

Abstract
The intrasubspecies diversity of six strains of Lactococcus lactis subsp. lactis was investigated at the genomic level and in terms of phenotypic and transcriptomic profiles in an ultrafiltration cheese model. The six strains were isolated from various sources, but all exhibited a dairy phenotype (growth in ultrafiltration cheese model and high acidification rate). The six strains exhibited similar behaviors in terms of growth during cheese ripening, while different acidification capabilities were detected. Even if all strains displayed large genomic similarities, sharing a large core genome of almost 2,000 genes, the expression of this core genome directly in the cheese matrix revealed major strain-specific differences that potentially could account for the observed different acidification capabilities. This work demonstrated that significant transcriptomic polymorphisms exist even among Lactococcus lactis subsp. lactis strains with the same dairy origin.
Lactococcus lactis is a lactic acid bacterium that is used as a dairy starter in the production of fermented milk products, particularly cheese. Starter cultures play crucial roles during all phases of the cheese ripening process. L. lactis converts sugars into lactic acid, thus inducing milk acidification and coagulation. Lactic acid production is of economic value within the dairy industry since acidification preserves milk products from the growth of unwanted bacteria and molds (9). L. lactis also produces small quantities of other metabolites and enzymes that contribute to the more-subtle aromas and flavors distinguishing different cheeses (23, 31).
In order to assess natural diversity within the L. lactis strains, L. lactis strains from nondairy niches, such as plant material, were isolated (21, 22, 27). L. lactis strains used for dairy production have been genetically divided into two subspecies. L. lactis subsp. lactis includes both plant-associated isolates and strains that originate with dairy products, while L. lactis subsp. cremoris includes only dairy strains. Genome sequencing studies of L. lactis and comparative genomics of both L. lactis subsp. lactis and L. lactis subsp. cremoris provided evidence of inter- and intrasubspecies genetic diversity (3, 12, 22, 29). In particular, in plant-associated L. lactis subsp. lactis, new genes have been identified and these genotypic differences were found to correspond to phenotypic characteristics involved in adaptive responses to environmental changes (22). More recently, genetic polymorphisms in L. lactis subsp. cremoris were studied, revealing significant divergence between the strains (24). However, the presence/absence of specific genes is not necessarily translated into different phenotypes and/or bacterial evolution. For example, recently a high degree of diversity of closely related L. lactis strains was detected by regulatory phenotyping of specific gene products (1). Similarly, reports related to Escherichia coli plasticity and evolution within E. coli/Shigella species showed that (intra- and interspecies) phenotypic divergence reflected not only genetic diversity but also gene expression diversity of common gene subsets (11, 28).
Understanding the determinants of bacterial diversity is thus complex and requires an integrated approach based on a multilevel analysis. Therefore, in this study, we investigated the intrasubspecies diversity of six strains of L. lactis subsp. lactis at the phenotypic, genomic, and transcriptomic levels. Although isolated from various sources, the six strains have a dairy phenotype (e.g., growth in an ultrafiltration cheese model and a high acidification rate): four were isolated from starters or manufactured products, one from raw milk, and one from grassland. Our results show that even if all strains share a large core genome of 1,915 genes, diversity at the gene expression level does exist between strains. This core transcriptomic diversity between the strains could account for the phenotypic differences observed in model cheese at the level of acidification properties.
MATERIALS AND METHODS
Strains and growth conditions.
Six strains of Lactococcus lactis subsp. lactis, UCMA5713, LD55, LD61, LL08, LL52, and S86 (Table 1), were selected from the Génoferment collection (Toulouse, France). This collection was used recently for the characterization of genome diversity of L. lactis subspecies (15). Three strains (S86, LD61, and LL52) were isolated from industrial starters, one from manufactured dairy product (LD55), and one from raw milk (LL08). Strain UCMA5713 was isolated in the French Normand meadow neighboring a dairy factory and showed features typical of many dairy starters (15). Strains S86, LD55, LD61, and LL08 belonged to the four major strain ribogroups of the collection (S1, S4, S6, and S7) (Table 1) and are representative of the dairy diversity of the collection. Therefore, these strains were chosen for the current study. In addition, we have selected two additional strains, belonging to the S1 and S7 ribogroups, the starter strain LL52, due to its large chromosome size (the highest value of the Génoferment collection; 2.677 ± 27 kb) and the strain UCMA5713, the single plant-associated strain of the collection able to ferment milk. LD61 was used as the reference in our study since it is the best transcriptionally characterized strain in our analysis (19).
TABLE 1.
Strain origins, genomic characteristics, and taxonomy
| Strain | Origin | EcoRI ribotype | Chromosome size (Mb) | Biovar diacetylactisa |
|---|---|---|---|---|
| S86 | Starter (France) | 1797-S-1 | 2.482 ± 35 | No |
| LD55 | Camembert (Normandy, France) | 1797-S-4 | 2.595 ± 93 | No |
| LD61 | Soft cheese starter (France) | 58-S-6 | 2.579 ± 59 | Yes |
| LL08 | Raw milk (Normandy, France) | 199-S-7 | 2.545 ± 33 | No |
| UCMA5713 | Grassland (Normandy, France) | 1797-S-1 | 2.362 ± 14 | No |
| LL52 | Soft cheese starter (France) | 199-S-7 | 2.677 ± 27 | No |
Results from the work of D. Passerini, V. Laroute, P. Le Bourgeois, P. Loubière, P. Ritzenthaler, M. Cocaign-Bousquet, and M. L. Daveran-Mingot (submitted for publication), who revisited the definition of Lactococcus lactis subsp. lactis biovar diacetylactis and positioned the six strains in this taxonomy.
Strains were cultured on skimmed raw milk ultrafiltration (UF) retentate (25). The UF retentate was preincubated overnight at 4°C and then for 45 min at 50°C and homogenized during 1.5 min at 24,000 rpm with an Ultra Turrax disperser system (Imlab, France). After addition of rennet (0.3 μl ml−1), 400 g UF retentate was inoculated at 2 × 106 CFU/g with L. lactis subsp. lactis strains. After incubation for 8 h at 30°C, the cheeses were transferred at 12°C during 7 days for simulation of ripening. Cultures (24 h) using chemically defined medium (CDM) (17), M17 (Difco M17 broth), and M13 (4) were also performed in anaerobic tubes at 30°C under a nitrogen atmosphere with an agitation speed of 250 rpm. In these different laboratory media and in UF retentates, at least three independent cultures of the six strains were performed.
Extractions of total RNA and genomic DNA.
Total RNA was extracted from cells grown for 24 h in UF-cheese (25). Total RNA quality and quantity were verified using a NanoDrop (ND1000) spectrophotometer (Thermo Scientific) and a Nanokit (Agilent RNA 6000) RNA kit. Ten micrograms of total RNA was used to perform retrotranscription. Genomic DNA was extracted from cells grown overnight on M17 as previously described (14). For comparative genomic hybridization (CGH) experiments, 2 μg of sonicated genomic DNA from each strain was used.
Labeling, hybridization, and detection.
Membrane spotting and analytical support were provided by the Biochips platform (Genopole, Toulouse, France). CGH and gene expression analyses were performed using nylon arrays containing the PCR fragments (Eurogentec) for 1,948 genes of L. lactis IL1403 (3). Synthesis of radiolabeled cDNA, nylon array hybridizations, and washings were performed as described previously (20). Membranes were exposed to a PhosphorImager screen for 3 days and scanned with a phosphofluoroimager (Storm 860; Molecular Dynamics). Sonicated DNA (2 μg) was labeled after denaturation by random priming according to the manufacturer's instructions (Biolabs). Random hexamer primers (10 ng/μl), specific L. lactis primers (10 ng/μl), dATP, dTTP, dGTP (0.3 M), 33P-labeled dCTP (1 μCi/μl), and 1 U/μl of Klenow fragment (Biolabs) were used. After 1 h at 37°C, dCTP (0.1 M) and the Klenow fragment (0.5 U/μl) were added and incubated for 1 additional hour at 37°C. Labeled DNA was purified through Microspin G25 columns (Amersham Biosciences). For CGH or gene expression analyses, three independent biological repetitions corresponding to independent cultures were carried out.
Data analysis and statistical treatment.
Signal intensities were quantified, tested statistically, and assigned to gene names with the Bioplot software program (developed by S. Sokol at the Plateforme Génomique, Toulouse, France [http://biopuce.insa-toulouse.fr/]). Local background was removed from all spot intensities.
For transcriptomic experiments, signals were normalized to the mean intensity of the corresponding membrane. Expression ratios were calculated between the strain of interest and strain LD61, which was considered the reference for expression studies with milk (19). In order to be compared, transcriptomic analyses were performed in parallel for all the strains in the same series and repeated three times independently. The statistical significance of expression ratios was evaluated as previously described (7) using false discovery rate (FDR) calculations and a statistical threshold of 7%. In order to determine expression changes at the level of the functional (sub)categories (global tendencies), over- and underexpressed gene enrichments in the different groups were calculated with the Wilcoxon test. This test was performed without any a priori selection of expression data, and we have considered a significant P value of 5%, as previously described (7). The classification of Bolotin et al. was used (3).
For CGH experiments, genes were declared absent or highly divergent if they were detected as absent by all three statistical methods (a, b, and c). In statistical method a, genes corresponding to spot intensities less than a cutoff value in at least two repetitions out of three were declared absent. For each membrane, a cutoff value was defined as the detection limit plus two times its standard deviation. The detection limit of each membrane was calculated as the mean intensity of the 178 empty spots (spots containing no probe). The b and c statistical treatments were performed on whole detected spots without any previous selection. In this case, the ratios of the signal intensities were calculated between the studied strain and the sequenced reference strain, IL1403 (3), after subtraction of the local background and signal normalization. Two normalization methods were used: either by the mean intensity of the corresponding membrane (method b) or by the intensities of a subset of conserved genes with minimal sequence divergence between the samples analyzed (method c) (26). We chose the subset of 57 conserved genes previously identified for L. lactis strains by van Hijum and coworkers (26) using three Streptococcus strains as reporter genomes, and we removed four genes (atpA, rplW, rpmC, and ychH) from this subset because they were missing on our membranes. Absent genes were those corresponding to ratios of normalized intensities (strain of interest compared to IL1403) lower than 0.5. This threshold of 0.5 was chosen because it was previously used in CGH data analyses (10, 24).
Genes were declared to be divergent when the ratios of their normalized intensities (compared to that of IL1403) by method c were lower than 0.76. The 0.76 criterion was chosen after estimation of the average technical variation of the signal ratios using three independent repetitions of each tested strain. However, it should be kept in mind that the number of divergent genes could be biased, since low CGH ratios could also result from artifact (cross-hybridization with another gene in the genome, small GC-rich region).
ORFs detected as absent with all statistical approaches (low spot intensity and low intensity ratios with both normalizations) were subjected to PCR amplification using the genomic DNA preparation already used for CGH experiments. Primers were those previously designed by Eurogentec. Taq polymerase (New England BioLabs) and Phusion polymerase (Finnzymes) were used, and the hybridization temperature ranged between 42 and 55°C.
Clustering analysis was performed with the R free statistical software program (http://www.r-project.org/).
Real-time PCR.
Ten micrograms of total mRNA was retrotranscribed as described previously (13). Primers for real-time PCR (Table 2) were designed using the Bio-Rad Beacon Designer software program to have lengths of 20 to 25 bases, GC contents of more than 50%, and melting temperatures of about 60°C and to amplify PCR products 88 to 145 bases long. The specificities of the primers for the genes of interest were controlled by using the L. lactis IL1403 genome with the Vector NTI software program (Invitrogen). Real-time PCR was carried out on a MyIQ unit with Sybr green supermix as described previously (13). The threshold was determined with a baseline at 137.5 relative fluorescence units above the background level. Three dilutions of the cDNA were performed to determine the PCR efficiency (ranging from 78 to 115%). The pbp2A and smc genes were chosen as internal normalization controls, since they did not show significant expression variation in these experiments. The Pfaffl analysis method (16) was used to calculate the change in transcript levels between strains UCMA5713, LD55, LL08, LL52, and S86 and the reference strain, LD61. Four genes (yaiA, feoA, ipd, and pyrR) were tested, and three independent measurements were performed for a given gene/strain couple. For direct comparison with transcriptomic data, quantitative reverse transcription-PCR results were expressed as ratios in transcript concentrations between the strain of interest and the reference strain, corrected by using the pbp2A or smc normalization ratios.
TABLE 2.
Sequences of primer pairs for qPCR experiments
| Gene | Primer sequence (5′-3′) |
|
|---|---|---|
| Forward | Reverse | |
| feoA | TCAGACGCCGCTTGATGGAC | AGTTCAAGAGGGTCGCCAAGTG |
| yaiA | CAGAAGAAGATGGGCATGGAGC | GTCGGTACACGGAATGAAGCAC |
| pyrR | GACCGTGGACATCGTGAATTGC | CATTGCCATCGTGTTCAGACATTTG |
| ipd | TGGCTGATGGCTATGCTCGTAC | CGGCGTAACTTCCTGCTAATCC |
| pbp2A | TGGAGCAGTCACAGGCAGATAC | GTCAGACCATACTTGCGTTCCG |
| smc | CGCAGTTGTCGGTCCAAATGG | ACGAAGTGCTTTAGCCGATTGC |
Microarray data accession numbers.
Raw CGH and transcriptomic data were deposited in the GEO public database (http://www.ncbi.nlm.nih.gov/geo/) and assigned accession numbers GSE23990 and GSE23987, respectively.
RESULTS
Phenotypic analysis of L. lactis strains.
Six strains of L. lactis subsp. lactis representative of the dairy diversity of the Génoferment collection were selected (see Materials and Methods). The dairy performances of the six strains were tested with a UF-cheese model (see Materials and Methods). Their abilities to grow and acidify this medium were compared throughout the cheese ripening process (Fig. 1). Cell population profiles were very similar for all strains, leading to a constant value ranging between 1.9× 109 and 2.3 × 109 CFU/g of cheese at the end of the culture (after 7 days). The growth rate, calculated in the growth phase between UF-cheese inoculation and the 8-h time point, confirmed the large growth similarities for all six strains, with values ranging between 0.78 and 0.88 h−1 (data not shown). However, acidification properties of six strains differed in UF-cheese, with end pH values between 4.58 (for UCMA5713) and 4.85 (for LD61). This end pH variation is considered to be significant for cheese quality in the dairy industry. In addition, strains were tested for their ability to grow with glucose as a carbon source on media commonly used in laboratories, i.e., synthetic CDM medium and rich M17 medium (see Materials and Methods). After 24 h of growth, strains exhibited only weak differences in terms of growth rate (1.11 to 1.22 h−1 on CDM and 0.75 to 0.95 h−1 on M17), final cell population (1.21 × 109 to 1.83 × 109 CFU/ml on CDM and 2.18 × 109 to 2.50 × 109 CFU/ml on M17), and pH (4.12 to 4.57 on CDM and 4.51 to 4.57 on M17). It was strain LL52 that reached the low final pH value of 4.12 on CDM medium. None of the six strains were able to grow on M13 medium (4), which differed from CDM medium by lacking 12 amino acids, 7 vitamins, and all bases, revealing auxotrophies of these strains.
FIG. 1.

Growth of the six L. lactis strains and pH profile during UF-cheese ripening (average of at least three independent experiments with standard deviations).
Comparative hybridization analysis.
Array-based comparative genome hybridization (CGH) was used to determine the core genome (similar genomic content) of the six strains of L. lactis subsp. lactis exhibiting a dairy phenotype. We have also included in these experiments the genome of the sequenced strain L. lactis subsp. lactis IL1403, which was used for microarray platform design. Using strain IL1403, we identified 19 genes with absolute intensities lower than the cutoff value, which were thus declared absent using statistical method a (see Materials and Methods). These genes were thus tagged as missing on the microarray platform and were omitted in the subsequent analyses. Therefore, 1,929 genes were analyzed in the CGH experiments. No control to identify false-positive genes (genes declared present while they are absent) was included in the CGH experiment.
In order to analyze genomic divergences between the strains, ratios of intensities were calculated between the tested strain and strain IL1403 as indicated in Materials and Methods (normalization method c using likely conserved genes). These ratios, given in Table S1 in the supplemental material, were plotted as a function of gene position on the IL1403 chromosome (Fig. 2). Overall, only a few genes had low ratios (no more than 10% of the genes exhibited ratios lower than 0.76 and were thus declared divergent), indicating strong similarities between the sequences of the six strains. One can observe, however, that the sequence of reference strain LD61 is very close to the sequence of strain IL1403, while the five other strains share diversity in the same regions. Seven diversity regions were detected: (i) regions 2, 3, 5, and 6, containing phage-related genes (i.e., pi1, pi2, pi3, and ps3), were visually enriched at low ratios in these five strains (S86, LD55, LL08, LL52, and UCMA5713), indicating that these transposable elements can be a source of sequence divergence between L. lactis subsp. lactis strains; and (ii) regions 1 and 7, containing in particular genes related to the cell envelope (ycbBDFHIJ genes in locus 1 and pspB in locus 7) and region 4, including genes involved in citrate and malate metabolism (the citRCDEF and mae genes, respectively). These divergent regions can include absent or highly divergent genes but also present genes with low genomic divergence.
FIG. 2.

CGH results normalized using likely conserved genes and expressed as ratios of the signal intensities of the studied strains against that of the reference strain, IL1403 (statistical method c; see Table S1 in the supplemental material). Ratios lower than 1 for the six strains were plotted as a function of gene position on the IL1403 chromosome. Locus 1, position ∼0.20 Mbp, ycbABCDKFGHIJ and ycaFG genes; locus 2, position ∼0.44 Mbp, pi1 genes; locus 3, position ∼1.03 Mbp, pi2 genes; locus 4, position ∼1.20 Mbp, citRCDEF, mae, ymbCDHIJK, and ymcABCDEF genes; locus 5, position ∼1.41 Mbp, pi3 genes; locus 6, position ∼2.01 Mbp, ps3 genes; locus 7, position ∼2.30 Mbp, pspAB and ywjABCDEFGH genes. Genes were declared divergent only when their ratios were lower than 0.76.
Utilization of these CGH ratios in order to predict gene deletions is not trivial when strains are genetically close. Low ratios can indeed correspond to absent genes as well as genes with low sequence identity with strain IL1403 and low hybridization efficiencies (26). Only sequencing experiments have the resolving power to give a definitive answer on the presence or absence of a specific gene in a tested strain. In our experiment, we identified absent or highly divergent genes when genes were declared absent by the three different statistical methods a, b, and c (see Materials and Methods). Method a is based on the absolute signal intensity with an empty spot threshold constraint. The two other procedures, b and c, use comparison to the IL1403 strain and are based on two different statistical normalizations, i.e., normalization by whole spot mean intensity (method b) or by a subset of conserved genes (method c). Of the 1,929 ORFs spotted on the microarray membranes, only 16 genes were identified as absent in at least one of the six strains tested in CGH experiment: in UCMA5713, pspB, citR, pi109, pi147, pi202, pi207, ps313, and ps315, in LD55, citR, pi147, pi202, pi203, pi204, pi207, pi213, pi225, and pi226, in LL08, citR, pi202, and ps315, in LL52, citR, pi204, ps305, ps310, ps311, and ps313, and in S86, pspB, citR, pi202, pi203, and pi204 were absent. In order to discriminate absent genes from highly divergent genes (false-negative genes), PCR amplifications specific to these 16 genes were also performed on genomic DNA using hybridization temperatures between 42 and 55°C. Under our PCR conditions, amplified fragments were analyzed for gene pi202 in strains UCMA5713, LD55, and S86, for genes pi109 and pi207 in strain UCMA5713, and for genes ps311 and ps313 in strain LL52. Following these PCR experiments, the number of genes declared absent in at least one of the six strains was therefore reduced to 14. No ORF was commonly absent in the six strains, meaning that no IL1403-specific ORF was detected. The number of absent ORFs ranged from 8 in strain LD55 to 0 in strain LD61 even though a divergent locus was observed in strain LD61 (Fig. 2). Among the 14 absent ORFs, 12 ORFs were involved in the phage-related functions and prophage functional category, more specifically related to prophages pi1, pi2, and ps3. Only two absent ORFs were linked to metabolic functions, pspB, encoding glucosyltransferase S of the cell envelope functional category, and citR, coding for a citrate lyase regulator. This experiment underlined a very large core genome with 1,915 genes of the IL1403 genome present in all of the six studied strains with a dairy phenotype.
Gene expression analysis.
The expression of the genes common to the six studied strains was compared under UF-cheese model conditions using DNA arrays corresponding to the IL1403 genome. The comparison was performed at 24 h of growth, since the dynamic study of the strain LD61 under similar conditions has shown that the number of differentially expressed genes did not change thereafter (6). All data were compared to those for the reference dairy strain LD61 (IL1403 did not grow in milk), and for each gene, expression ratios were calculated for the five strains S86, LD55, LL08, LL52, and UCMA5713. These ratios were similar to those observed by quantitative PCR (qPCR) analyses (Table 3). Transcriptomic ratios are given in Table S1 in the supplemental material. Significant ratios corresponding to genes differentially expressed in each strain in comparison to expression in the LD61 strain were selected with the usual statistical criterion of an FDR of ≤7%.
TABLE 3.
Comparison of gene expression ratios between the strain of interest and the reference strain, LD61, in transcriptomic and qPCR analysesa
| Relative gene expression | Gene name | Strain | Transcriptomic ratio | qPCR ratio normalized with: |
|
|---|---|---|---|---|---|
| pbp2A | smc | ||||
| Increased | feoA | UCMA5713 | 3.5 | 5.2 | 6.8 |
| feoA | LD55 | 3.3 | 4.7 | 4.8 | |
| feoA | LL52 | 4.9 | 1.3 | 1.2 | |
| yaiA | LL52 | 5.6 | 2.8 | 2.7 | |
| Unchanged | feoA | LL08 | 1.0 | 1.0 | 1.0 |
| Decreased | ipd | UCMA5713 | 0.1 | 0.2 | 0.3 |
| ipd | LD55 | 0.2 | 0.1 | 0.1 | |
| ipd | LL52 | 0.1 | 0.1 | 0.1 | |
| ipd | S86 | 0.1 | 0.1 | 0.1 | |
| pyrR | LL52 | 0.2 | 0.5 | 0.4 | |
| pyrR | S86 | 0.3 | 0.1 | 0.1 | |
Corrected in qPCR experiments by using pbp2A or smc normalization ratios.
In order to check if the transcriptomic ratios were underestimated for genes with low hybridization efficiencies (corresponding to low CGH ratios), we calculated the correlation coefficients between all the 1,915 expression ratios and CGH ratios using the LD61 strain as the reference. With the Pearson, Spearman, or Kendall method (result not shown), correlation coefficients were lower than 0.05, indicating that such a bias is not significantly present in our experiments. To establish the relationships of the different strains, the expression data set of the 1,915 core genome genes of the five strains S86, LD55, LL08, LL52, and UCMA5713 was classified by hierarchical clustering. Clustering parameters were based on the agglomeration method of Ward. The strain dendrogram (Fig. 3), based on gene expression ratios, grouped strains LD55 and UCMA5713, while strain LL08 was the most divergent compared with the other four strains. To establish a link between genomic composition and gene regulation, we measured if genes declared as divergent were statistically overrepresented in the group of genes whose expression was significantly different from that for strain LD61 (ratio selected with FDR ≤ 7%). By using a hypergeometrical distribution, probabilities higher than 12% were observed for each tested strain showing no significant enrichment of divergent genes in the group of differentially regulated genes.
FIG. 3.
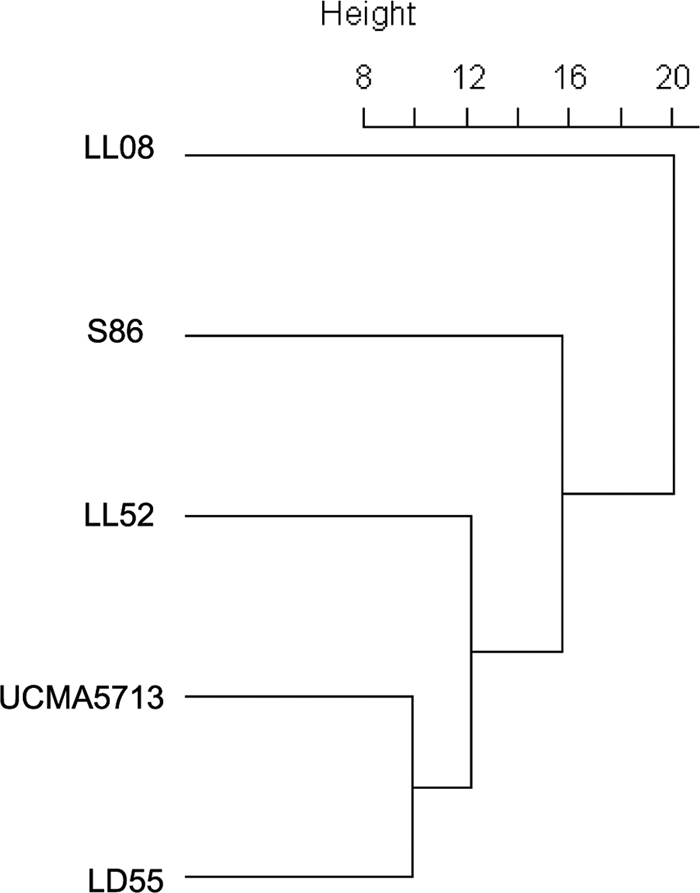
Strain dendrogram based on hierarchical clustering using gene expression ratios (using strain LD61 as a reference) of the 1,915 common genes of the five strains.
A total of 968 different genes were differentially regulated in at least one of the five strains compared to the LD61 strain. For each strain, genes were equally partitioned between higher and lower expression levels compared to that for strain LD61. The strains UCMA5713, LD55, and S86 exhibited 254, 332, and 270 differentially expressed genes, while the other two strains, LL08 and LL52, revealed even higher numbers (respectively, 562 and 586 genes). In order to analyze the contributions of individual gene expression regulation to the metabolic traits of the different strains, we determined, for each strain, the over- and underexpressed gene enrichments in the various functional categories by the Wilcoxon test for the whole category (P < 0.05 for significance) as previously described (7). The significantly regulated (sub)categories compared to strain LD61 are reported in Table 4. Strong divergence of the five strains compared to strain LD61 is observed within many regulated categories distributed throughout metabolism. Even if some (sub)categories showed a similar type of regulation for the five strains, most of the functional categories exhibited different regulation between the strains, underlining the large transcriptomic expression variability between the five strains. Table 5 lists significant differentially regulated genes, which will be discussed below.
TABLE 4.
Significantly regulated functional categories for each of the five strains (compared to strain LD61) according to a Wilcoxon testa
| Functional category or subcategory | Expression level in strain: |
||||
|---|---|---|---|---|---|
| UCMA5713 | LD55 | LL08 | LL52 | S86 | |
| Amino acid biosynthesis | L | L | L | L | L |
| Aromatic amino acid family | L | L | L | ||
| Aspartate family | L | L | L | ||
| Branched-chain family | L | L | L | L | L |
| Histidine family | L | L | L | L | L |
| Cellular processes | L | ||||
| Chaperones | H | ||||
| Transformation | H | ||||
| Protein and peptide secretion | L | ||||
| Biosynthesis of cofactors, prosthetic groups, and carriers | |||||
| Menaquinone and ubiquinone | H | H | |||
| Thioredoxin, glutaredoxin, and glutathione | L | ||||
| Fatty acid and phospholipid metabolism | H | H | |||
| Central intermediary metabolism | L | ||||
| Other | L | L | L | L | |
| Energy metabolism | L | ||||
| Anaerobic | L | L | L | ||
| ATP-PMF conversion | L | L | L | ||
| Fermentation | L | ||||
| Sugars | H | H | H | H | |
| TCA cycleb | H | ||||
| Other categories | L | ||||
| Adaptations and atypical conditions | H | ||||
| Transposon related functions | H | ||||
| Purines, pyrimidines, nucleosides and nucleotides | L | L | L | L | L |
| Purine ribonucleotide biosynthesis | L | L | L | L | |
| Pyrimidine ribonucleotide biosynthesis | L | L | L | L | L |
| Salvage of nucleosides and nucleotides | L | ||||
| Sugar-nucleotide biosynthesis and interconversions | H | H | |||
| Regulatory functions | L | L | L | L | |
| AraC family regulators | L | ||||
| General | L | ||||
| LysR family regulators | L | L | |||
| MarR family regulators | L | L | |||
| Two-component systems | L | ||||
| Replication | |||||
| Degradation of DNA | H | H | H | H | |
| Translation | L | ||||
| Translation factors | L | ||||
| Ribosomal proteins: synthesis and modification | H | ||||
| Amino acyl tRNA synthetases | L | ||||
| Transport and binding proteins | L | L | L | L | |
| Amino acids, peptides and amines | L | L | |||
| Anions | L | ||||
| Carbohydrates, organic alcohols and acids | H | ||||
P < 0.05. “H” and “L” indicate expression levels that are higher and lower, respectively, than those in LD61.
Tricarboxylic acid cycle.
TABLE 5.
Differentially regulated genes sustaining regulated functional categories in the five strains (compared to strain LD61, as shown in Table 4)a
| Functional category or subcategory | Differentially regulated gene(s)b in strain: |
||||
|---|---|---|---|---|---|
| UCMA5713 | LD55 | LL08 | LL52 | S86 | |
| Amino acid biosynthesis | |||||
| Aromatic amino acid | trpA B | aroH | trpAB | aroCDH | aroH |
| Branched chain | ilvCD, leuC | ilvD, leuC | ilvBCD, leuBD | ilvBD, leuBC | ilvBCDN, leuBC |
| Histidine | hisZ | hisCZ | hisACDGHIKZ | hisDHIZ | hisCZ |
| Transport and energy metabolism | |||||
| Sugars | galKMT, glk, lacCZ, thgA | galM, glk, gntZ, lacCZ | bglA, galKMT, gntZ, lacCZ, scrK, thgA, uxaC, ypdBD, yrcA | galEKMT, lacZ, thgA yidC | galKM, thgA, uxaC |
| Glutamate/GABA | gadC, gadB | ||||
| Glutamate/ornithine | gltQS, argBCDEJ | ||||
| Arginine deiminase pathway | arcD2, arcBC1 | arcABC1 | arcBC1 | ||
| Biogenic amine synthesis | potAD | ||||
| Citrate metabolism | citCDEF, icd | ||||
| Glycolysis | ldh | yjhF, pgmB | |||
| Fermentation | als | ackA1A2, frdC, butB | |||
| Alternative sugar metabolism | scrK, yrcA ypdBD | ||||
| ATP-PMF conversion | atpDEH | ||||
| PTS and carbohydrate transport | celB, ptcABC, rgpC, ypdA, yngFypcGH | ||||
| Biosynthesis of cofactors, prosthetic groups, and carrier | |||||
| Thioredoxin, glutaredoxin, and glutathione | gshR, trxB1 | ||||
| Cell envelope | |||||
| Adaptations and atypical conditions | tpx | ||||
| Polysaccharide | rgpBEF, ycbBDFHIJ | ||||
| Peptidoglycan | murA1BC | ||||
| Translation | |||||
| Ribosomal proteins and translation factors | rplBCNT, rpsCDGQS,rpmI, rnpA, infC | ||||
| Regulatory functions | |||||
| AraC family regulators | xylR | ||||
| DeoR family regulators | lacR | ||||
| General | purR | birA2, rmeD, ydcG, yjaJ, ypfD,yrtfA ysfD | argR, birA1, copR, tenA,yabA, ybeD, ydcGyecA, ykhIyliA | glnR, rmeBymiA, ynaB, | |
| GntR family regulators | rgrB | ||||
| GTP-binding proteins | yphL,yyaL | ylqL, yyaL | |||
| LacI family regulators | rliB | rliC | |||
| LysR family regulators | fhuR | ||||
| MarR family regulators | rmaG | rmaA,zitR | rmaC, rmaIJ | ||
| Two-component systems | llrG | ||||
The expression ratios are listed in Table S1 in the supplemental material. Genes that are underlined exhibited increased expression compared to that in strain LD61, while genes that are not underlined showed expression lower than that in LD61.
Student test; FDR ≤ 7% for significance.
The efficient proteolysis of L. lactis LD61 in UF-cheese has been reported to lead to accumulation of free amino acids (in particular, glutamate) and to a low expression of nitrogen metabolism (6). In our study, after 24 h of growth in a UF-cheese model, the five strains displayed lower expression levels of the amino acid biosynthesis pathways (e.g., aromatic amino acids [aro and trp genes], branched-chain amino acids [ilv and leu genes], and amino acids of the histidine family [his genes]) than the LD61 reference strain.
In UF-cheese, it was shown that strain LD61 induced specific responses to counteract different sources of stress, including acidic and oxidative stresses and carbon limitation (6). Compared to LD61, the four strains LL08, LL52, S86, and UCMA5713 exhibited modification of expression of genes involved in acidic stress resistance. Several genes of the arginine deiminase pathway, which produces NH3 through the conversion of arginine into ornithine, were more highly expressed in strains LL08 (arcBC1 and the arginine/ornithine antiporter arcD2), LL52 (genes arcABC1), and S86 (arcBC1). In strain LL08, concomitantly, genes involved in glutamate transport and conversion into ornithine (gltQS and argBCDEJ) were more highly expressed than in LD61. In this strain, the genes citCDEF, involved in citrate metabolism, which is an alternative acid stress response, were also more highly expressed. In strain LL52, the genes atpDEH, encoding the different subunits of the ATPase, were expressed at lower levels than in the LD61 strain. This enzyme catalyzes proton expulsion and is thus directly involved in resistance to acidic stress. In the plant-associated strain UCMA5713, a net consumption of protons could be provided via the glutamate decarboxylase converting glutamate into the biogenic amine γ-aminobutyrate (GABA). Glutamate transporter (gadC, coding for a glutamate/GABA antiporter) and glutamate decarboxylase (gadB) expression was specifically increased in strain UCMA5713 compared to that in the other strains. Generally, it was found that the lactococcal gadC and gadB genes were maximally expressed at low pH (5), agreeing with the lower final pH observed for this particular strain.
In response to oxidative stress, strain LL52 exhibited lower expression of genes involved in maintaining a favorable redox balance, such as gshR and trxB1, which code for glutathione reductase and thioredoxin reductase, respectively. Significantly higher transcription of the gene tpx, coding for a peroxidase, was observed in strain S86.
Concerning carbon limitation, a global overexpression of genes involved in carbon metabolism and sugar transport was observed in the strain LD61 (6). Here, all five strains induced a higher expression of the Leloir pathway, which is dedicated to lactose assimilation (genes of the gal and lac families), than the LD61 strain. In addition, expression of several genes involved in alternative sugar metabolism, such as scrK, ypdBD, and yrcA, was specifically more highly transcribed in strain LL08 than in strain LD61. Consistent with this observation, increased expression was observed for genes related to sugar transport, such as the PTS system for cellobiose (celB and ptcABC) or genes coding for permease, binding proteins, and transporter (rgpC, ypdA, ypcGH, and yngF) involved in the general carbohydrate transport system for N-galactosides, sugars, and polysaccharides in strain LL08. In strain LL08, increased expression of genes involved in carbon transport is consistent with the increased expression of the ldh gene, coding for lactate dehydrogenase, which catalyzes lactate production from pyruvate, the last step of carbon catabolism in homolactic bacteria. More surprisingly, the pathway of pyruvate conversion into acetolactic acid was also highly expressed, with an increased transcription of the als gene in strain LL08 compared to that in the LD61 strain. Inversely, genes involved in central carbon metabolism in strain LL52 were specifically transcribed to a lower level in this strain (glycolytic genes yjhF and pgmB and those involved in the functional category fermentation).
Finally, some strains exhibited specific traits compared to the LD61 strain. The production of biogenic amines (i.e., putrescine) from ornithine is likely in strain LL08, since increased expression of the genes potA and potD, involved in spermidine/putrescine transport, was observed. Another important peculiarity of this strain is the overexpression of the functional category ribosomal proteins (encoded by the rpl and rps genes) and of the infC gene, coding for a translation initiation factor. Cell envelope modifications were suggested in both strains LL08 and LL52, since higher expression was observed in LL08 for the rgpBEF and ycbBDFHIJ genes (potentially involved in biosynthesis, assembly, and transport of cell wall polysaccharides and important for phage adsorption [3, 8, 30]) and in LL52 for three genes, murA1BC, involved in aminosugar metabolism. Finally, the significant strain-specific expression of regulators belonging to different families (except in the case of strain UCMA5713) can be underlined (Table 5).
DISCUSSION
In this article, we have explored the genomic similarity and transcriptomic diversity of six L. lactis subsp. lactis strains exhibiting a dairy phenotype (growth in a UF-cheese model and high acidification rate). The microarray platform was designed based on the sequenced strain IL1403 (3). This strain has lost its dairy phenotype due to plasmid curation and uncontrolled laboratory evolution but has a dairy background and is thus a good reference for the study. Since no plasmid-borne gene was targeted with this microarray platform, the genomic and transcriptomic diversity linked to the plasmid content of the tested strains was not examined in this study.
The six L. lactis subsp. lactis isolates are part of the Génoferment collection. They were isolated from various sources, and all showed a dairy phenotype. During growth in UF-cheese, no significant difference in terms of growth rate and final cell density was observed between the strains. However, phenotypic differences were detected when final cheese pHs were compared, since a 0.3-pH-unit variation can lead to different cheese quality.
Analysis of the genome content by CGH showed that the six strains shared a large core genome of 1,915 genes, corresponding to more than 99% of the IL1403 ORFs spotted on the membranes. Significantly lower core genome values are obtained for other species (18). Here, the core genome could be named a “dairy” core genome since it is representative of the very homogeneous group of strains sharing a dairy phenotype (growth in a UF-cheese model and high acidification rate). However, we cannot exclude a certain genomic variability between the strains related to their accessory genomic content, notably for strain LL52, harboring an additional 260 kb compared to the IL1403 strain. Using CGH analysis, we were able to analyze the accessory genome (or noncore) of strain IL1403 (defined as genes present in the IL-403 strain but not in all strains). Fourteen genes constituting part of the accessory genome can be divided into two groups as defined by Berger and coworkers (2): the mobilome (phage-related genes) and the diversity region involved in strain-specific fitness (genes involved in citrate metabolism, citR, and surface polysaccharide, pspB). This small accessory genome does not contain any gene unique to strain IL1403.
Nevertheless, we demonstrated diversity between the six strains at the level of gene expression of their core genome (1,915 genes). Such a level of regulation was already reported in the literature between two species, E. coli/Shigella (in adaptation to their environment), or two closely related E. coli K-12 substrains (during growth) (11, 28). Variation in gene expression was present between plant- and dairy-associated strains but, more interestingly, also between strains of the same dairy origin (starters and raw milk).
Specific gene regulation in the plant-associated strain UCMA5713 was mainly restricted to the increased expression of genes involved in glutamate uptake and conversion into GABA. Siezen et al. (22) did not report modification of the genomic content of two L. lactis plant isolates, KF147 and KF282, related to GABA metabolism, even if two polyamine transporter systems were present in these strains. Although being a plant-associated strain, UCMA5713 exhibited a dairy phenotype and was shown to be closely related to the dairy-origin strains (expression dendrogram, Fig. 3). This could be explained by the observation that strain UCMA5713 was isolated in grassland neighboring a dairy factory, not excluding the fact that UCMA5713 is not a real grassland strain but is a strain of dairy origin found on the plant. Among the dairy-origin strains, strain LL08 displayed the largest transcriptomic divergence, as shown by its separated branch in the dendrogram and its large amount of strain-specific transcriptomic features. Increased expression of genes involved in central metabolism, glutamate consumption, ribosomal protein synthesis, and translation factors and the stronger induction of alternative sugar metabolism and transport could indicate that this strain is still able to maintain metabolic activity even after 24 h of growth in UF-cheese. From an industrial point of view, the potential capabilities of strain LL08 to produce diacetyl, a butter flavor compound, and polysaccharides, potentially involved in cheese texture, are interesting properties even if a concomitant synthesis of undesired biogenic amines is also possible.
We have tried to link the different observation levels, phenotypes (gowth on UF-cheese matrix and acidification capability), genomic contents and transcriptomic profiles. After 24 h of growth in a UF-cheese model, the transcriptomic profiles (common for the five strains or strain specific) are in agreement with the genomic content of the strains and correspond to responses previously observed for the LD61 strain (6), i.e., acid and oxidative stresses and carbon but not nitrogen limitation. In the case of citrate utilization, however, a discrepancy was observed. Even if citrate is present in the cheese matrix, the five strains (UCMA5713, LD55, LL08, LL52, and S86) should not be able to metabolize citrate since they do not belong to Lactococcus lactis subsp. lactis biovar diacetylactis. The absence of the citR gene in the five strains is consistent with this observation. However, more surprisingly, a citDEF operon was present in all five strains, and increased expression of citDEF was observed in strain LL08 compared to that in strain LD61, belonging to Lactococcus lactis subsp. lactis biovar diacetylactis. In addition, using PCR (results not shown), we observed that neither the citP gene (coding for the citrate permease) nor the citM-citRCDEFGX region was present in strains UCMA5713, LD55, LL08, LL52, and S86. One explanation could be the presence of cross-hybridization of other genes (plasmid-borne genes) with the microarray probes designed against the IL1403 citDEF genes.
Despite a strong similarity of the core genome between the strains, a large transcriptomic polymorphism is observed. We have checked that there was no significant bias in expression change determination linked to low hybridization efficiencies. In addition, for each tested strain, we showed that genes declared divergent were not overrepresented in the group of genes differentially regulated compared to the LD61 strain. The strain-specific expression of a similar core genome could be related to the ability of accessory genome expression to interact with core genome expression. However, the strain with the largest chromosome (LL52) and the potentially largest accessory genome is not the most divergent strain at the transcriptomic level. The discrepancy between genomic similarity and transcriptomic diversity could more probably reveal strain-dependent regulatory networks. This conclusion is supported by variable regulation within the functional category regulatory functions (Table 4) between the strains, related to 37 regulatory genes exhibiting strain-specific expression differences (Table 5).
In conclusion, this study showed significant diversity of the level of gene expression between six strains of L. lactis subsp. lactis (even between strains with the same dairy origin) despite their close genetic content and their large core genome of almost 2,000 genes. We have demonstrated that integration of phenotypic/genomic and transcriptomic results is thus needed to access intrasubspecies diversity. The potential role of transcriptome polymorphism in adaptation of these strains to specific growth conditions will have to be investigated in the future.
Supplementary Material
Acknowledgments
This research was financially supported by the French National Research Agency (ANR), project GenoFerment (ANR-05-PNRA-020). P. Tan-a-ram was the recipient of a French government scholarship.
We thank R. Perrin and C. Beltramo (SOREDAB). We also thank N. D. Lindley for useful discussions.
Footnotes
Published ahead of print on 3 December 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Bachmann, H., et al. 2009. Regulatory phenotyping reveals important diversity within the species Lactococcus lactis. Appl. Environ. Microbiol. 75:5687-5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger, B., et al. 2007. Similarity and differences in the Lactobacillus acidophilus group identified by polyphasic analysis and comparative genomics. J. Bacteriol. 189:1311-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolotin, A., et al. 2001. The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp. lactis IL1403. Genome Res. 11:731-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cocaign-Bousquet, M., C. Garrigues, P. Novak, and P. Loubiere. 1995. Rational development of a simple synthetic medium for the sustained growth of Lactococcus lactis. J. Appl. Bacteriol. 79:108-116. [Google Scholar]
- 5.Cotter, P. D., and C. Hill. 2003. Surviving the acid test: responses of gram-positive bacteria to low pH. Microbiol. Mol. Biol. Rev. 67:429-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cretenet, M., et al. 2011. Dynamic analysis of the Lactococcus lactis transcriptome in cheeses made from milk concentrated by ultrafiltration reveals multiple strategies of adaptation to stresses. Appl. Environ. Microbiol. 77:247-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dressaire, C., et al. 2008. Growth rate regulated genes and their wide involvement in the Lactococcus lactis stress responses. BMC Genomics 9:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dupont, K., T. Janzen, F. K. Vogensen, J. Josephsen, and B. Stuer-Lauridsen. 2004. Identification of Lactococcus lactis genes required for bacteriophage adsorption. Appl. Environ. Microbiol. 70:5825-5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Even, S., N. D. Lindley, and M. Cocaign-Bousquet. 2003. Transcriptional, translational and metabolic regulation of glycolysis in Lactococcus lactis subsp. cremoris MG 1363 grown in continuous acidic cultures. Microbiology 149:1935-1944. [DOI] [PubMed] [Google Scholar]
- 10.Fukiya, S., H. Mizoguchi, T. Tobe, and H. Mori. 2004. Extensive genomic diversity in pathogenic Escherichia coli and Shigella strains revealed by comparative genomic hybridization microarray. J. Bacteriol. 186:3911-3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Gall, T., P. Darlu, P. Escobar-Paramo, B. Picard, and E. Denamur. 2005. Selection-driven transcriptome polymorphism in Escherichia coli/Shigella species. Genome Res. 15:260-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makarova, K. S., and E. V. Koonin. 2007. Evolutionary genomics of lactic acid bacteria. J. Bacteriol. 189:1199-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maligoy, M., M. Mercade, M. Cocaign-Bousquet, and P. Loubiere. 2008. Transcriptome analysis of Lactococcus lactis in coculture with Saccharomyces cerevisiae. Appl. Environ. Microbiol. 74:485-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nouaille, S., et al. 2004. Influence of lipoteichoic acid D-alanylation on protein secretion in Lactococcus lactis as revealed by random mutagenesis. Appl. Environ. Microbiol. 70:1600-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Passerini, D., et al. 2010. When genes but not genomes reveal bacterial domestication: an example from Lactococcus lactis. PLoS One 5:e15306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poolman, B., and W. N. Konings. 1988. Relation of growth of Streptococcus lactis and Streptococcus cremoris to amino acid transport. J. Bacteriol. 170:700-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen, T. B., et al. 2008. Streptococcus thermophilus core genome: comparative genome hybridization study of 47 strains. Appl. Environ. Microbiol. 74:4703-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raynaud, S., R. Perrin, M. Cocaign-Bousquet, and P. Loubiere. 2005. Metabolic and transcriptomic adaptation of Lactococcus lactis subsp. lactis biovar diacetylactis in response to autoacidification and temperature downshift in skim milk. Appl. Environ. Microbiol. 71:8016-8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redon, E., P. Loubiere, and M. Cocaign-Bousquet. 2005. Role of mRNA stability during genome-wide adaptation of Lactococcus lactis to carbon starvation. J. Biol. Chem. 280:36380-36385. [DOI] [PubMed] [Google Scholar]
- 21.Siezen, R. J., et al. 2010. Complete genome sequence of Lactococcus lactis subsp. lactis KF147, a plant-associated lactic acid bacterium. J. Bacteriol. 192:2649-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siezen, R. J., et al. 2008. Genome-scale genotype-phenotype matching of two Lactococcus lactis isolates from plants identifies mechanisms of adaptation to the plant niche. Appl. Environ. Microbiol. 74:424-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smit, G., B. A. Smit, and W. J. Engels. 2005. Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. FEMS Microbiol. Rev. 29:591-610. [DOI] [PubMed] [Google Scholar]
- 24.Taibi, A., N. Dabour, M. Lamoureux, D. Roy, and G. Lapointe. 23 February 2010. Evaluation of the genetic polymorphism among Lactococcus lactis subsp. cremoris strains using comparative genomic hybridization and multilocus sequence analysis. Int. J. Food Microbiol. [Epub ahead of print.] doi: 10.1016/j.ijfoodmicro.2010.02.020. [DOI] [PubMed]
- 25.Ulve, V. M., et al. 2008. RNA extraction from cheese for analysis of in situ gene expression of Lactococcus lactis. J. Appl. Microbiol. 105:1327-1333. [DOI] [PubMed] [Google Scholar]
- 26.van Hijum, S. A., et al. 2008. Supervised Lowess normalization of comparative genome hybridization data—application to lactococcal strain comparisons. BMC Bioinform. 9:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Hylckama Vlieg, J. E., et al. 2006. Natural diversity and adaptive responses of Lactococcus lactis. Curr. Opin. Biotechnol. 17:183-190. [DOI] [PubMed] [Google Scholar]
- 28.Vijayendran, C., et al. 2007. The plasticity of global proteome and genome expression analyzed in closely related W3110 and MG1655 strains of a well-studied model organism, Escherichia coli-K12. J. Biotechnol. 128:747-761. [DOI] [PubMed] [Google Scholar]
- 29.Wegmann, U., et al. 2007. Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp. cremoris MG1363. J. Bacteriol. 189:3256-3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashita, Y., Y. Tsukioka, K. Tomihisa, Y. Nakano, and T. Koga. 1998. Genes involved in cell wall localization and side chain formation of rhamnose-glucose polysaccharide in Streptococcus mutans. J. Bacteriol. 180:5803-5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yvon, M., E. Chambellon, A. Bolotin, and F. Roudot-Algaron. 2000. Characterization and role of the branched-chain aminotransferase (BcaT) isolated from Lactococcus lactis subsp. cremoris NCDO 763. Appl. Environ. Microbiol. 66:571-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.