malT(Con)(T949A) pstB::Frt | This study |
| AJW3946 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt | This study |
| AJW3954 | AJW678 ompR::Tn10malT(Con)(T949A) pstB::Frt phnC::Km | This study |
| AJW3955 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt phnC::Km | This study |
| AJW3956 | AJW678 ompR::Tn10malT(Con)(T949A) pstB::Frt phoA::Km | This study |
| AJW3957 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt phoA::Km | This study |
| AJW3958 | AJW678 ompR::Tn10malT(Con)(T949A) pstB::Frt ugpB::Km | This study |
| AJW3959 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt ugpB::Km | This study |
| AJW4000 | AJW678 ompR::Tn10malT(Con)(T949A) rseA::Frt hfq::Km | This study |
| AJW4196 | AJW678 ompR::Tn10malT(Con)(T949A) pstB::Frt phoE::Km | This study |
| AJW4197 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt phoE::Km | This study |
| AJW4245 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt psiF::Km | This study |
| AJW4248 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt phnD::Km | This study |
| AJW4264 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt phnF::Km | This study |
| AJW4266 | AJW678 ompR::Tn10malT(Con)(T949A) pstC::Frt phnO::Km | This study |
| Plasmids | ||
| pRL27 | Tn5 delivery vector; Kmr | 38 |
| pRC7 | Mini-F (mF) plasmid, carries lacZ under control of the IPTG-inducible lac promoter; Ampr | 20 |
| mF-ompR | ompR ORF in pRC7, Ampr | 58 |
| pCA24N | Expression vector, IPTG inducible; Cmr | 32 |
| pTrc99a | Expression vector, IPTG inducible; Ampr | Amersham Pharmacia, Piscataway, NJ |
| pLC245 | pTrc99a plasmid, carries rpoE; Ampr | 60 |
| pphoBCAE11K | Allelic-exchange vector pIB307, temp sensitive, pSC101 replicon, carries the constitutive phoBCA(E11K) allele; Cmr | Bill McCleary, personal communication |
| Fusions | ||
| rpoHP3-lacZ | λΦ(rpoHP3-lacZ) | 41 |
| malEpΔ92-lac | trp::(Kanr-malEpΔ92-lac)op | 62 |
The transcriptional malEpD92-lac fusion was a generous gift from Winfried Boos (Universität Konstanz, Germany) and was described earlier (62, 71). The pTrc99a vector carrying rpoE (pLC245) and the transcriptional Φ(rpoHP3-lacZ) fusion were generous gifts of Carol Gross (University of California at San Francisco) and have been described previously (41, 60).
Induction of λ prophage by UV light, amplification of the resultant phage, and construction and verification of monolysogens were performed as described previously (52, 73, 74).
The malT(Con) allele (malTc-1) used in this study was a generous gift from Linda Kenney (University of Illinois at Chicago). It harbors a T949A base substitution and encodes the MalT(Con)(W317R) protein.
Unless otherwise mentioned, deletion alleles or plasmids were derived from the Keio or ASKA collection, respectively (4, 32). To obtain nonpolar deletion alleles, resistance cassettes were removed using Flp recombinase, according to the previously described protocol (18).
Media and growth conditions.
Because the ompR malT(Con) mutant is conditionally lethal, cells were grown overnight under permissive conditions: 22°C in M63 minimal salts (73) with 22 mM sorbitol as the sole carbon source and supplemented with 100 μg/ml l-threonine, l-histidine, l-leucine, l-methionine, l-tryptophan, and 10 μg/ml thiamine. Whenever required, kanamycin (40 μg/ml), chloramphenicol (25 μg/ml), ampicillin (100 μg/ml), tetracycline (15 μg/ml), 5-bromo-4-chloro-3-indoxyl-β-d-galactopyranoside (X-Gal) (32 μg/ml), or isopropyl-β-d-thiogalactoside (IPTG) (concentrations as indicated) was added.
For tests of lethality, an inoculum from the overnight culture was subcultured at 37°C in LB (1% [wt/vol] tryptone, 0.5% [wt/vol] yeast extract, 0.5% [wt/vol] NaCl). LB agar plates also contained 1.5% (wt/vol) Bacto agar. These growth conditions were considered nonpermissive. For experiments with inorganic phosphate, sodium chloride was replaced with 100 mM sodium phosphate (pH 7) or with 100 mM sucrose. Cell growth was monitored spectrophotometrically (DU640; Beckman Instruments, Fullerton, CA) by optical density at 600 nm (OD600).
Promoter activity assays.
To monitor Φ(rpoHP3-lacZ) promoter activity, cells were grown aerobically with agitation at 250 rpm at 37°C. At various time points during growth, 50 μl of culture was harvested and added to 50 μl All-in-One β-galactosidase reagent (Pierce Biotechnology, Rockford, IL). β-Galactosidase activity was determined quantitatively in a microtiter format, as described previously (5). To avoid misleading results caused by lysing cells that spill β-galactosidase into the growth medium, we considered β-galactosidase measurements only before the onset of cell death.
Microscopic methods.
FM4-64 (Molecular Probes, Eugene, OR), LIVE/DEAD (Invitrogen, Carlsbad, CA), and Hoechst 33342 (Sigma-Aldrich, St. Louis, MO) fluorescent stains were used according to the manufacturer's protocol. Nile red staining was performed as described earlier (26). Imaging was conducted with a Leica DM IRB fluorescence microscope (Leica, Bannockburn, IL) and an Optronics camera and MagnaFire2.1C imaging software (Optronics, Goleta, CA). Phase-contrast microscopy was performed with a Nikon Optiphot microscope (Nikon, Melville, NY). For transmission electron microscopy, cells were grown aerobically with agitation at 250 rpm under nonpermissive conditions. Cells were collected by centrifugation and fixed overnight in a solution containing 4% glutaraldehyde and 0.1 M cacodylate buffer (pH 7.3). After fixation, cells were washed, postfixed with 1% osmium tetroxide for 1 h, washed again, and subjected to serial dehydration with ethanol. Samples were embedded in EMbed812 resin (EMS, Hatfield, PA), thin sectioned, and stained with 2% uranyl acetate. Finally, the samples were examined with a transmission electron microscope (TEM) (Hitachi H-600) operating at an accelerating voltage of 75 kV.
Cytoplasmic membrane permeability assay procedure.
To assess the integrity of the cytoplasmic membrane, we monitored cytoplasmic β-galactosidase activity using the normally cytoplasmic membrane-impermeative chromogenic substrate ortho-nitrophenyl-β-galactoside (ONPG). The assay was carried out as previously described (88) with minor modifications. In brief, cells carrying a malEpD92-lac reporter fusion (71) as the source of β-galactosidase were grown in LB at 37°C. At regular time intervals, two aliquots of 1 ml were harvested and centrifuged. The pellets were washed in 10 mM Na-phosphate-100 mM NaCl (pH 7.4) and resuspended in 1 ml of the same buffer. For determination of total β-galactosidase activity, cells of one aliquot were lysed by adding 50 μl chloroform. In both lysed and unlysed aliquots, the reaction was started by adding 100 μl of 10 mM Na-phosphate containing 0.4 mg/ml ONPG. After incubation for 30 min at 37°C, the reaction was stopped by adding 500 μl of 1 M NaCO3, and β-galactosidase activity was determined at 405 nm. To account for malEpD92-lac expression differences between cells carrying wild-type (WT) malT versus malT(Con) alleles, the total enzyme activity in each chloroform-lysed sample was normalized to 100%. The β-galactosidase activity of untreated cells was calculated as percentage of the total activity.
AP activity.
Alkaline phosphatase (AP) activity was measured according to the protocol of Zundel et al. (89) with minor modifications. Cells were grown in LB to an OD of approximately 0.3, and 1 ml of culture was centrifuged and the pellet resuspended in 1 ml of 1 M Tris (pH 9), 50 μl of chloroform, and 50 μl of 0.1% SDS. After vigorous vortexing, 500 μl cells were mixed with 50 μl 40 mM para-nitrophenylphosphate (p-NPP) in 1 M Tris (pH 9). The reaction mixture was incubated at 37°C until it turned yellow, at which time 500 μl of 1 M KH2PO4 was added to stop the reaction. Arbitrary alkaline phosphatase units were calculated using the following equation: AP units = (OD405 nm × 1,000)/(OD600 nm × time).
Outer membrane preparations.
Outer membrane preparations were performed as described previously (44). Outer membrane proteins were separated using 12% SDS-PAGE containing 4.8 M urea and stained with Coomassie brilliant blue (70).
Transposon mutagenesis.
To generate transposon insertions that suppress synthetic lethality, ompR malT(Con) mutants were transformed with the pRL27 vector (38) under permissive conditions, the resultant transformants were grown for 48 h under nonpermissive conditions, and colonies were screened for viability. To identify the location of the transposon insertion, we performed tail-arbitrary PCR. The first step in this protocol aims to amplify the genomic region of interest. This promotes the formation of desired PCR products and simultaneously reduces the probability of unspecific primer pairing during subsequent steps. The two subsequent PCRs amplify the DNA region adjacent to the transposon insertion (17). The reaction mixture for the amplification step and the first round of PCR contained 1 μg genomic DNA, 1× PCR buffer, 0.2 μM deoxynucleoside triphosphates (dNTPs), 6 mM MgCl2, 0.2 μM specific external primer Text (5′-CAGCAACACCTTCTTCACGA-3′), 0.6 μM degenerate primer arb1 (5′-GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT-3′) (51), and 1 U Taq polymerase in a total volume of 25 μl. Template amplification and first-round PCR conditions were as follows: 94°C for 30 s, 54°C for 30 s, and 72°C for 120 s (6 cycles); 94°C for 30 s, 30°C for 30 s, and 72°C for 120 s (5 cycles); 94°C for 30 s, 45°C for 30 s, and 72°C for 120 s (30 cycles); and 72°C for 5 min. The reaction mixture for the second round of PCR contained 1 μl first-round PCR product, 1× PCR buffer, 0.2 μM dNTPs, 6 mM MgCl2, 0.2 μM specific internal primer Tint (5′-GAGTCGACCTGCAGGCATGC-3′) (40), 0.6 μM primer arb2 (5′-GGCCACGCGTCGACTAGTAC-3′) (51), and 1 U Taq polymerase in a total volume of 25 μl. Second-round PCR conditions were as follows: 94°C for 30 s, 54°C for 30 s, and 72°C for 120 s (30 cycles), followed by 72°C for 5 min. The PCR products were subsequently sequenced.
σE regulon member screen.
To screen σE regulon members, ASKA collection strains (32) that harbor plasmids with open reading frames (ORFs) of reported σE regulon members (see Table S1 in the supplemental material) were grown individually to an OD600 of 3. Plasmids were isolated using the GeneJET plasmid miniprep kit (Fermentas, Glen Burnie, MD) and transformed into chemically competent ompR malT(Con) mutants under permissive conditions. Transformants were subsequently screened for viability during growth under nonpermissive conditions using 10 μM IPTG to induce gene expression.
RESULTS
ompR malT(Con) mutants lyse due to a dysfunctional cell envelope.
In a previous paper, we reported that the ompR malT(Con) mutant (strain AJW3098 [Table 1]) exhibits a conditional lethal phenotype. Colonies grown on LB plates at 37°C (nonpermissive conditions) displayed a translucent morphology and rapidly developed papillae (Fig. 1A) (58).
FIG. 1.

Death phenotypes of ompR malT(Con) mutants. (A) Colony phenotype of ompR malT(Con) mutants (strain AJW3098) grown on LB plates at 37°C for 48 h. Note the translucent colony appearance and the formation of papillae. (B and C) Colony phenotype of ompR malT(Con) mutants on LB plates supplemented with X-Gal at 37°C. ompR malT(Con) mutants were transformed with the empty mF vector (B) (20) or the vector carrying ompR (C). Note the halo around the colony in panel B. (D and E) FM4-64 fluorescent membrane stain (D) and LIVE/DEAD stain with propidium iodide (E) of ompR malT(Con) mutants during late exponential phase in LB at 37°C.
Complementation studies provided a first indication concerning the nature of the death phenotype. On plates supplemented with X-Gal, ompR malT(Con) mutants transformed with the empty mF vector (which constitutively expresses the lacZ gene) (20) formed dead, dark blue colonies that were often accompanied by a blue halo. In contrast, ompR malT(Con) colonies complemented with mF-ompR were viable and light blue and formed no halo (58) (Fig. 1B and C). These observations indicate increased envelope permeativity of the ompR malT(Con) mutant.
To gain insight into the mechanism of cell death, we investigated cells microscopically during growth under nonpermissive conditions. Both phase-contrast microscopy (data not shown) and fluorescence microscopy using the membrane stain FM4-64 revealed no obvious alterations in cell shape even after death occurred (Fig. 1D). Thus, at this level of inspection, both the outer membrane and the peptidoglycan of the ompR malT(Con) mutant appear to be largely intact. Yet, as growth progressed, LIVE/DEAD staining with propidium iodide (Fig. 1E) and phase-contrast microscopy (data not shown) revealed an increasing percentage of dead and phase-dark cells, respectively. The change in light diffraction indicates a change in the composition of cytoplasmic material. We conclude that these cytoplasmic changes are not caused by a significant loss or degradation of nucleic acids because propidium iodide, which is generally excluded by viable cells, readily stained the nucleic acids of ompR malT(Con) double mutant cells even after the onset of death. This conclusion was confirmed using the fluorescent stain Hoechst 33342, which permits visualization of DNA and RNA in viable and nonviable cells (data not shown).
To confirm changes in the content of cytoplasmic material, we used transmission electron microscopy. We grew ompR malT(Con) mutants under nonpermissive conditions and harvested cells at various time points during the growth curve (Fig. 2A). During early exponential phase, the cells appeared normal. As growth progressed, an increasing number of cells lost their electron-dense cytoplasmic material, giving them a ghost-like appearance (Fig. 2B to D and G). At this point, the outer membrane displayed no visible disruptions and the cell shape remained unaltered, confirming our light and fluorescence microscopic observations. However, many cells showed a slight enlargement of the periplasmic space and/or disruptions of the cytoplasmic membrane (Fig. 2G). During further growth, cell debris and membrane fragments could be observed (Fig. 2E). Much later, the percentage of ghost-like cells diminished. They were replaced by viable cells, which we later demonstrated to carry suppressor mutations (see below). While most of the viable, suppressor-containing cells appeared normal, some cultures contained a substantial percentage of cells with plasmolytic bays (Fig. 2F). These results confirm our previous report that the synthetic phenotype is bactericidal (58). Furthermore, the observed envelope disruptions indicate that cell death coincides with the loss of cytoplasmic material.
FIG. 2.

Time course of ompR malT(Con) mutant phenotypes. (A) Growth curve of the ompR malT(Con) mutant (strain AJW3098) in LB at 37°C. Arrows and letters refer to time points depicted in panels B to F. (B to F) TEM images of the ompR malT(Con) mutant harvested after 90 min (B), 216 min (C), 320 min (D), 500 min (E), and 1,440 min (F) of growth. Note the plasmolytic bays in panel F. (G) Enlargement of an ompR malT(Con) mutant cell at 320 min. The arrows point to disruptions of the cytoplasmic membrane.
The apparent loss of cytoplasmic material prompted us to test the integrity of both the outer and cytoplasmic membranes. To test cytoplasmic membrane integrity, we used a cytoplasmic membrane permeability assay. Uptake of the chromogenic substrate ONPG into the cells requires the lactose permease LacY. Consequently, LacY-deficient cells harboring an intact cytoplasmic membrane are impermeable to ONPG (64). However, ONPG can cross a damaged cytoplasmic membrane and access the cytoplasm, where it is hydrolyzed into galactose and the yellow compound ortho-nitrophenol by the cytoplasmic enzyme β-galactosidase (24, 88). Using this assay, we compared the cytoplasmic membrane permeabilities of WT cells (strain AJW678) and of ompR (strain AJW2050), malT(Con) (strain AJW3499), and ompR malT(Con) (strain AJW3098) mutants, each carrying a malEpD92-lac reporter fusion (71) as the source of β-galactosidase. We found that WT cells and the ompR and malT(Con) single mutants displayed low β-galactosidase activity, whereas the ompR malT(Con) double mutant showed increasing activity approximately coincident with the loss of turbidity (Fig. 3A). In combination with the cytoplasmic membrane disruptions observed by transmission electron microscopy, these data led us to conclude that ompR malT(Con) mutants lose their cytoplasmic membrane impermeability.
FIG. 3.
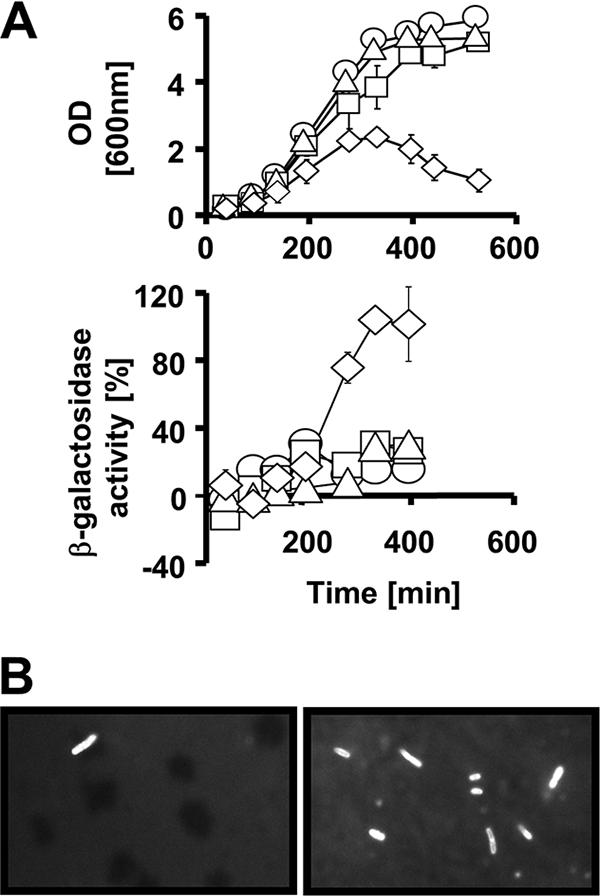
Envelope permeability of ompR malT(Con) mutants. (A) Inner membrane permeability of WT (AJW678) (circles), ompR (AJW2050) (squares), malT(Con) (AJW3499) (triangles), and ompR malT(Con) (AJW3098) (diamonds) mutants. Cells were grown in LB at 37°C. At regular intervals, samples were taken for OD readings and β-galactosidase measurements as described in Materials and Methods. Upper panel, growth curves. Lower panel, percent β-galactosidase activity. Values represent the mean of triplicates. Error bars indicate standard deviations and are shown only when greater than the size of the symbol. (B) Outer membrane permeativity of ompR malT(Con) mutants. Cells were grown in LB at 37°C. After 150 min (left panel) and 400 min (right panel), cells were harvested, stained with Nile red, and observed under a fluorescence microscope. The total cell numbers in the two images are identical.
To test outer membrane integrity, we stained cells with Nile red, a fluorescent stain that is excluded by cells with an intact outer membrane. Nile red is colorless when solubilized in water but fluoresces strongly when it interacts with the lipid bilayer of the cytoplasmic membrane (23). We grew WT cells, the ompR and malT(Con) single mutants, and the ompR malT(Con) double mutant under nonpermissive conditions and, at regular intervals, stained the cells with Nile red. In WT cells and the ompR and malT(Con) single mutants, we observed fewer than 1% fluorescent cells throughout the growth curve. In contrast, as growth progressed, an increasing number of ompR malT(Con) double mutants fluoresced. By the time the culture lost turbidity, nearly 100% of the cells fluoresced (Fig. 3B). We conclude that the ompR malT(Con) double mutant has an outer membrane defect. Taken together, our results show that ompR malT(Con) double mutants have an envelope impermeability defect that coincides with the onset of cell death.
Some suppressors of lethality reduce mal gene transcription and/or LamB expression.
To identify the cause(s) of death, we performed a transposon mutagenesis. ompR malT(Con) double mutants were transformed with the Tn5 delivery vector pRL27 (38) and screened for survivors under nonpermissive conditions. Among 16,000 colonies, we obtained 27 independent viable colonies. Thirteen of the 27 candidates grew poorly or not at all on M63 minimal plates with maltose as the sole carbon source, indicating that these insertions disrupted MalT regulon expression.
To identify the locations of these transposon insertions, we isolated genomic DNA and performed tail-arbitrary PCR in 8 of the 13 candidates with impaired growth on maltose. Six insertions disrupted genes of the maltose system: one insertion disrupted lamB, two insertions disrupted malK (the gene immediately upstream of lamB), and three insertions disrupted malT. Other insertions that reduced growth on maltose were found in cya (one insertion) and hns (one insertion), which encode known positive regulators of the MalT regulon (11, 12, 30). These results confirm our earlier report that reduced expression of LamB permits survival of the ompR malT(Con) double mutant (58).
Activation of the σE regulon suppresses lethality.
To identify the non-Mal-related suppressors, we turned our attention to the transposon mutants that retained a Mal+ phenotype. Three of these insertions disrupted genes encoding the anti-σE factors rseA (two insertions) and rseB (one insertion) (Fig. 4A and data not shown). To confirm the role of anti-σE factors in survival and to exclude the possibility that suppression resulted from spontaneous acquisition of uncharacterized suppressors during mutagenesis, we constructed an rseA ompR malT(Con) triple mutant (strain AJW3818). In contrast to its ompR malT(Con) parent, this strain formed viable colonies when grown under nonpermissive conditions (data not shown) and displayed WT-like growth characteristics in liquid medium (Fig. 4B).
FIG. 4.
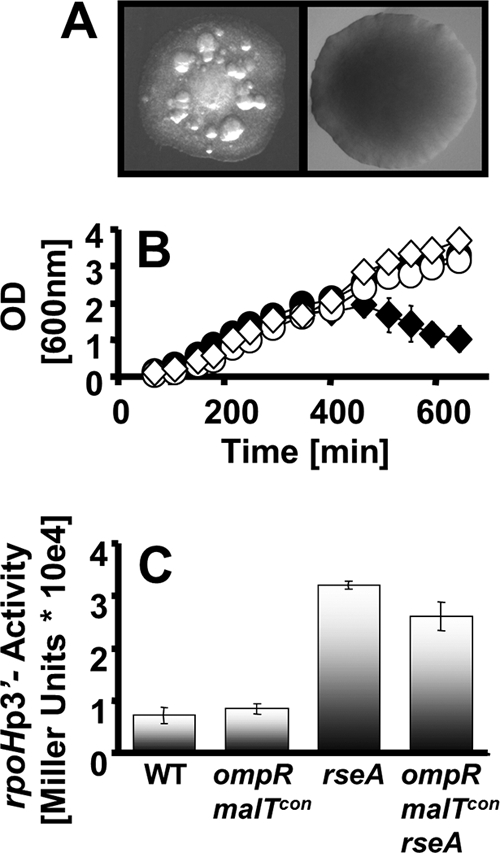
Effect of σE activation on ompR malT(Con) survival. (A) Colony phenotypes of the ompR malT(Con) mutant (AJW3098) (left) and the ompR malT(Con) rseA::Tn5 mutant (strain AJW3855) (right). Colonies were grown in LB at 37°C. (B) Growth curves of WT (AJW678) (closed circles), ompR malT(Con) (AJW3098) (closed diamonds), rseA (AJW3815) (open circles), and ompR malT(Con) rseA (AJW3818) (open diamonds) strains. Cells were grown in LB at 37°C. Values represent the means of triplicates, and error bars indicate standard deviations and are shown only when greater than the size of the symbol. (C) β-Galactosidase activities of WT (AJW678), ompR malT(Con) (AJW3098), rseA (AJW3815), and ompR malT(Con) rseA (AJW3818) strains carrying a Φ(rpoHP3-lacZ) promoter fusion grown under nonpermissive conditions. Cells were harvested at an OD600 of 1. Values represent the means and standard deviations for triplicates.
Since RseA and RseB inhibit σE, we hypothesized that σE regulon activation promotes cell viability. To test this hypothesis, we used Φ(rpoHP3-lacZ) to measure activity from the σE-dependent rpoHP3 promoter (41). We observed substantially increased promoter activity in the rseA single, rseA ompR double, rseA malT(Con) double, and rseA ompR malT(Con) triple mutants (strains AJW3815, AJW3816, AJW3817, and AJW3818, respectively) relative to their RseA+ parents (strains AJW678, AJW2050, AJW3499, and AJW3098, respectively) (Fig. 4C and data not shown).
To further test our hypothesis, we expressed rpoE from the IPTG-inducible plasmid pTrc99a in the ompR malT(Con) mutant. When induced, rpoE restored viability to cells grown under nonpermissive conditions (Fig. 5A), likely due to a substantial increase in σE regulon transcription as confirmed by rpoHP3 activity (Fig. 5B). Taken together, our results indicate that activation of σE can prevent cell death.
FIG. 5.
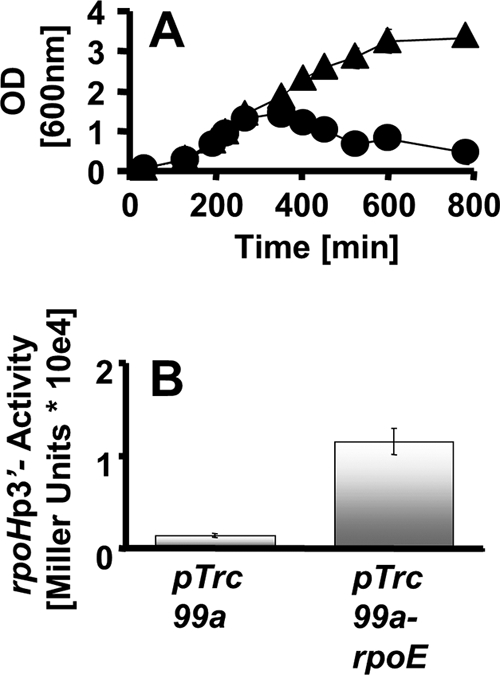
Effect of σE overexpression on ompR malT(Con) survival. (A) Growth curves of the ompR malT(Con) mutant (strain AJW3098) transformed with the empty pTrc99a vector (circles) or the vector carrying rpoE (triangles) under nonpermissive conditions. Expression of rpoE was induced with 50 μM IPTG. Values represent the means of triplicates, and error bars indicate standard deviations and are shown only when greater than the size of the symbol. (B) β-Galactosidase activity of the ompR malT(Con) mutant carrying an Φ(rpoHP3-lacZ) promoter fusion transformed with the empty pTrc99a vector (left bar) or the vector expressing rpoE (right bar). Cells were grown under nonpermissive conditions, and expression of rpoE was induced with 50 μM IPTG. Cells were harvested at an OD600 of 1. Values represent the means and standard deviations for triplicates.
Like σE, the Cpx and Rcs two-component systems combat stresses associated with the cell envelope (39, 56, 63). We therefore asked if the ompR malT(Con) double mutant experiences a general envelope stress or one specifically relieved by σE. We activated the Rcs system by introducing an ackA mutation into the ompR malT(Con) mutant (strain AJW2051). This mutation causes an accumulation of acetyl phosphate, which can activate several two-component systems, including the Rcs phosphorelay (22, 86). The ackA ompR malT(Con) triple mutant, however, was not viable (data not shown). Similarly, an ompR malT(Con) mutant that expresses the constitutively active cpxA* allele was not viable (data not shown). We conclude that the envelope stress experienced by ompR malT(Con) double mutants is specifically relieved by the σE regulon.
Identification of σE regulon members that support survival.
We used two different approaches to seek σE regulon members involved in promoting cell survival. First, we overexpressed σE regulon members (see Table S1 in the supplemental material) (16, 47, 63) in the ompR malT(Con) double mutant and screened the resulting transformants for viability under nonpermissive conditions. We found that expression of lhr, narW, ybfG, and lptB permitted survival (data not shown). These genes encode a putative helicase, a chaperone belonging to the group of redox enzyme maturation proteins, a TqsA-regulated pseudogene associated with biofilm formation, and the ATPase subunit of the LPS transporter, respectively. The finding that expression of lptB permits cell survival suggests that the stress experienced by ompR malT(Con) double mutants might be associated with an outer membrane imbalance.
Second, we determined whether σE exerts its effect by regulating small RNAs (sRNAs). In both Salmonella enterica and E. coli, the σE-regulated MicA and RybB sRNAs have been reported to facilitate degradation of certain OMP mRNAs in an Hfq-dependent manner (29, 80, 82). MicA, particularly, has been shown to cause degradation of lamB mRNA in Salmonella enterica (8). Indeed, we found that the rseA ompR malT(Con) triple mutant has a reduced outer membrane protein profile as determined by outer membrane preparations (data not shown). This reduction in OMPs is likely the cause for survival of these cells. To test whether σE exerts its effect on cell survival by upregulating sRNAs and thus reducing LamB levels, we deleted hfq in the rseA ompR malT(Con) triple mutant (strain AJW4000) and found that these cells survived under nonpermissive conditions (data not shown). Thus, σE-regulated Hfq-dependent sRNAs do not promote cell survival.
Induction of σE by LamB is not sufficient to permit cell survival.
Since the increased production of LamB in ompR malT(Con) double mutants causes cell death, we hypothesized that these cells fail to sufficiently activate σE. To test this hypothesis, we measured activity of the rpoHP3 promoter in malT(Con) single and ompR malT(Con) double mutants and found that, similar to the case for WT cells, neither of the mutants induced transcription from the rpoHP3 promoter (data not shown). Similarly, the overexpression of lamB from the pCA24N vector in WT cells resulted in cell death but only weakly activated rpoHP3 transcription (data not shown). We conclude that, similar to the case for OmpF or OmpC (41), the overexpression of LamB can activate σE; however, this activation is not strong enough to permit survival.
Activation of the Pho regulon suppresses lethality.
Other transposon insertions that displayed a Mal+ phenotype were located in the pstSCAB-phoU operon. The phoU and pstC genes, as well as the pstSC intergenic region, were each disrupted by one insertion, while three insertions disrupted pstB. To confirm that disruptions in the pstSCAB-phoU operon indeed permit cell survival, we constructed pstB, pstC, and phoU mutants in an ompR malT(Con) double mutant background (strains AJW3782, AJW3783, and AJW3780, respectively) and grew the cells under nonpermissive conditions. On solid media and in liquid culture, deletion of pstB or pstC permitted survival (Fig. 6A). Likewise, deletion of phoU restored viability, despite its previously reported poor growth (Fig. 6A) (77). In contrast, the deletion of either pstA or pstS (strains AJW3781 and AJW3785, respectively) delayed death but did not permit survival (Fig. 6A).
FIG. 6.

Effect of PhoB activation on ompR malT(Con) mutant survival. (A) Deletion of the pst-phoU genes rescues lethality. Growth curve of the WT (black circles) (AJW678), ompR malT(Con) (black diamonds) (AJW3098), ompR malT(Con) pstS (gray diamonds) (AJW3785), ompR malT(Con) pstC (gray triangles) (AJW3783), ompR malT(Con) pstA (gray circles) (AJW3781), ompR malT(Con) pstB (gray squares) (strain AJW3782), and ompR malT(Con) phoU (white triangles) (AJW3780) strains under nonpermissive conditions are shown. Values represent the mean of triplicates, and error bars indicate standard deviations and are shown only when greater than the size of the symbol. (B) Expression of constitutive PhoBCA(E11K) delays death. Growth curves of the ompR malT(Con) mutant (black diamonds) (AJW3098) and the ompR malT(Con) mutant expressing phoBCA(E11K) (white triangles) are shown. For comparative purposes, the growth curve of the suppressed ompR malT(Con) rseA strain (black circles) is included. Values represent the means of triplicates, and error bars indicate standard deviations and are shown only when greater than the size of the symbol. (C) Activation of the PhoB regulon increases alkaline phosphatase activity in the ompR malT(Con) mutant. Cells were grown under nonpermissive conditions and harvested at an OD of approximately 0.3. Alkaline phosphatase activity was measured using p-NPP as a substrate and is expressed as arbitrary units as described in Materials and Methods. Error bars indicate the standard deviations for triplicates.
Because mutations in the pstSCAB-phoU operon activate transcription of the PhoB regulon (45, 77), we hypothesized that this activation permits cell survival. We tested this hypothesis by overexpressing the phoBCA(E11K) allele, which constitutively activates the PhoB regulon (William McCleary, personal communication). That ompR malT(Con) mutants carrying this allele displayed a delayed death phenotype (Fig. 6B) provides further evidence that activation of the PhoB regulon rescues lethality.
The observation that some PhoB regulon-inducing mutations permit cell survival, while others merely delay death, prompted us to ask whether varying levels of PhoB regulon activation cause these differences. To assess activation levels, we measured alkaline phosphatase (AP) activity. We found that a deletion of each of the pstSCAB-phoU genes caused increased AP activity in ompR malT(Con) mutants (Fig. 6C). Deletion of pstB, pstC, and phoU, which permitted viability, led to a substantial increase in AP activity, with the ompR malT(Con) pstB triple mutant displaying the highest activity (Fig. 6C). In contrast, deletion of pstS and pstA, which only delayed death, caused only a moderate increase of AP activity (Fig. 6C). Similarly, ompR malT(Con) double mutants transformed with the phoBCA(E11K) plasmid showed moderate AP activity (Fig. 6C). Taken together, these data indicate that activation of the PhoB regulon promotes viability of the ompR malT(Con) double mutant and that the strength of induction correlates with the degree of viability.
Our earlier observation that activation of σE can promote cell survival prompted us to test whether the activation of the PhoB regulon indirectly promoted viability by inducing the σE stress response. We measured rpoHP3 activity in ompR malT(Con) pstB and ompR malT(Con) pstC triple mutants and found that the promoter activity was not increased compared to that of the ompR malT(Con) double mutant (data not shown), showing that PhoB regulon activation permits viability independently of σE.
To identify the PhoB regulon member(s) that suppresses lethality in ompR malT(Con) mutants, we first turned our attention to the OMP PhoE. Since ompR malT(Con) mutants display an altered OMP composition, we hypothesized that increased levels of PhoE could compensate for the lack of OmpF and OmpC. If this hypothesis was correct, then a deletion of phoE would be expected to cause death of the otherwise viable ompR malT(Con) pstB and ompR malT(Con) pstC triple mutants. When we grew the resulting ompR malT(Con) pstB phoE (strain AJW4196) and ompR malT(Con) pstC phoE (strain AJW4197) quadruple mutants under nonpermissive conditions, we indeed found these strains to be nonviable (Fig. 7A and B). In contrast, the overexpression of OmpF or OmpC in the ompR malT(Con) double mutant did not influence the lethal phenotype (data not shown). To check the status of PhoE (Fig. 7C) or the presence of OmpF or OmpC (data not shown) in these mutants, we performed outer membrane preparations. Taken together, these data show that PhoE makes a major contribution to suppression of the ompR malT(Con) lethality.
FIG. 7.
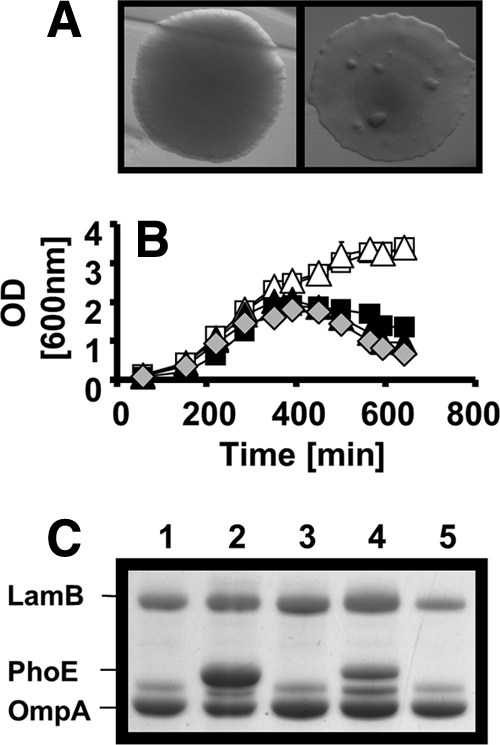
Effect of phoE on survival of ompR malT(Con) pstB and ompR malT(Con) pstC mutants. (A) Colony phenotype of the ompR malT(Con) pstC (AJW3946) (left) and the ompR malT(Con) pstC phoE (AJW4197) (right) mutants. Colonies were grown in LB at 37°C. (B) Growth curves of the ompR malT(Con) (AJW3098) (gray diamonds), ompR malT(Con) pstB (AJW3945) (white squares), ompR malT(Con) pstB phoE (AJW4196) (black squares), ompR malT(Con) pstC (AJW3946) (white triangles), and ompR malT(Con) pstC phoE (AJW4197) (black triangles) mutants. Cells were grown in LB at 37°C, and samples were taken at regular intervals. Values represent the means of triplicates, and error bars indicate standard deviations and are shown only when greater than the size of the symbol. (C) Deletion of phoE was confirmed using outer membrane preparations. Cells were grown in LB at 37°C and harvested during late exponential phase. Gels were stained with Coomassie brilliant blue. Lane 1, ompR malT(Con) mutant (AJW3098); lane 2, ompR malT(Con) pstB mutant (AJW3945); lane 3, ompR malT(Con) pstB phoE mutant (AJW4196); lane 4, ompR malT(Con) pstC mutant (AJW3946); lane 5, ompR malT(Con) pstC phoE mutant (AJW4197).
To test whether any of the other PhoB regulon members also play a role in viability, we overexpressed the monocistronic phoH gene in the ompR malT(Con) double mutant and found that it did not permit viability (data not shown). We then tested the multicistronic operons by inactivating the phoA-psiF, ugpBAECQ, or phnCDEFGHIJKLMNOP operon with marked polar deletions of the first gene in each operon. We found that polar mutations in phoA, phnC, or ugpB did not cause lethality in the ompR malT(Con) pstC or ompR malT(Con) pstB triple mutant parent strain (strains AJW3957, AJW3955, AJW3959, AJW3956, AJW3954, and AJW3958). Similarly, deletions of psiF, phnD, phnF, or phnO did not cause lethality in the ompR malT(Con) pstC triple mutant (strains AJW4248, AJW4245, AJW3964, and AJW3966) (data not shown). These results indicate that activation of these operons is not required for survival of the ompR malT(Con) mutant.
Inorganic phosphate suppresses lethality.
The observation that increased expression of PhoE can promote cell survival prompted us to ask whether inorganic phosphate, a substrate transported by PhoE, can support cell viability. We grew the ompR malT(Con) mutant in LB supplemented with 100 mM sodium phosphate instead of 80 mM NaCl and observed that the mutant was viable (Fig. 8). To ensure that survival was caused by the presence of inorganic phosphate and not the lack of sodium chloride, we supplemented the medium with 100 mM sucrose instead of NaCl and found that the cells did not survive (data not shown). We then tested whether survival in the presence of inorganic phosphate required PhoE. We supplemented the ompR malT(Con) pstC phoE mutant, which dies in LB without inorganic phosphate, and found that these cells did not survive (data not shown). We conclude that inorganic phosphate can promote cell survival and that it does so in a PhoE-dependent manner.
FIG. 8.
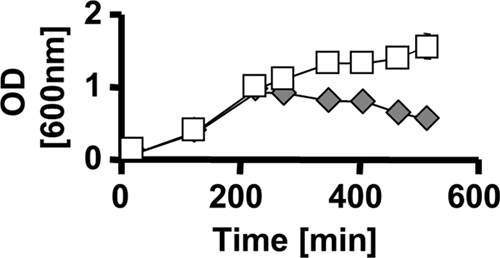
Effect of inorganic phosphate on ompR malT(Con) survival. Growth curves of the ompR malT(Con) mutant (AJW3098) in LB medium containing 80 mM NaCl (gray diamonds) and in LB containing 100 mM sodium phosphate (pH 7) instead of NaCl (white squares) are shown. Cells were grown at 37°C, and samples were taken at regular intervals. Values represent the means of triplicates, and error bars indicate standard deviations and are shown only when greater than the size of the symbol.
DISCUSSION
To survive, cells must respond to multiple environmental stimuli simultaneously. The proper response to one stress, however, could make the cell more susceptible to a second coexistent stress. To cope, a cell must possess a mechanism that balances the need to respond simultaneously to both stresses.
Our current and recent studies of ompR malT(Con) double mutants of E. coli show that elevated expression of LamB, the OMP responsible for maltose and maltodextrin uptake, causes cell death when the osmoregulator OmpR is disabled (58). To gain insight into the behavior of ompR malT(Con) double mutants, we first described the death process and then sought mutations that suppress death under nonpermissive conditions.
Characterization of death.
ompR malT(Con) double mutants exhibit a fascinating behavior. Under nonpermissive conditions, they initially grow at the same rate as WT cells and appear normal (Fig. 2A). Upon entering late exponential phase, however, these cells experience vast changes in cell morphology characterized by severe disruption of their cytoplasmic membranes (Fig. 2B to G and 3A) and aberrant outer membrane permeability (Fig. 3B). These morphological changes occur approximately coincident with the appearance of ghost-like cells (Fig. 2B to E) and a precipitous drop in CFU (58). During further growth, the percentage of ghosts diminishes (Fig. 2F), membrane fragments and cellular debris appear (Fig. 2E), and the culture rapidly loses turbidity (Fig. 2A) (58). Ultimately, the culture is taken over by suppressor mutants with intact cell envelopes (Fig. 2F). Some of these cultures include a subpopulation of cells with plasmolytic bays (Fig. 2F, arrows), which are often formed by cells exposed to high-osmolarity conditions (33, 46). The development of plasmolytic bays in certain suppressor strains might be an attempt by cells that lack OmpR to cope with elevated osmolality.
It is clear that death ensues because of membrane damage, but what actually damages the membranes? Substantial evidence supports the hypothesis that LamB expression in the absence of a functional OmpR is the major contributing factor, as mutations that eliminate or conditions that diminish the expression of LamB permit survival of the ompR malT(Con) double mutant (58). To understand why LamB causes such problems, we performed a genome-wide unbiased search for suppressor mutations. The resultant suppressor mutations fell into three distinct classes: (i) those that eliminate or diminish LamB expression, (ii) suppressors that activate the σE regulon, and (iii) suppressors that activate the PhoB regulon (Fig. 8). Each class is discussed below.
Suppressors that eliminate or diminish LamB expression.
About half of all suppressor mutants, whether of spontaneous origin or caused by transposon insertion, showed weak or no growth on maltose as the sole carbon source. The identities of the transposon-disrupted genes (lamB, malK, malT, cya, and hns) confirm our earlier report that LamB expression permits survival of the ompR malT(Con) double mutant (58) and support our hypothesis that LamB is the primary cause of death.
Suppressors that activate σE.
Perturbations in cell envelope compartments, such as misincorporated OMPs, an imbalance in the porin composition, or a skewed LPS-to-porin ratio, are reported to activate the σE envelope stress response (1, 21, 25, 55). In the absence of such perturbations, the anti-σE factors RseA and RseB act in concert to sequester σE at the inner membrane and thus inhibit σE-dependent transcription (1, 21, 25, 55). In response to those perturbations, a proteolytic cascade, mediated by the proteases RseP and DegS, releases σE (1, 2), which associates with core RNA polymerase to transcribe a set of genes that combat envelope stress (16, 47, 60). This set of genes primarily includes periplasmic folding catalysts and proteases that assist in the assembly of outer membrane proteins or the degradation of misfolded polypeptides. It also includes genes involved in outer membrane biogenesis.
Activation of the σE regulon, either by disruption of rseA or rseB or by overexpression of rpoE, supported survival of the ompR malT(Con) double mutant (Fig. 4 and 5). The finding that activation of this envelope stress response suppresses lethality supports our microscopic and biochemical findings that increased LamB expression in an ompR mutant background causes a severe envelope stress that ultimately results in death. This stress could arise during translocation of excess LamB across the cytoplasmic membrane, during its folding and assembly in the periplasm, or by its presence in the outer membrane.
It is unlikely that the ompR malT(Con) double mutant dies because its Sec translocon has difficulty secreting WT LamB induced by MalT(Con). We base this conclusion on the observation that YccA, an inhibitor of the FtsH protease that degrades jammed Sec transporters, did not affect the death phenotype (81; S. A. Reimann and A. J. Wolfe, unpublished data). It is also unlikely that death ensues because of folding and assembly problems. Several σE regulon members facilitate folding, assembly, and/or degradation of OMPs. For example, DegP, SurA, and Skp exhibit general chaperone activities (21) and are thought to assist in the delivery of the porins to their final destination (21, 54, 65, 67, 75). Similarly, expression of CpxA* has been reported to cause the degradation of the misfolded LamB-LacZ-PhoA tripartite fusion protein by activating the protease/chaperone DegP. However, viability was not promoted by overexpression of these chaperones (Reimann and Wolfe, unpublished data) or of CpxA* (data not shown). We conclude that LamB does not exert its detrimental effect by accumulating or by misfolding in the periplasm.
Outer membrane preparations of the ompR malT(Con) rseA triple mutant revealed somewhat reduced amounts of its major OMPs, OmpA and LamB (data not shown). This reduction does not appear to act through the σE regulon members MicA and RybB, two sRNAs that degrade certain OMP mRNAs in an Hfq-dependent manner (29, 80, 82), because deletion of hfq in the ompR malT(Con) rseA triple mutant did not cause lethality (data not shown). Thus, we conclude that σE does not exert its effect on LamB-induced stress through Hfq-dependent sRNAs. However, the reduced porin concentration in the outer membrane of the ompR malT(Con) rseA triple mutant does suggest that death results from some imbalance between outer membrane components. This hypothesis is further supported by the observation that overexpression of lptB suppresses death. LptB, a protein with a nucleotide-binding domain typically found in ABC transporters, is located at the cytoplasmic face of the inner membrane (78). It is thought to assist in extracting LPS from the outer leaflet of the inner membrane for subsequent transport to the outer membrane (68, 76). LPS is an essential component of the E. coli outer membrane, and imbalances between LPS and OMPs have been shown to cause lethality (66, 69, 87). Intriguingly, lptB is located in a locus that includes ptsN, whose overexpression suppresses general envelope stress (25), including the stress experienced by the ompR malT(Con) double mutant (Reimann and Wolfe, unpublished data). Thus, we propose that induction of σE suppresses death by performing two functions: decreasing overall porin concentration while increasing LPS transport.
Suppressors that activate the PhoB regulon.
The majority of Mal+ suppressors were disruptions of the pstSCAB-phoU operon. Because the products of this operon inhibit the two-component system PhoBR, mutations in the PstSCAB transporter and the modulator protein PhoU (27, 45, 61) activate the PhoB regulon, whose characterized members function in the uptake and metabolism of phosphate sources (57, 79, 84).
The PhoB regulon includes three transport systems that facilitate the uptake of inorganic phosphate or phosphorous compounds into the cytoplasm. The Pst, Ugp, and Phn transport systems (encoded by pstSCAB, ugpBACEQ, and phnCDEFGHIJKLMNOP, respectively) facilitate uptake and metabolism of inorganic phosphate, glycerol-3-phosphate, and phosphonates, respectively (9, 13, 14, 72, 83). In addition to these transporters, the PhoB regulon includes two proteins that assist in phosphate uptake: the periplasmic alkaline phosphatase and the OMP PhoE (encoded by phoA and phoE, respectively) (35, 85). The functions of two further PhoB regulon members, PhoH and PsiF, are currently unknown (31, 42).
Intriguingly, we found that mutations in certain genes of the pstSCAB-phoU operon permit survival of the ompR malT(Con) mutant, while mutations in other genes only delay death. Furthermore, we found that the level of PhoB regulon activation correlates with the degree of viability. These observations argue against the simple hypothesis that transcription of this operon is driven by a single upstream promoter. Instead, they argue for a more complex architecture that includes several internal promoters. Indeed, an additional promoter upstream of pstB has been suggested (3).
When activated, the response regulator PhoB positively regulates all regulon members, including the OMP PhoE (27). PhoE belongs to the general porin family, which also includes OmpF and OmpC. In contrast to the more substrate-specific porins, e.g., the maltose/maltodextrin-specific LamB, these general porins exhibit much less preference with regard to their substrates. PhoE, also called phosphoporin because of its preference for phosphate, tends to prefer the uptake of anions, whereas OmpF and OmpC preferentially admit cations (34, 48). That increased PhoE expression permits viability of the ompR malT(Con) double mutant may be attributed to (i) its increased presence in the outer membrane or (ii) its function as a general OMP. Both possibilities are discussed below.
Imbalances between outer membrane components have been reported to cause cell damage, and genetic interactions between proteins that assist in LPS assembly and proteins that assist in outer membrane protein assembly have been demonstrated. These studies suggest that the integrity of the outer membrane requires an appropriate balance between LPS and OMP assembly (66, 69, 87). The ompR malT(Con) double mutant clearly has an altered OMP composition, since it lacks the OMPs OmpF and OmpC while possessing an abundance of LamB. The fact that increased PhoE can rescue viability in these cells might lead to the assumption that PhoE can compensate for the lack of OmpF and/or OmpC. If this were the case, then expression of OmpF or OmpC in the ompR malT(Con) double mutant also should permit viability, but this was not the case (data not shown). Thus, more than a simple imbalance between the OMPs is responsible for cell death.
An alternative explanation could involve the function of PhoE. That PhoE specifically suppresses ompR malT(Con) death could lie in its preference for phosphorylated and anionic compounds. If cell survival requires the transport of a phosphorylated anionic compound, it would explain why OmpF and OmpC do not suppress death. Indeed, exposure to inorganic phosphate promoted survival of the ompR malT(Con) mutant in a PhoE-dependent manner (Fig. 8). It is unlikely that exposure to 100 mM inorganic phosphate promotes viability by increasing PhoE levels, because the PhoB regulon is induced in response to reduced phosphate concentrations, not elevated ones (27, 84). Yet, a small amount of PhoE appears to be sufficient for inorganic phosphate to enter the cell and exert its effect. Whether phosphate is utilized directly in the periplasm or is further transported into the cytoplasm by the constitutive Pit transporter remains unclear. It is, however, possible that increased cytoplasmic phosphate levels are utilized for the assembly of phosphorylated cell envelope components such as LPS or phospholipids, which subsequently promote cell survival.
Intriguingly, PhoE is reported to be an OmpR regulon member (10, 50, 53). Those studies, however, reported that OmpR represses phoE transcription (Fig. 9). Thus, deletion of ompR would be expected to enhance PhoE expression. However, we were unable to detect PhoE in the ompR malT(Con) mutant (Fig. 7C). We therefore conclude that the ability of OmpR to protect malT(Con) mutants from death does not involve PhoE and that PhoE suppresses cell death independently of OmpR.
FIG. 9.

The multifactorial role of OmpR in the prevention of cell death caused by increased LamB levels in malT(Con) mutants. The main cause of ompR malT(Con) synthetic lethality is increased LamB expression in the absence of OmpR. Cell death can be prevented by several avenues, as follows. (i) Reduced expression of LamB eliminates the main cause of lethality. (ii) The activation of σE by OmpR leads to increased abundance of LPS in the outer membrane, restoring the porin/LPS balance. (iii) The increased abundance of the OMP PhoE restores the porin/LPS balance. (iv) The uptake of inorganic phosphate by PhoE enables phosphate or a derivative to prevent cell death.
Why does MalT(Con)-induced LamB expression cause death when OmpR is disabled?
We propose that death of the ompR malT(Con) mutant results from two imbalances: one between LPS and OMP and a second involving inorganic phosphate or a derivative. We base this proposal on the following observations: (i) the viable ompR malT(Con) rseA triple mutant carries a diminished OMP load, (ii) elevated expression of the σE-regulated LPS transport-associated protein LptB supports viability, (iii) induction of PhoE specifically suppresses death without inducing σE, and (iv) exposure to inorganic phosphate suppresses death. We further propose that these imbalances result in reduced outer membrane integrity, which either immediately precedes or coincides with cytoplasmic membrane disintegration and the consequent spillage of cytoplasmic contents.
Supplementary Material
Acknowledgments
We thank Winfried Boos, Tom Silhavy, Linda Kenney, Carol Gross, William McCleary, Tom Bernhardt, William Metcalf, and the National Institute for Genetics (Japan) for providing strains, reporter fusions, plasmids, and phage; Noriko Shibata and Linda Fox for help with transmission electron microscopy; Bozena Zemaitaitis for performing experiments with inorganic phosphate; Karen Visick for fruitful discussions; members of the Wolfe and Visick labs for critical reading of the manuscript; and the National Institute of General Medical Sciences (GM066130) and the Loyola University Potts Foundation (LU 11200) for funding.
Footnotes
Published ahead of print on 3 December 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Alba, B. M., and C. A. Gross. 2004. Regulation of the Escherichia coli sigma-dependent envelope stress response. Mol. Microbiol. 52:613-619. [DOI] [PubMed] [Google Scholar]
- 2.Alba, B. M., J. A. Leeds, C. Onufryk, C. Z. Lu, and C. A. Gross. 2002. DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma(E)-dependent extracytoplasmic stress response. Genes Dev. 16:2156-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amemura, M., K. Makino, H. Shinagawa, A. Kobayashi, and A. Nakata. 1985. Nucleotide sequence of the genes involved in phosphate transport and regulation of the phosphate regulon in Escherichia coli. J. Mol. Biol. 184:241-250. [DOI] [PubMed] [Google Scholar]
- 4.Baba, T., et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beatty, C. M., D. F. Browning, S. J. Busby, and A. J. Wolfe. 2003. Cyclic AMP receptor protein-dependent activation of the Escherichia coli acsP2 promoter by a synergistic class III mechanism. J. Bacteriol. 185:5148-5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boos, W., and A. Bohm. 2000. Learning new tricks from an old dog: MalT of the Escherichia coli maltose system is part of a complex regulatory network. Trends Genet. 16:404-409. [DOI] [PubMed] [Google Scholar]
- 7.Boos, W., and H. Shuman. 1998. Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol. Mol. Biol. Rev. 62:204-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossi, L., and N. Figueroa-Bossi. 2007. A small RNA downregulates LamB maltoporin in Salmonella. Mol. Microbiol. 65:799-810. [DOI] [PubMed] [Google Scholar]
- 9.Brzoska, P., and W. Boos. 1988. Characteristics of a ugp-encoded and phoB-dependent glycerophosphoryl diester phosphodiesterase which is physically dependent on the Ugp transport system of Escherichia coli. J. Bacteriol. 170:4125-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Case, C. C., B. Bukau, S. Granett, M. R. Villarejo, and W. Boos. 1986. Contrasting mechanisms of envZ control of mal and pho regulon genes in Escherichia coli. J. Bacteriol. 166:706-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapon, C. 1982. Role of the catabolite activator protein in the expression of the maltose regulon of Escherichia coli. Ann. Microbiol. (Paris) 133A:77-80. [PubMed] [Google Scholar]
- 12.Chapon, C., and A. Kolb. 1983. Action of CAP on the malT promoter in vitro. J. Bacteriol. 156:1135-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox, G. B., H. Rosenberg, J. A. Downie, and S. Silver. 1981. Genetic analysis of mutants affected in the Pst inorganic phosphate transport system. J. Bacteriol. 148:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox, G. B., D. Webb, and H. Rosenberg. 1989. Specific amino acid residues in both the PstB and PstC proteins are required for phosphate transport by the Escherichia coli Pst system. J. Bacteriol. 171:1531-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dardonville, B., and O. Raibaud. 1990. Characterization of malT mutants that constitutively activate the maltose regulon of Escherichia coli. J. Bacteriol. 172:1846-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dartigalongue, C., D. Missiakas, and S. Raina. 2001. Characterization of the Escherichia coli sigma E regulon. J. Biol. Chem. 276:20866-20875. [DOI] [PubMed] [Google Scholar]
- 17.Das, S., J. C. Noe, S. Paik, and T. Kitten. 2005. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J. Microbiol. Methods 63:89-94. [DOI] [PubMed] [Google Scholar]
- 18.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debarbouille, M., H. A. Shuman, T. J. Silhavy, and M. Schwartz. 1978. Dominant constitutive mutations in malT, the positive regulator gene of the maltose regulon in Escherichia coli. J. Mol. Biol. 124:359-371. [DOI] [PubMed] [Google Scholar]
- 20.de Boer, P. A., R. E. Crossley, and L. I. Rothfield. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56:641-649. [DOI] [PubMed] [Google Scholar]
- 21.Duguay, A. R., and T. J. Silhavy. 2004. Quality control in the bacterial periplasm. Biochim. Biophys. Acta 1694:121-134. [DOI] [PubMed] [Google Scholar]
- 22.Fredericks, C. E., S. Shibata, S. Aizawa, S. A. Reimann, and A. J. Wolfe. 2006. Acetyl phosphate-sensitive regulation of flagellar biogenesis and capsular biosynthesis depends on the Rcs phosphorelay. Mol. Microbiol. 61:734-747. [DOI] [PubMed] [Google Scholar]
- 23.Greenspan, P., and S. D. Fowler. 1985. Spectrofluorometric studies of the lipid probe, Nile red. J. Lipid Res. 26:781-789. [PubMed] [Google Scholar]
- 24.Guven, K., M. Yolcu, R. Gul-Guven, S. Erdogan, and D. D. Pomerai. 2005. The effects of organic pesticides on inner membrane permeability in Escherichia coli ML35. Cell Biol. Toxicol. 21:73-81. [DOI] [PubMed] [Google Scholar]
- 25.Hayden, J. D., and S. E. Ades. 2008. The extracytoplasmic stress factor, SigmaE, is required to maintain cell envelope integrity in Escherichia coli. PLoS One 3:e1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herrera, G., A. Martinez, M. Blanco, and J. E. O'Connor. 2002. Assessment of Escherichia coli B with enhanced permeability to fluorochromes for flow cytometric assays of bacterial cell function. Cytometry 49:62-69. [DOI] [PubMed] [Google Scholar]
- 27.Hsieh, Y. J., and B. L. Wanner. 2010. Global regulation by the seven-component Pi signaling system. Curr. Opin. Microbiol. 13:198-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Igo, M. M., J. M. Slauch, and T. J. Silhavy. 1990. Signal transduction in bacteria: kinases that control gene expression. New Biol. 2:5-9. [PubMed] [Google Scholar]
- 29.Johansen, J., A. A. Rasmussen, M. Overgaard, and P. Valentin-Hansen. 2006. Conserved small non-coding RNAs that belong to the SigmaE regulon: role in down-regulation of outer membrane proteins. J. Mol. Biol. 364:1-8. [DOI] [PubMed] [Google Scholar]
- 30.Johansson, J., B. Dagberg, E. Richet, and B. E. Uhlin. 1998. H-NS and StpA proteins stimulate expression of the maltose regulon in Escherichia coli. J. Bacteriol. 180:6117-6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kazakov, A. E., O. Vassieva, M. S. Gelfand, A. Osterman, and R. Overbeek. 2003. Bioinformatics classification and functional analysis of PhoH homologs. In Silico Biol. 3:3-15. [PubMed] [Google Scholar]
- 32.Kitagawa, M., et al. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12:291-299. [DOI] [PubMed] [Google Scholar]
- 33.Koch, A. L. 1998. The biophysics of the gram-negative periplasmic space. Crit. Rev. Microbiol. 24:23-59. [DOI] [PubMed] [Google Scholar]
- 34.Koebnik, R., K. P. Locher, and P. Van Gelder. 2000. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol. Microbiol. 37:239-253. [DOI] [PubMed] [Google Scholar]
- 35.Korteland, J., J. Tommassen, and B. Lugtenberg. 1982. PhoE protein pore of the outer membrane of Escherichia coli K12 is a particularly efficient channel for organic and inorganic phosphate. Biochim. Biophys. Acta 690:282-289. [DOI] [PubMed] [Google Scholar]
- 36.Kullman, L., M. Winterhalter, and S. M. Bezrukov. 2002. Transport of maltodextrins through maltoporin: a single-channel study. Biophys. J. 82:803-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumari, S., et al. 2000. Regulation of acetyl coenzyme A synthetase in Escherichia coli. J. Bacteriol. 182:4173-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsen, R. A., M. M. Wilson, A. M. Guss, and W. W. Metcalf. 2002. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 178:193-201. [DOI] [PubMed] [Google Scholar]
- 39.Laubacher, M. E., and S. E. Ades. 2008. The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J. Bacteriol. 190:2065-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lauro, F. M., et al. 2008. Large-scale transposon mutagenesis of Photobacterium profundum SS9 reveals new genetic loci important for growth at low temperature and high pressure. J. Bacteriol. 190:1699-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mecsas, J., P. E. Rouviere, J. W. Erickson, T. J. Donohue, and C. A. Gross. 1993. The activity of sigma E, an Escherichia coli heat-inducible sigma-factor, is modulated by expression of outer membrane proteins. Genes Dev. 7:2618-2628. [DOI] [PubMed] [Google Scholar]
- 42.Metcalf, W. W., P. M. Steed, and B. L. Wanner. 1990. Identification of phosphate starvation-inducible genes in Escherichia coli K-12 by DNA sequence analysis of psi::lacZ(Mu d1) transcriptional fusions. J. Bacteriol. 172:3191-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizuno, T., and S. Mizushima. 1990. Signal transduction and gene regulation through the phosphorylation of two regulatory components: the molecular basis for the osmotic regulation of the porin genes. Mol. Microbiol. 4:1077-1082. [DOI] [PubMed] [Google Scholar]
- 44.Morona, R., and P. Reeves. 1982. The tolC locus of Escherichia coli affects the expression of three major outer membrane proteins. J. Bacteriol. 150:1016-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muda, M., N. N. Rao, and A. Torriani. 1992. Role of PhoU in phosphate transport and alkaline phosphatase regulation. J. Bacteriol. 174:8057-8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mulder, E., and C. L. Woldringh. 1993. Plasmolysis bays in Escherichia coli: are they related to development and positioning of division sites? J. Bacteriol. 175:2241-2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mutalik, V. K., G. Nonaka, S. E. Ades, V. A. Rhodius, and C. A. Gross. 2009. Promoter strength properties of the complete sigma E regulon of Escherichia coli and Salmonella enterica. J. Bacteriol. 191:7279-7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nikaido, H., and E. Y. Rosenberg. 1983. Porin channels in Escherichia coli: studies with liposomes reconstituted from purified proteins. J. Bacteriol. 153:241-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oshima, T., et al. 2002. Transcriptome analysis of all two-component regulatory system mutants of Escherichia coli K-12. Mol. Microbiol. 46:281-291. [DOI] [PubMed] [Google Scholar]
- 50.Oshima, T., S. Ishikawa, K. Kurokawa, H. Aiba, and N. Ogasawara. 2006. Escherichia coli histone-like protein H-NS preferentially binds to horizontally acquired DNA in association with RNA polymerase. DNA Res. 13:141-153. [DOI] [PubMed] [Google Scholar]
- 51.O'Toole, G. A., and R. Kolter. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449-461. [DOI] [PubMed] [Google Scholar]
- 52.Powell, B. S., M. P. Rivas, D. L. Court, Y. Nakamura, and C. L. Turnbough, Jr. 1994. Rapid confirmation of single copy lambda prophage integration by PCR. Nucleic Acids Res. 22:5765-5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pruss, B. M., C. Besemann, A. Denton, and A. J. Wolfe. 2006. A complex transcription network controls the early stages of biofilm development by Escherichia coli. J. Bacteriol. 188:3731-3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raivio, T. L. 2005. Envelope stress responses and Gram-negative bacterial pathogenesis. Mol. Microbiol. 56:1119-1128. [DOI] [PubMed] [Google Scholar]
- 55.Raivio, T. L., and T. J. Silhavy. 2001. Periplasmic stress and ECF sigma factors. Annu. Rev. Microbiol. 55:591-624. [DOI] [PubMed] [Google Scholar]
- 56.Raivio, T. L., and T. J. Silhavy. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179:7724-7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rao, N. N., and A. Torriani. 1990. Molecular aspects of phosphate transport in Escherichia coli. Mol. Microbiol. 4:1083-1090. [DOI] [PubMed] [Google Scholar]
- 58.Reimann, S. A., and A. J. Wolfe. 2009. A critical process controlled by MalT and OmpR is revealed through synthetic lethality. J. Bacteriol. 191:5320-5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reyes, M., and H. A. Shuman. 1988. Overproduction of MalK protein prevents expression of the Escherichia coli mal regulon. J. Bacteriol. 170:4598-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rhodius, V. A., W. C. Suh, G. Nonaka, J. West, and C. A. Gross. 2006. Conserved and variable functions of the SigmaE stress response in related genomes. PLoS Biol. 4:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rice, C. D., J. E. Pollard, Z. T. Lewis, and W. R. McCleary. 2009. Employment of a promoter-swapping technique shows that PhoU modulates the activity of the PstSCAB2 ABC transporter in Escherichia coli. Appl. Environ. Microbiol. 75:573-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richet, E. 1996. On the role of the multiple regulatory elements involved in the activation of the Escherichia coli malEp promoter. J. Mol. Biol. 264:852-862. [DOI] [PubMed] [Google Scholar]
- 63.Rizzitello, A. E., J. R. Harper, and T. J. Silhavy. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 183:6794-6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rotman, B. 1977. On the rate limiting step in downhill transport via the LacY permease of Escherichia coli. J. Supramol. Struct. 7:29-35. [DOI] [PubMed] [Google Scholar]
- 65.Rouviere, P. E., and C. A. Gross. 1996. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 10:3170-3182. [DOI] [PubMed] [Google Scholar]
- 66.Ruiz, N., B. Falcone, D. Kahne, and T. J. Silhavy. 2005. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121:307-317. [DOI] [PubMed] [Google Scholar]
- 67.Ruiz, N., D. Kahne, and T. J. Silhavy. 2006. Advances in understanding bacterial outer-membrane biogenesis. Nat. Rev. Microbiol. 4:57-66. [DOI] [PubMed] [Google Scholar]
- 68.Ruiz, N., D. Kahne, and T. J. Silhavy. 2009. Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat. Rev. Microbiol. 7:677-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruiz, N., T. Wu, D. Kahne, and T. J. Silhavy. 2006. Probing the barrier function of the outer membrane with chemical conditionality. ACS Chem. Biol. 1:385-395. [DOI] [PubMed] [Google Scholar]
- 70.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratroy manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 71.Schlegel, A., O. Danot, E. Richet, T. Ferenci, and W. Boos. 2002. The N terminus of the Escherichia coli transcription activator MalT is the domain of interaction with MalY. J. Bacteriol. 184:3069-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schweizer, H., M. Argast, and W. Boos. 1982. Characteristics of a binding protein-dependent transport system for sn-glycerol-3-phosphate in Escherichia coli that is part of the pho regulon. J. Bacteriol. 150:1154-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Silhavy, T. J., M. L. Berman, and L. W. Enquist. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Cold Spring Har- bor, NY.
- 74.Simons, R. W., F. Houman, and N. Kleckner. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85-96. [DOI] [PubMed] [Google Scholar]
- 75.Sklar, J. G., T. Wu, D. Kahne, and T. J. Silhavy. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 21:2473-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sperandeo, P., et al. 2007. Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J. Bacteriol. 189:244-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steed, P. M., and B. L. Wanner. 1993. Use of the rep technique for allele replacement to construct mutants with deletions of the pstSCAB-phoU operon: evidence of a new role for the PhoU protein in the phosphate regulon. J. Bacteriol. 175:6797-6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stenberg, F., et al. 2005. Protein complexes of the Escherichia coli cell envelope. J. Biol. Chem. 280:34409-34419. [DOI] [PubMed] [Google Scholar]
- 79.Torriani, A. 1990. From cell membrane to nucleotides: the phosphate regulon in Escherichia coli. Bioessays 12:371-376. [DOI] [PubMed] [Google Scholar]
- 80.Udekwu, K. I., and E. G. Wagner. 2007. Sigma E controls biogenesis of the antisense RNA MicA. Nucleic Acids Res. 35:1279-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Stelten, J., F. Silva, D. Belin, and T. J. Silhavy. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vogel, J., and K. Papenfort. 2006. Small non-coding RNAs and the bacterial outer membrane. Curr. Opin. Microbiol. 9:605-611. [DOI] [PubMed] [Google Scholar]
- 83.Wanner, B. L., and J. A. Boline. 1990. Mapping and molecular cloning of the phn (psiD) locus for phosphonate utilization in Escherichia coli. J. Bacteriol. 172:1186-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wanner, B. L., and B. D. Chang. 1987. The phoBR operon in Escherichia coli K-12. J. Bacteriol. 169:5569-5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wanner, B. L., and P. Latterell. 1980. Mutants affected in alkaline phosphatase, expression: evidence for multiple positive regulators of the phosphate regulon in Escherichia coli. Genetics 96:353-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wanner, B. L., and M. R. Wilmes-Riesenberg. 1992. Involvement of phosphotransacetylase, acetate kinase, and acetyl phosphate synthesis in control of the phosphate regulon in Escherichia coli. J. Bacteriol. 174:2124-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu, T., et al. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235-245. [DOI] [PubMed] [Google Scholar]
- 88.Zhang, L., R. Benz, and R. E. Hancock. 1999. Influence of proline residues on the antibacterial and synergistic activities of alpha-helical peptides. Biochemistry 38:8102-8111. [DOI] [PubMed] [Google Scholar]
- 89.Zundel, C. J., D. C. Capener, and W. R. McCleary. 1998. Analysis of the conserved acidic residues in the regulatory domain of PhoB. FEBS Lett. 441:242-246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.