

Abstract
In Corynebacterium glutamicum, the ArgR protein, a transcriptional repressor, affects the expression level of the argB gene through binding to its promoter region. The argB promoter region (positions −77 to −25) has been found by in vitro electrophoretic mobility shift assay (EMSA) results and in silico analysis to be important for the DNA binding of ArgR. Proline supplementation prevented the DNA binding of ArgR to the argB promoter region and triggered an increase of the argB mRNA level. Additional mutational analyses of the argB promoter region found nucleotides critical for ArgR binding (G located at position −58, C at position −55, and A at position −41 of the argB promoter) in that region. Another transcriptional repressor, FarR, was also demonstrated to bind to the argB promoter region. This binding was delimited to positions −57 to −77 on the argB promoter. FarR has only one putative binding domain located at positions −57 to −77, but this region exactly overlapped with the binding region located from positions −55 to −77 for the binding of ArgR within the argB promoter; thus, if ArgR bound with the argB promoter first, the binding of FarR was not observed in this region. However, if FarR bound to the binding domain located at positions −57 to −77 first, ArgR could bind other binding sites located at positions −49 to −25 within the argB promoter. Finally, this study suggests that ArgR can affect FarR binding to the argB promoter region, as protein binding is dominated by the protein most able to do so.
The main regulator of a set of arg regulons (19), arginine repressor (ArgR), acts as a transcriptional repressor by binding to the hexameric structure of its target sequences, known as “ARG boxes”(7). Detailed studies of ArgR have been conducted with many bacteria, for example, Escherichia coli (19), Pseudomonas aeruginosa (22), and Bacillus stearothermophilus (6). In addition, the mechanism of ArgR is unusually well conserved across a wide range of divergent bacteria: both Gram-negative and Gram-positive bacteria (35).
Corynebacterium glutamicum is a Gram-positive soil bacterium widely used in the production of amino acids (18, 32). Bioinformatics tools have recently been used to detect the potential transcription regulators of winged helix-turn-helix (HTH) binding proteins, including ArgR, that were previously predicted from the genome sequence (1, 2, 11). Experimental data and in silico analyses of a diverse range of bacteria show a surprising conservation of the arginine repressor proteins and their respective target sites. For instance, the identical 18.8-kDa polypeptides and the folded structure derived from the amino acid sequence of ArgR molecules from C. glutamicum closely match the winged-helix structures and N-terminal DNA-binding domains of several other species (14, 20).
Previous studies have shown that the biosynthesis of ornithine, an intermediate molecule of arginine's biosynthesis, depends upon the DNA binding of ArgR to the operating regions of arg genes (14). In addition to its regulatory function, ArgR has a particular activity for the upstream region of the argB gene, encoding the N-acetylglutamate kinase enzyme, an enzyme relevant to ornithine biosynthesis in C. glutamicum (14, 16).
Fatty acyl-responsive regulator (FarR) (30), a previously uncharacterized transcription factor of the HTH GntR family (27) similar to HutC/FarR, seems to be involved in the regulation of amino acid biosynthesis in C. glutamicum (8). The transcription of the argB gene was influenced by FarR. Interestingly, both ArgR and FarR control ornithine and arginine levels by repressing the transcription of the arg genes (8, 14). However, the mechanism by which FarR stimulates argB transcription remains unclear.
This paper's focus is the ArgR-mediated regulation of argB expression in C. glutamicum. To establish how ArgR's operating site acts on the argB gene, the effects of ArgR on DNA-binding affinity were examined in vitro. The effects of the DNA-binding sites of FarR on the promoters of arg genes were also analyzed by in vivo chromatin immunoprecipitation (ChIP) assays of C. glutamicum. Furthermore, a new relationship of the two transcriptional regulators FarR and ArgR to the argB promoter is provided through the detailed analysis of the interaction of FarR with the promoter site of the argB gene.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
Table 1 lists the bacterial strains and plasmids used in this study. Wild-type strain C. glutamicum ATCC 13032 (American Type Culture Collection, Manassas, VA) and mutant strain C. glutamicum SJC 8074 (provided by Sangji University, South Korea) (10) were grown at 30°C in Luria-Bertani medium (29) and, for the production of ornithine and arginine, in mineral medium containing yeast extract (MMY) [0.8 g KH2PO4, 10 g (NH4)2SO4, 1 g MgSO4·7H2O, 1.2 g Na2HPO4, 20 mg MnSO4·H2O, 20 mg FeSO4·7H2O, 10 mg ZnSO4·7H2O, 10 g yeast extract, 20 g CaCO3, and 60 g glucose liter−1) (17). In a 250-ml shake flask, a 1-ml preculture was inoculated into 50 ml of MMY medium. Cultivation was performed at 30°C at 150 rpm on a rotary shaker. E. coli BL21(DE3) cells (Novagen Chemicals, Inc., Germany) were grown at 37°C in Luria-Bertani medium (29). Selection for the presence of plasmids was carried out by using ampicillin (50 μg ml−1 for E. coli). Shake flask culture growth was monitored by measuring the optical density at 600 nm (OD600) using a UV/Vis spectrophotometer (Mecasys Co., Ltd., South Korea).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| C. glutamicum | Wild type | ATCC 13032 |
| C. glutamicum SJC 8074 | Deletion of the argR, argF, and proB genes | 10 |
| E. coli BL21(DE3) | F−ompT hsdSB(rB− mB−) gal dcm (DE3) | Novagen, Madison, WI |
| Plasmids | ||
| pET-21a | Apr; f1 origin; 6× histidyl fusion vector | Novagen, Madison, WI |
| pEMBTL-SY0 | pET-21a containing the argR structural gene | 14 |
| pEMBTL-SY5 | pET-21a containing the farR structural gene | 13 |
Genetic manipulations.
Chromosomal DNA was prepared from C. glutamicum by using a Wizard SV genomic DNA kit (Promega, Madison, WI). Plasmid DNA was prepared from E. coli cells using an alkaline lysis technique with a QIAspin miniprep kit (Qiagen, Germany). DNA modification, analysis by agarose gel electrophoresis, and ligation were performed according to standard procedures (29). The PCR experiments were carried out by using a T Gradient thermocycler (Biometra, Germany), Ex Taq DNA polymerase (Takara Bio, Inc., Japan), and chromosomal DNA as the template. The PCR products were purified by using a QIAquick PCR purification kit (Qiagen, Germany). The oligonucleotides used for PCR amplification were purchased from AccuOligo (Bioneer Co., South Korea).
Purification of histidine-tagged ArgR and FarR.
The construction of pEMBTL-SY0 carrying the argR coding region and pEMBTL-SY5 carrying the farR coding region were described in previous studies (Table 1) (13, 14). pEMBTL-SY5 and pEMBTL-SY0, were used to transform E. coli BL21(DE3) cells by electroporation. The syntheses of ArgR and FarR fused with a six-histidine tag were induced in recombinant E. coli BL21 cells (pEMBTL-SY0 and pEMBTL-SY5, respectively) by the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) after the culture had reached an OD600 of 0.6. The cells were grown for 3 h, harvested, and then disrupted by using a Vibra Cell Sonic disruptor (Sonics & Materials, Inc.). Purification of the fusion proteins was carried out by Ni-nitrilotriacetic acid (NTA) affinity chromatography according to the instructions provided by Qiagen (Hilden, Germany). The purified fusion proteins were used directly for the production of polyclonal rabbit antibodies for ChIP assays (Ab Frontier, Inc., South Korea) and electrophoretic mobility shift assay (EMSA) experiments.
EMSAs.
Short DNA probes that that were approximately 30 bp long were generated by the annealing of complementary single oligonucleotides. For the labeling of the DNA and the setup of the reaction mixture containing purified His6-ArgR or His6-FarR for the EMSA, a digoxigenin (DIG) gel shift kit (Roche, Mannheim, Germany) was used according to the manufacturer's instructions. The oligonucleotides were blunt ended, since labeling were performed with terminal transferase and digoxigenin-11-dUTP (DIG-ddUTP). The labeled and unlabeled probes were incubated with His6-ArgR or His6-FarR. Binding reactions were performed with binding buffer [20 mM HEPES (pH 7.6), 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM dithiothreitol (DTT), 0.2% (wt/vol) Tween 20, 30 mM KCl, 1 μg poly(dI-dC), 0.2 μg poly-l-lysine] for 30 min at room temperature. Separation by gel electrophoresis was performed with native 6% polyacrylamide gels using 0.5× Tris-borate-EDTA (TBE) buffer as a running buffer. Subsequently, the labeled DNA was blotted onto a positively charged nylon membrane (Roche, Mannheim, Germany) by electroblotting. For the detection of the labeled DNA, X-ray films were used. Each sample was analyzed in triplicate.
DNA-protein cross-linking and ChIP assays.
A previously described ChIP protocol (14) was adapted as follows. Individual strains were grown at 30°C for several generations in MMY medium. At an OD600 of 1.9 to 2.0, samples (10 ml) from 100-ml cultures were transferred into new tubes and treated with formaldehyde to a final concentration of 1% and incubated for 10 min at 30°C with gentle agitation. The cells were harvested by centrifugation (10 min at 1,618 × g at 4°C) and washed twice in chilled phosphate-buffered saline. The cells were resuspended in 0.5 ml of a solution containing 1% sodium dodecyl sulfate, 10 mM EDTA, 50 mM Tris-HCl (pH 8.1), 1 mM phenylmethylsulfonyl fluoride, and 5 μg ml−1 RNase A. The cells were then incubated at 30°C for 10 min and chilled on ice. The lysates were sonicated for 10 cycles, each lasting 20 s. The chromosomal DNA of the lysate was sheared to give a mean fragment size of 200 to 1,000 bp. The cell debris was used for the ChIP assay. The experiments were performed by using a ChIP assay kit (Upstate) according to the manufacturer's protocol, and the DNA-protein complexes in the supernatant were immunoprecipitated by using 3 μg ml−1 affinity-purified polyclonal rabbit antibodies raised against the purified hexahistidine-tagged ArgR or FarR protein. Subsequently, PCR amplification of the immunoprecipitated DNA was carried out for 27 cycles using the primers designed to amplify the six arg genes (Table 2). Each sample was analyzed in triplicate.
TABLE 2.
Oligonucleotides used in this study
| Primer and purpose | Sequence (5′-3′) | Description (reference) |
|---|---|---|
| RT-PCR | ||
| argB-F | ATATTGGTTTGGTCGGAGA | Amplification of argB cDNA (this study) |
| argB-R | TACAGTTCCCCATCCTTGT | |
| 16S rRNA-F | TCCTGGTGTAGCGGTGAAA | Amplification of 16S rRNA (this study) |
| 16S rRNA-R | CCCACCTTCCTCCGAGTTA | |
| ChIP assay | ||
| argC-F | TGCACTTCCAGGTGGT | PCR primers for ChIP detection of arg genes (14) |
| argC-R | AGTTACACCATACACG | |
| argJ-F | CTTAAGCGTTGGTTTTG | |
| argJ-R | CGGTAATGCCTTTTTCT | |
| argB-F | TCGAACCACTGACCTGA | |
| argB-R | CAGCGAGGACATTTGCG | |
| argF-F | TGGTGATCACCGACGAA | |
| argF-R | AAACCTCTGCCTGCTCT | |
| argG-F | GCACCACTTAAAGCG | |
| argG-R | AGAACGATGCGGTTAG | |
| argH-F | CTCCAAGATCGCTAACA | |
| argH-R | TCCATGTGGTGTTCTTC |
RT-PCR.
The levels of argB mRNA were quantified by real-time reverse transcription (RT)-PCR using SYBR green PCR master mix (ABI 7700; Applied Biosystems, CA). In brief, the total RNA from the same biomass of C. glutamicum was extracted by using TRIzol reagent (Gibco-BRL) according to the manufacturer's instructions. Reverse transcription was first performed to synthesize cDNA using total RNA (0.5 μg), random primers (16-mers) (Bioneer Co., Daejeon, South Korea), deoxynucleoside triphosphates (dNTPs) (1 mmol/liter), 4.5 units of avian myeloblastosis virus (AMV) reverse transcriptase (Promega Co.), and 20 units of RNase inhibitor (Promega Co.). cDNA corresponding to 50 ng of RNA was then added to SYBR green Ampli Taq master mix and 0.9 μmol/liter each specific primer in a total volume of 50 μl. Table 2 lists the primer pairs used for argB and the 16S rRNA gene (internal standard). PCR was carried out with a real-time PCR cycler (ABI 700; Applied Biosystems, CA). The thermal cycling conditions were 48°C for 30 min, 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 1 min. At the end of each phase at 60°C, the fluorescence was measured and used for quantitation. Each sample was analyzed in triplicate.
Determination of amino acid concentration.
High-performance liquid chromatography (HPLC) (Waters Alliance 2690 analytical HPLC system; Waters Co.) was used to determine amino acids. The system was equipped with a Nova-Pak C14 column and a Waters 747 scanning fluorescence detector (Waters Co.). All the results represent the data from at least three independent experiments and include a mean value.
RESULTS
In vitro binding of ArgR to the argB promoter region.
ArgR is a transcriptional regulator that controls ornithine biosynthesis through an interactive pathway of arginine biosynthesis in C. glutamicum (19). As previously reported, DNA sites for ArgR binding occur at four genes, located at the upstream region of argC, argB, argF, and argG. It was also shown previously that the DNA-binding affinity of ArgR for the upstream region of the argB gene is affected only by proline supplementation (14, 16). It was reported that ArgR not only recognizes but also binds weakly conserved operator sequences of the form 5′-TNTGAATwwwwATTCANW-3′ (where N is any base and W/w is A or T [with w in the linker region between two palindromic sequences of ArgR binding sites]) in E. coli, located in the three known arginine-inducible promoters (3, 19, 20). These promoters contain ARG boxes: the promoters of artJ (PartJ Ec), artPIQM (PartP Ec), and the hisJQMP operon (PhisJ Ec) (3). An alignment of the three ARG boxes with the argB promoter region of C. glutamicum showed the 27-bp consensus sequence and highlighted the conserved nucleotides at each position (Fig. 1). To corroborate this finding further, EMSA was explored to see whether ArgR bound to the argB promoter region in vitro (Fig. 2). The two subfragments (subfragments B1 and B2) of the promoter regions of the argB gene were incubated with His6-ArgR (Fig. 2A). Competition and supershift assays showed that ArgR did not bind to two other sites located at positions −102 to −72 and −27 to +3 under the promoter regions of argB (data not shown). These results defined the ArgR-responsive sequence as subfragments B1 and B2 located at positions −77 to −25 of the argB promoter. As shown in Fig. 2B (right), His6-ArgR has clearly bound to the two subfragments. However, competitive assays showed that His6-ArgR did not bind to the two subfragments. Therefore, this suggests that an important site for the DNA binding of ArgR is between positions −77 and −25 of the argB promoter region.
FIG. 1.

Putative ARG box sequences. An alignment of the argB promoter region with the ARG box sequences in the argJ, artP, and hisJ promoter from E. coli (Ec) (3) using the ClustalX program (31) and a picture representing the conservation of bases at each position in the inferred consensus operator sequence (generated by WebLogo 2.8.2 software) (4) are shown. The conserved nucleotides in at least eight boxes are shaded and asterisked. Bases substituted in the argB promoter region used for EMSAs are indicated, and the new base is shown above. Cg, C. glutamicum.
FIG. 2.

Binding of ArgR to the putative ARG box sequence in the argB promoter region. (A) Schematic representation of the upstream region of the argB gene. The numbers indicate the ends of the fragments relative to the proposed translational start site (position +1) in this study. Vertical arrows indicate the positions of base substitutions (Fig. 1). The box indicates the putative ARG box, the gray bar represents the putative recognition region of FarR binding, PargB indicates the DNA fragment used for the in vivo DNA-binding affinities of ArgR (14) and FarR by ChIP assays, arrows indicate the bases substituted in the argB promoter region, and asterisks represent missing contact probing of ArgR. (B) EMSAs were performed with two subfragments (subfragments B1 and B2) of the promoter region. His6-ArgR (2,400 nM) was incubated with the DIG-labeled probe, and the protein-DNA complexes were resolved by electrophoresis on native 6% polyacrylamide gels. Comp. denotes competitor assays (40-fold excess DIG-unlabeled oligonucleotides) of ArgR.
The argB promoter region is regulated in response to proline.
Previous studies showed that a reduced DNA-binding affinity of ArgR for the upstream region of the argB gene provokes an increase in levels of ornithine biosynthesis (14, 16). Thus, the DNA-binding activity of His6-ArgR in vitro was analyzed by using the electrophoretic mobility shift assay (EMSA) technique, testing subfragments (subfragments B1 and B2) of the argB promoter region to verify whether the DNA binding of ArgR to the argB promoter is affected directly by proline (Fig. 3 A). Consistent with previously reported results of in vivo experiments (14), the addition of proline had an effect on DNA binding by His6-ArgR although dissolving the affinity of His6-ArgR. Furthermore, to test whether proline influences the expression of the argB gene, the transcriptional activation of argB by proline was examined. Concurring with data from previous studies, it was clear that under the 10 mM proline-supplemented condition, the amount of argB mRNA increased (Fig. 3B). In conclusion, the results indicate that proline supplementation prevents the DNA binding of ArgR to subfragments B1 and B2 but triggers an increase in the level of argB transcription. Therefore, proline is a potent inducer of argB gene transcription regulated by ArgR.
FIG. 3.

Proline response of ArgR to the upstream regions of the argB gene. (A) DNA-binding activity of His6-ArgR (2,400 nM) for subfragments B1 and B2 by EMSA in the presence of 0 mM, 1 mM, 5 mM, and 10 mM proline. (B) RT-PCR analysis of the argB gene in 10 mM proline-treated C. glutamicum. The mRNA expression level of the argB gene was calculated as a ratio of 16S rRNA gene expression. The results are reported as the means of data from three experiments.
Mutational analysis of ArgR-binding sites within the argB promoter region.
Furthermore, to investigate which nucleotides within the consensus sequence were necessary for ArgR binding, single-nucleotide exchanges were made in the argB promoter region (Fig. 1). The most conserved bases were substituted to introduce transversion from AT to CG and from CG to AT: T1→G, G6→T, C9→A, A10→C, T15→G, A21→C, A23→C, and A27→C (Table 2). In addition, C5, which was previously identified as the major position for ArgR binding by DNase I footprinting in E. coli (3), was also replaced with A. As shown in Fig. 4, His6-ArgR clearly bound all DNA fragments except G6T, C9A, and A23C. In particular, the fragments that contained C9A and A23C belonging to the highlighted and conserved nucleotides of Fig. 1 showed a very weak, or no, band shift compared with subfragments B1 and B2 as wild-type promoter regions of the argB gene (Fig. 2B). These results define the C9 and A23 nucleotides as major points of binding of ArgR to the argB promoter.
FIG. 4.

EMSAs were performed with substituted DNA fragments of subfragment B1 and B2 regions of the argB promoter. His6-ArgR (2,400 nM) was incubated with the DIG-labeled probe. ArgR-DNA complexes are indicated by arrows; Comp. denotes competitor assays (40-fold excess mutated DIG-unlabeled oligonucleotides) of ArgR. Boxes represent alignments of subfragments B1 and B2 and their substituted DNA fragments.
FarR is related to the biosynthesis of ornithine and arginine in C. glutamicum.
Another transcriptional regulator, FarR, was found previously to be involved in the glutamate and arginine metabolic pathway in C. glutamicum (8). As shown in Fig. 5 B, ChIP assays were used to measure the level of FarR binding to promoter regions of arg genes in vivo. For this purpose, putative promoter regions of six arg genes (argC, argJ, argB, argF, argG, and argH) were selected in accordance with previously reported research (14). Compared with ChIP results and the growth curve of C. glutamicum, the DNA-binding affinity of FarR showed a gradual increase in trends at all target regions until an early stationary growth phase (Fig. 5). Interestingly, 12 to 14 h after inoculation, the productions of both ornithine and arginine reached a plateau (Fig. 5A). Additionally, the argB promoter region is more repressed by FarR than by other promoters during 12 h to 14 h due to the relative amount of DNA of FarR for each promoter region. In previous studies, another transcriptional repressor, ArgR, was shown to regulate ornithine biosynthesis by binding to the upstream regions of arg genes (15, 16). In particular, ornithine biosynthesis is highly regulated when ArgR acts on the argB promoter region under conditions of proline supplementation (14). This finding suggests that FarR also governs ornithine and arginine biosynthesis and the DNA binding of FarR to the argB promoter region, which is important for the physiological mechanisms.
FIG. 5.

Time profiles of in vivo DNA binding of ArgR. (A) Cell growth and ornithine and arginine production during cultivation of C. glutamicum. Cell growth is indicated by circles, and ornithine and arginine concentrations are indicated by open and closed triangles, respectively. The results are reported as the means ± standard deviations (SD) (n = 3). (B) In vivo binding of FarR to individual promoter regions of arg genes analyzed by ChIP assays. The C. glutamicum wild-type strain was treated with formaldehyde to cross-link FarR to promoters and lysed, and FarR complexes were immunoprecipitated for analysis.
In vitro binding of FarR to the argB promoter region.
According to data from previous studies, FarR binding to the gdh promoter region was confirmed by gel retardation experiments (positions −444 to −469 from the transcriptional start site) (8, 9). This study showed that FarR binds strongly to the gdh promoter region in vivo (data not shown). This site consists of a highly palindromic region with the sequence 5′-GCCAGGTTATATAACCAGTC-3′ (8). Surprisingly, putative FarR-binding sites were located at positions −57 to −77 from the proposed translation start site (position +1) of the argB promoter (5′-ACGTGGAGATCAACTCCGCGT-3′), analyzed by multiple-sequence alignment with the FarR-binding site of the gdh promoter region and the consensus binding motif (Fig. 2A). HutC/FarR-type regulators of the GntR family bind by dimerization at the palindromic region of the DNA sequence in an inverted-repeated manner (27). Thus, a comparison of the sequence suggests that FarR may regulate the argB promoter region by its direct interaction with the DNA site for ornithine and arginine biosynthesis. Subsequently, to confirm the putative FarR-binding site, an EMSA was performed on purified His6-FarR. Figure 6 shows that the presence of FarR, at a 1,169 nM concentration, led to a significant retardation of the DNA fragment (subfragment B1) carrying the argB promoter region between positions −50 and −77, suggesting that FarR binds to the argB promoter region. The precise DNA binding of FarR was determined by competitive assays. As shown in Fig. 6, the binding of FarR to the argB promoter region was specific, as this binding was significantly inhibited in a dose-dependent manner by DIG-unlabeled subfragment B1.
FIG. 6.

EMSAs were performed with a subfragment (subfragment B1) of the promoter region in the argB promoter region. The His6-FarR protein was incubated with a DIG-labeled probe containing subfragment B1 and subjected to EMSA. (Left) Binding of His6-FarR. The concentrations of FarR were 501 nM (lane 1), 835 nM (lane 2), 1,169 nM (lane 3), and 1,670 nM (lane 4). Lane 0, no protein. (Right) Competitor assay of His6-FarR (1,670 nM) using excess DIG-unlabeled oligonucleotides.
Relationship of binding of ArgR and FarR to the argB promoter region.
From the mutational analysis of ArgR binding, the results indicate that G6T, C9A, and A23C are the three critical sites for ArgR binding to the argB promoter region (Fig. 2A and 4). Among them, G6T at sites at position −58 of the argB promoter is included in the putative FarR-binding site, located at positions −57 to −77 within the argB promoter region. Additionally, C9A (at sites at position −55 of the argB promoter) was located in the vicinity of the putative FarR-binding site (Fig. 2A). This finding suggests that ArgR and FarR have some relation to regulate argB through the binding of the argB promoter region as a transcriptional regulator. Therefore, the binding of ArgR and FarR to the argB promoter region was further investigated by comparing in vivo ChIP assays with in vitro EMSAs. First, the DNA binding of FarR to the arg genes was examined by ChIP at 14 h after C. glutamicum SJC 8074 (argR mutant) inoculation, in order to elucidate a correlation between the binding of FarR and the binding of ArgR to the argB promoter region (Fig. 7 A). The DNA binding of FarR to the argB promoter region was also clearly observed in the absence of ArgR in C. glutamicum, and this phenomenon was observed for the promoter region of the argB, argF, argG, and argH genes. This means that FarR did directly bind to the argB promoter in C. glutamicum and not indirectly through ArgR.
FIG. 7.
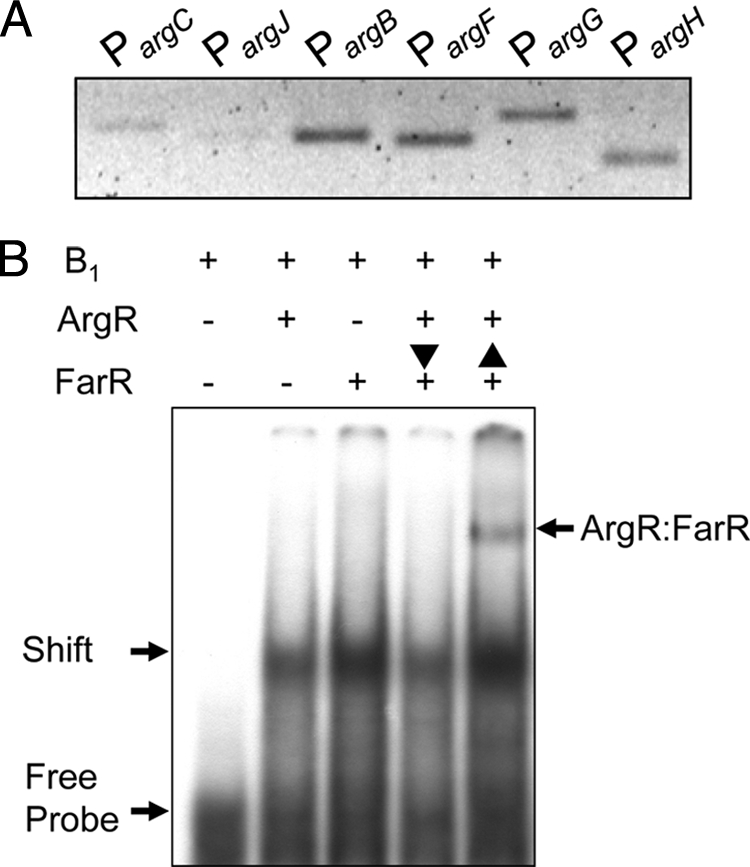
Comparative analysis of in vivo and in vitro bindings of ArgR and FarR to the argB promoter region. (A) ChIP assay of FarR binding to promoter regions of the argB gene. C. glutamicum strain SJC 8074 (argR mutant) was treated with formaldehyde to cross-link FarR to promoters and lysed, and FarR complexes were immunoprecipitated for analysis. (B) EMSAs of His6-ArgR and His6-FarR. EMSA was done with subfragment B1 as described in the legend of Fig. 2. The vertical arrows indicate the order of incubation of His6-FarR (1,670 nM) and His6-ArgR (2,400 nM) with subfragment B1.
In addition, the effect of the binding of purified ArgR and FarR to the argB promoter region was tested by EMSAs (Fig. 7B). Formations of protein-DNA complexes were observed for the individual EMSAs of ArgR and FarR, using DIG-labeled subfragment B1 (positions −50 to −77). To further corroborate a relationship between these regulators, a different reaction order of ArgR and FarR for contact with subfragment B1 was performed. In this assay, the ArgR-DNA complex was observed only when FarR was incubated with the ArgR-DNA complex after first binding ArgR with subfragment B1, meaning that FarR did not bind to the ArgR-DNA complex. Whereas both ArgR and FarR formed a complex with the DNA fragment (ArgR-FarR), they formed protein-DNA complexes individually when ArgR was incubated with the FarR-DNA complex after first binding FarR with subfragment B1. This suggests that FarR has only one putative binding domain located at positions −57 to −77, but this region exactly overlapped with subfragment B1 for the binding of ArgR within the argB promoter; thus, if ArgR bound with the argB promoter first, the binding of FarR would be difficult. However, if FarR bound to the binding domain located at positions −57 to −77 first, ArgR could bind other binding sites located at positions −49 to −25 within the argB promoter.
DISCUSSION
Previous in vivo studies suggested that the upstream region of the argB gene on the arg operon plays an important role in interacting with ArgR under proline-supplemented conditions in C. glutamicum (14, 16). Clarification of the specific effect of the transcriptional repressor ArgR on the action of another transcriptional regulator, FarR, on the argB promoter region in C. glutamicum is the aim of this work.
In bacteria, the argB gene encodes N-acetylglutamate kinase, a key enzyme for ornithine biosynthesis. The enzyme's importance comes from its feedback inhibition control: it regulates its own pathway (28, 34). The C. glutamicum argB gene can be transcribed from an internal promoter located in its upstream region (28). In the consensus C. glutamicum promoter, the prominent feature is a conserved extended −10 region, tgngnTA(c/t)aaTgg (with the less-conserved nucleotides lowercase), while the −35 region is much less conserved (23). Caldara et al. previously described how liganded ArgR and RNA polymerase effectively compete in vivo by binding to partially overlapping sites (3). Sequence comparison allows the observation that the hypothesized −35 region of the argB promoter does not overlap the putative ARG box deduced by EMSAs (data not shown). However, the argB gene is considered a member of the C. glutamicum ArgR regulon, even though none of the core promoter elements overlap the ARG boxes (3, 24).
The protein building block proline has other important functions, including being a source of energy, carbon, and nitrogen and being an osmolyte (21, 25). The first and controlling step of the synthesis of proline from glutamate is catalyzed by glutamate-5-kinase (G5P), and it was reported previously that this enzyme is feedback inhibited by proline (25). A particularly interesting finding from previous studies, which relates to this work, was that proline also expedited ornithine biosynthesis, a catalyzed synthesis with glutamate as a primary metabolite in C. glutamicum (14, 16). In this study, proline was observed to improve argB gene transcription due to its ability to decrease the efficacy of the binding of ArgR to the argB promoter region (Fig. 3). The combined results support the hypotheses that the enhancement of ornithine biosynthesis under conditions of proline supplementation might be mediated by two physiological effects: (i) metabolic flux from glutamate favors ornithine biosynthesis rather than proline biosynthesis, as G5P is feedback inhibited by proline supplementation (25), and (ii) proline acts as an antirepressor by binding to a specific region of the ArgR structure (14). The determination of the three-dimensional (3-D) structure of the ArgR-proline complex, essential for clarifying this issue, remains but a sought-after goal.
It has been known for some time that FarR is a fatty acid- and fatty acyl coenzyme A (acyl-CoA)-responsive DNA-binding protein. Its new function might be connected to amino acid biosynthesis and central carbon energy metabolism (8), since the farR gene in the tricarboxylic acid (TCA) cycle gene cluster of E. coli (gltA-sdhCDAB-sucABCD-farR) is autoregulated by the FarR protein (26). The ChIP results provide evidence that FarR binds to the upstream regions of arg genes (Fig. 6) as well as to the gdh promoter, which converts an intermediate of the TCA cycle to glutamate (15). Indeed, glutamate biosynthesis is closely related to fatty acid synthesis (5). The inactivation of DtsR (detergent sensitivity rescuer), which is assumed to be involved in fatty acid synthesis, triggers glutamate overproduction in coryneform bacteria (12, 36). Thus, it would be beneficial to clarify how FarR participates in fatty acid and amino acid synthesis in C. glutamicum.
The ArgR and FarR proteins have calculated molecular masses of 29.3 and 18.8 kDa, respectively (16, 33). In addition, the FarR protein is predicted to form dimers, in accordance with the behavior of other members of the GntR family of transcriptional regulators (27). Therefore, it may be presumed that ArgR and FarR in C. glutamicum have differing structures as hexamers and dimers, respectively. This is a likely cause of the distinct patterns of binding of ArgR and FarR to the argB promoter region (Fig. 7B). This study tries to provide insight into the complex regulation of argB expression. The regulator proteins ArgR and FarR likely bind to the argB upstream region in a manner similar to the previously reported upstream binding of FarR to gdh (8). In addition to FarR, the expression of gdh in C. glutamicum was also found to be regulated by the binding of the global transcriptional regulator protein, AmtR, to two separate sites in the gdh upstream region, at positions −184 to −209 and −334 to −359 from the site of the start of transcription, in a recent study of the stringent response (9). Among the latter positions is a putative ARG box sequence (14) as well as contact with the −10 to −35 region of the gdh promoter (9). In this study, FarR has only one putative binding domain located at positions −57 to −77, but this region exactly overlapped with subfragment B1 for binding ArgR within the argB promoter; thus, if ArgR bound with the argB promoter first, the binding of FarR would be difficult. However, if FarR bound to the binding domain located at positions −57 to −77 first, ArgR could bind other binding sites located at positions −49 to −25 within the argB promoter. Therefore, both ArgR and FarR formed a complex with the DNA fragment (ArgR-FarR) and formed protein-DNA complexes individually when ArgR was incubated with the FarR-DNA complex after first binding FarR with subfragment B1.
In conclusion, these results demonstrate that C. glutamicum ArgR regulates argB gene transcription as a repressor and that this repression is regulated by the intracellular molecule proline. Moreover, this study extends our understanding of the molecular mechanisms involved in the transcription regulation of the argB gene by demonstrating the interaction of ArgR with another transcription factor, FarR. The findings have a number of hopeful implications for future studies of ArgR that may elucidate its regulatory mechanisms in greater detail.
Acknowledgments
This study was supported by the 21C Frontier Microbial Genomics and Applications Center Program, grant no. 11-2008-10-002-00, Ministry of Education, Science & Technology, Republic of Korea. We are grateful for their support.
Footnotes
Published ahead of print on 29 November 2010.
REFERENCES
- 1.Brinkrolf, K., I. Brune, and A. Tauch. 2007. The transcriptional regulatory network of the amino acid producer Corynebacterium glutamicum. J. Biotechnol. 129:191-211. [DOI] [PubMed] [Google Scholar]
- 2.Brune, I., K. Brinkrolf, J. Kalinowski, A. Pühler, and A. Tauch. 2005. The individual and common repertoire of DNA-binding transcriptional regulators of Corynebacterium glutamicum, Corynebacterium efficiens, Corynebacterium diphtheriae and Corynebacterium jeikeium deduced from the complete genome sequence. BMC Genomics 6:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caldara, M., P. N. Minh, S. Bostoen, J. Massant, and D. Charlier. 2007. ArgR-dependent repression of arginine and histidine transport genes in Escherichia coli K-12. J. Mol. Biol. 373:251-267. [DOI] [PubMed] [Google Scholar]
- 4.Crooks, G. E., G. Hon, J. M. Chandonia, and S. E. Brenner. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eggeling, L., and M. Bott. 2005. Handbook of Corynebacterium glutamicum. CRC Press, Taylor & Francis Group, Boca Raton, FL.
- 6.Ghochikyan, A., et al. 2002. Arginine operator binding by heterologous and chimeric ArgR repressors from Escherichia coli and Bacillus stearothermophilus. J. Bacteriol. 184:6602-6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grandori, R., et al. 1995. The DNA-binding domain of the hexameric arginine repressor. J. Mol. Biol. 254:150-162. [DOI] [PubMed] [Google Scholar]
- 8.Hänßler, E., et al. 2007. FarR, a putative regulator of amino acid metabolism in Corynebacterium glutamicum. Appl. Environ. Microbiol. 76:625-632. [DOI] [PubMed] [Google Scholar]
- 9.Hänßler, E., et al. 2009. A game with many players: control of gdh transcription in Corynebacterium glutamicum. J. Biotechnol. 142:114-122. [DOI] [PubMed] [Google Scholar]
- 10.Hwang, J. H., G. H. Hwang, and J. Y. Cho. 2008. Effect of increased glutamate availability on L-ornithine production in Corynebacterium glutamicum. J. Microbiol. Biotechnol. 18:704-710. [PubMed] [Google Scholar]
- 11.Ikeda, M., and S. Nakagawa. 2003. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 62:99-109. [DOI] [PubMed] [Google Scholar]
- 12.Kimura, E., C. Abe, Y. Kawahara, T. Nakamatsu, and H. Tokuda. 1997. A dtsR gene-disrupted mutant of Brevibacterium lactofermentum requires fatty acids for growth and efficiently produces L-glutamate in the presence of an excess of biotin. Biochem. Biophys. Res. Commun. 234:157-161. [DOI] [PubMed] [Google Scholar]
- 13.Lee, S. Y., et al. 2010. Utilization of phenol and naphthalene affects synthesis of differential amino acids in Corynebacterium glutamicum. Curr. Microbiol. 61:596-600. [DOI] [PubMed] [Google Scholar]
- 14.Lee, S. Y., H. S. Shin, J.-S. Park, Y. H. Kim, and J. Min. 2010. Proline reduces the binding of transcriptional regulator ArgR to upstream of argB in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 86:235-242. [DOI] [PubMed] [Google Scholar]
- 15.Lee, S. Y., Y. H. Kim, and J. Min. 2010. Conversion of phenol to glutamate and proline in Corynebacterium glutamicum is regulated by transcriptional regulator ArgR. Appl. Microbiol. Biotechnol. 85:713-720. [DOI] [PubMed] [Google Scholar]
- 16.Lee, S. Y., Y. H. Kim, and J. Min. 2009. The effect of ArgR-DNA binding affinity on ornithine production in Corynebacterium glutamicum. Curr. Microbiol. 59:483-488. [DOI] [PubMed] [Google Scholar]
- 17.Lee, Y. J., and J. Y. Cho. 2006. Genetic manipulation of a primary metabolic pathway for L-ornithine production in Escherichia coli. Biotechnol. Lett. 28:1849-1856. [DOI] [PubMed] [Google Scholar]
- 18.Leuchtenberger, W., K. Huthmacher, and K. Drauz. 2005. Biotechnological production of amino acids and derivatives: current status and prospects. Appl. Microbiol. Biotechnol. 69:1-8. [DOI] [PubMed] [Google Scholar]
- 19.Maas, W. K. 1994. The arginine repressor of Escherichia coli. Microbiol. Rev. 58:631-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makarova, K. S., A. A. Mironov, and M. S. Gelfand. 2001. Conservation of the binding site for the arginine repressor in all bacterial lineages. Genome Biol. 2:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marco-Marín, C., et al. 2007. A novel two-domain architecture within the amino acid kinase enzyme family revealed by the crystal structure of Escherichia coli glutamate 5-kinase. J. Mol. Biol. 367:1431-1446. [DOI] [PubMed] [Google Scholar]
- 22.Park, S. M., C. D. Lu, and A. T. Abdelal. 1997. Cloning and characterization of argR, a gene that participates in regulation of arginine biosynthesis and catabolism in Pseudomonas aeruginosa PAO1. J. Bacteriol. 179:5300-5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pátek, M., J. Nešvera, A. Guyonvarch, O. Reyes, and G. Leblon. 2003. Promoters of Corynebacterium glutamicum. J. Biotechnol. 10:311-323. [DOI] [PubMed] [Google Scholar]
- 24.Paul, L., P. K. Mishra, R. M. Blumenthal, and R. G. Matthews. 2007. Integration of regulatory signals through involvement of multiple global regulators: control of the Escherichia coli gltBDF operon by Lrp, IHF, Crp, and ArgR. BMC Microbiol. 7:1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pérez-Arellano, I., V. Rubio, and J. Cervera. 2006. Mapping active site residue in glutamate-5-kinase. The substrate glutamate and the feed-back inhibitor proline bind at overlapping sites. FEBS Lett. 580:6247-6253. [DOI] [PubMed] [Google Scholar]
- 26.Quail, M. A., C. E. Dempsey, and J. R. Guest. 1994. Identification of a fatty acyl responsive regulator (FarR) in Escherichia coli. FEMS Lett. 356:183-187. [DOI] [PubMed] [Google Scholar]
- 27.Rigali, S., A. Derouaux, F. Giannotta, and J. Dusart. 2002. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J. Biol. Chem. 277:12507-12515. [DOI] [PubMed] [Google Scholar]
- 28.Sakanyan, V., et al. 1996. Genes and enzymes of the acetyl cycle of arginine biosynthesis in Corynebacterium glutamicum: enzyme evolution in the early steps of the arginine pathway. Microbiology 142:99-108. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 30.Sampaio, M., et al. 2004. Phosphotransferase-mediated transport of the osmoolyte 2-O-α-mannosyl-D-glycerate in Escherichia coli occurs by the product of the mngA (hrsA) gene and is regulated by the mngR (farR) gene product acting as repressor. J. Biol. Chem. 279:5537-5548. [DOI] [PubMed] [Google Scholar]
- 31.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The Clustal X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Udaka, S. 1960. Screening method for microorganisms accumulating metabolites and its use in the isolation of Micrococcus glutamicus. J. Bacteriol. 79:754-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Duyne, G. D., G. Ghosh, W. K. Maas, and P. B. Sigler. 1994. Structure of the oligomerization and L-arginine binding domain of the arginine repressor of Escherichia coli. J. Mol. Biol. 256:377-391. [DOI] [PubMed] [Google Scholar]
- 34.Xu, Y., B. Labedan, and N. Glansdorff. 2007. Surprising arginine biosynthesis: a reappraisal of the enzymology and evolution of the pathway in microorganisms. Microbiol. Mol. Biol. Rev. 71:36-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu, Y., et al. 2003. Regulation of arginine biosynthesis in the psychropiezophilic bacterium Moritella profunda: in vivo repressibility and in vitro repressor-operator contact probing. J. Mol. Biol. 326:353-369. [DOI] [PubMed] [Google Scholar]
- 36.Yao, W., et al. 2009. Double deletion of dtsR1 and pyc induce efficient L-glutamate overproduction in Corynebacterium glutamcium. J. Ind. Microbiol. Biotechnol. 36:911-921. [DOI] [PubMed] [Google Scholar]