

Abstract
The 1,914-bp open reading frame of xylC from Thermoanaerobacterium saccharolyticum JW/SL-YS485 encodes a calculated 73-kDa β-xylosidase, XylC, different from any glycosyl hydrolase in the database and representing a novel glycohydrolase family. Hydrolysis occurred under retention of the anomeric configuration, and transglycosylation occurred in the presence of alcohols as acceptors. With the use of vector pHsh, expression of XylC, the third β-xylosidase in this bacterium, increased approximately 4-fold when a loop within the translational initiation region in the mRNA was removed by site-directed mutagenesis. The increased expression of xylCm is due to removal of a stem-loop structure without a change of the amino acid sequence of the heterologously expressed enzyme (XylCrec). When gel filtration was applied, purified XylC had molecular masses of 210 kDa and 265 kDa using native gradient gel electrophoresis. The protein consisted of 78-kDa subunits based on SDS gel electrophoresis and contained 6% carbohydrates. XylC and XylCrec exhibited maximum activity at 65°C and pH65°C 6.0, a 1-h half-life at 67°C, a Km for p-nitrophenyl-β-d-xyloside of 28 mM, and a Vmax of 276 U/mg and retained 70% activity in the presence of 200 mM xylose, suggesting potential for industrial applications.
Xylans—the major hemicellulose fractions in plants—are, after cellulose, the most abundant renewable polysaccharide. Complete degradation of xylan requires the action of endoxylanases (EC 3.2.1.8) and related xylanolytic enzymes, including various esterases such as acetyl xylan esterases (EC 3.1.1.6) and feruloyl esterases (EC 3.1.1.73). Glycosidases, such as 1,4-β-xylosidase (in short, β-xylosidase) (EC 3.2.1.37), α-arabinofuranosidase (EC 3.2.1.55), and α-glucuronisidase (EC 3.2.1.31), play a crucial role in a complete and rapid degradation of branched and substituted xylans (1). The characterized β-xylosidases presently belong to five glycohydrolase (GH) families (http://www.cazy.org/Glycoside-Hydrolases.html, accessed April 2010). Many bacterial and fungal xylan 1,4-β-xylosidases (EC 3.2.1.37) have been purified and characterized. However, compared to thermophilic fungal enzymes and enzymes from mesophilic bacteria, relatively few β-xylosidases from thermophilic (anaerobic) Bacteria and Archaea have been reported (4). Most of the characterized β-xylosidases from aerobic thermophiles are from Geobacillus stearothermophilus (various strains; belonging to GH 39, 43, and 52) and other Geobacillus species (references 3 and 21 and literature cited therein) but also from Bacteria such as Deinococcus geothermalis (5) and Rhodothermus marinus (16). The β-xylosidases from the anaerobic thermophilic Bacteria include those from Thermotoga maritima (35), Thermotoga sp. FjSS3-B.1 and Thermotoga neapolitana (31), and Thermoanaerobacterium brockii and Thermoanaerobacter ethanolicus JW200 (15, 26) (all GH 3); Clostridium stercorarium (GH 3 and 39) (22); Thermoanaerobacterium saccharolyticum (GH 39) (11, 13); Caldicellulosiruptor saccharolyticus (GH 43) (14); and Thermoanaerobacter italicus and Thermoanaerobacter mathrani (GH 52) (cited at InterPro protein matches [http://www.ebi.ac.uk/interpol/]). The thermostable enzymes are of interest due to their structural features which confer thermostability as well as for potential advantages in industrial processes.
Thermoanaerobacterium saccharolyticum JW/SL-YS485 is an anaerobic thermophile that grows at pH values ranging from 3.85 to 6.35 and at temperatures ranging from 30 to 66°C and can use xylan as its sole energy source. From this strain, two acetyl xylan esterases (27) and an α-O-methyl-glucuronidase (25), as well as a high-molecular-weight, cell-associated endoxylanase (24), have been purified and characterized. Furthermore, Liu et al. (12) and Lorenz and Wiegel (13) have reported the isolation and expression of the genes encoding an endoxylanase, an acetyl xylan esterase, and the β-xylosidase XylB.
In this paper, we report on a novel type of β-xylosidase (XylC), encoded by xylC from T. saccharolyticum JW/SL-YS485, and on significantly enhancing the overexpression of this enzyme in Escherichia coli by site-directed mutations of the mRNA and by using the expression vector pHsh. The properties of this enzyme, especially its high tolerance to elevated concentrations of substrates and the product xylose, make it potentially useful for various biotechnological applications, especially in combination with a high-affinity β-xylosidase to effectively cover a wide range of substrate concentrations.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Escherichia coli JM109 (Promega, Madison, WI) was used as the host for cloning and expression of the xylosidase gene. E. coli cells were grown at 37°C in Luria-Bertani (LB) broth containing ampicillin (100 mg/ml). T. saccharolyticum JW/SL-YS485 was cultivated as described previously (24). Plasmid pMD19-T vector (Takara, Dalian, China) was employed for cloning. The Hsh system, a new series of gene expression vectors, is regulated by the alternative σ32 factor of E. coli, and plasmid pHsh was employed for site-directed mutagenesis and gene expression (28). For overexpression of the recombinant β-xylosidase, XylCrec cells of E. coli JM109 harboring pHsh-xylCm were grown with vigorous aeration at 30°C in LB broth containing 100 μg of ampicillin per ml until an optical density at 600 nm (OD600) of 0.6 to 0.8 was reached. The expression of XylCrec was induced by a rapid increase of the culture temperature to 42°C, and growth was continued for 8 h at this temperature. The pHsh system is described elsewhere (34).
Purification of enzyme.
Using the same procedure, the parental and the recombinant xylosidase were purified from the cells of T. saccharolyticum JW/SL-YS485 and E. coli containing pHsh-xylCI, respectively. Bacterial cells were resuspended in 50 mM phosphate buffer (pH 7.0) and disrupted with a French press of 1.25 × 105 kPa. Cell extracts were obtained by centrifugation for 60 min at 100,000 × g and 4°C using a Beckman L8-M ultracentrifuge (Beckman Instruments, Inc., Palo Alto, CA). All purification steps were performed at room temperature in the presence of 0.02% (wt/vol) sodium azide, which was added to prevent microbial growth. Chromatography media and prepacked columns were purchased from Pharmacia Biotech Inc. (Piscataway, NJ), except phenyl 650 M, which was purchased from TosoHaas (Montgomeryville, PA).
Ion-exchange chromatography on DEAE-Sepharose.
Cell extracts were loaded onto a DEAE-Sepharose column (2.6 by 56 cm) which had been previously equilibrated with 1 liter of 25 mM Bis-Tris propane {1,3-bis[tris(hydroxymethyl)-methylamino]propane} buffer, pH 7.0. Proteins were eluted with a linear NaCl gradient (0 to 1 M) in 1.12 liters of Bis-Tris propane buffer at a flow rate of 4 ml/min and collected in 8-ml fractions.
Hydrophobic interaction chromatography.
The pooled, xylosidase activity-containing fractions from the DEAE-Sepharose column were mixed with a 4 M (NH4)2SO4 solution to give a final concentration of (NH4)2SO4 of about 1.5 M. This mixture was loaded onto a phenyl 650 M column (1 by 18 cm) which had been previously equilibrated with 25 mM Bis-Tris propane buffer containing 1.5 M (NH4)2SO4. Proteins were eluted using a linear gradient of (NH4)2SO4 from 1.5 to 0 M in 240 ml of Bis-Tris propane buffer at a flow rate of 1 ml/min and collected in fractions of 4 ml. Two separate peaks with xylosidase activity were obtained (termed XylA and XylC), pooled separately, and concentrated by a 2-h centrifugation at 3,750 × g and 4°C (model J2-21 centrifuge; Beckman Instruments, Inc., Palo Alto, CA) using Microsep, a centrifugal microconcentrator (Filtron Technology Corporation, Northborough, MA).
Ion-exchange chromatography on DEAE-Sephacel.
Fractions with xylosidase activity from the phenyl 650 M column were separately dialyzed each against 2 liters of 25 mM sodium citrate buffer (pH 6.4) overnight and applied onto a DEAE-Sephacel column (0.5 by 9 cm) equilibrated to pH 6.4 with 25 mM sodium citrate buffer, pH 6.4. Proteins were eluted with a linear pH gradient of 25 mM sodium citrate buffer from pH 6.4 to 3.0. The flow rate was 0.4 ml/min. The corresponding fractions with xylosidase activity from XylA and XylB were separately pooled in dialysis tubing and concentrated 20-fold using polyethylene glycol 10000 (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China).
Gel filtration.
The concentrated enzyme samples (0.2 ml) were applied each onto prepacked Superose 6 columns (10/30). Bis-Tris propane buffer (25 mM) containing 0.2 M NaCl was used for elution of proteins at a flow rate of 0.4 ml/min, and proteins were collected in 0.4-ml fractions.
Determination of molecular masses and pIs.
The apparent molecular masses of purified enzymes were determined by gel filtration on Superose 6 (10/30) and by native and SDS-polyacrylamide gel electrophoresis. The gel filtration molecular weight markers were MW-GF-200 (Sigma, St. Louis, MO). A native gradient (4 to 30%, wt/vol) polyacrylamide gel was cast using the Mini-Protean II multicasting chamber (Bio-Rad, Richmond, CA) according to the supplier's instructions. Gel electrophoresis was performed at 4°C for 16 h with a buffer consisting of 90 mM Tris, 80 mM boric acid, and 2.5 mM Na2-EDTA using the Mini-Protean II dual slab cell (Bio-Rad, Richmond, CA) and 160 V. SDS gradient gel (8 to 25%, wt/vol) electrophoresis was performed using the PhastSystem as suggested in the manual (Pharmacia Biotech Inc., Piscataway, NJ). Molecular weight markers from Pharmacia Biotech Inc. (Piscataway, NJ) and SDS-PAGE molecular weight standards (broad range) from Bio-Rad (catalog number 161-0317; Richmond, CA) were used as standards for native gel electrophoresis and SDS gradient gel (8 to 25%, wt/vol) electrophoresis, respectively. Isoelectric focusing was carried out on PhastGel IEF 4-6.5 (Pharmacia Biotech Inc., Piscataway, NJ) employing glucose oxidase (pI 4.2), trypsin inhibitor (pI 4.6), β-lactoglobulin A (pI 5.1), and carbonic anhydrase II (pI 5.4 and 5.9) purchased from Sigma (St. Louis, MO) as pI markers. Proteins in the polyacrylamide gels were stained with Coomassie blue (R-250).
Estimation of carbohydrate content.
The carbohydrate content of the purified enzymes was determined by the phenol-sulfuric acid assay using glucose as the standard (7). Fifty milliliters of enzyme solution was mixed with 50 ml of 5% (vol/vol) phenol and 250 ml of H2SO4; A490 was read after 25 min at 25°C.
NH2-terminal sequence.
The NH2-terminal sequences of XylA and XylC were determined by automated Edman degradation (protein sequencer model 470A or 477A; Applied Biosystems, Inc., Foster City, CA) at the Molecular Genetics Instrumentation Facility, University of Georgia.
Gene cloning, analysis, and mutagenesis.
Routine DNA manipulations were carried out essentially as described previously (23). Plasmid DNA and PCR products were purified using the Qiagen plasmid kit and PCR purification kit (Qiagen). PCRs were performed in a PE Applied Biosystems 9700 thermal cycler (Foster City, CA) using standard reaction conditions. Chemical reagents were purchased from Sigma (St. Louis, MO). DNA-modifying enzymes and polymerases were obtained from New England Biolabs (Beverly, MA) and Promega. Plasmid DNA was isolated using Qiagen (Valencia, CA) Miniprep or Tip 100 columns as described by the manufacturer. Oligonucleotide primers were synthesized from materials from Sangon (Shanghai, People's Republic of China). DNA sequencing was performed by the Biological Services Unit of Shanghai.
Nucleotide and amino acid sequences were analyzed with Dnaman, version 6.0, of the sequence analysis software package (Lynnon Biosoft). BLAST searches were done using the NCBI website (http://www.ncbi.nlm.nih.gov/, February 2010). The secondary structure of mRNA in the translation initiation region (TIR) of pHsh-xylC was predicted by the Mfold algorithm available online (http://mfold.rna.albany.edu/?=mfold/RNA-Folding-Form, accessed December 2010) (38).
Degenerate primers D1, D2, D3, and D4 (Table 1) were designed on the basis of NH2-terminal amino acid sequence MEYHVAKTGS from the purified XylC from T. saccharolyticum JW/SL-YS485. The gene encoding xylC was amplified from genomic DNA using restriction site-dependent PCR (RSD-PCR) (10) and ligated into pMD19-T. The xylC gene was amplified with primers P1 and P2 (Table 1) and subcloned into plasmid pHsh at StuI and HindIII sites to obtain pHsh-xylC.
TABLE 1.
Nucleotide sequences of primers used in this worka
| Primer | Nucleotide sequence |
|---|---|
| D1 | 5′-ATGGA(a/g)TA(t/c)CA(t/c)GTAGC-3′ |
| D2 | 5′-ATGGA(a/g)TA(t/c)CA(t/c)GTTGC-3′ |
| D3 | 5′-ATGGA(a/g)TA(t/c)CA(t/c)GTCGC-3′ |
| D4 | 5′-ATGGA(a/g)TA(t/c)CA(t/c)GTGGC-3′ |
| P1 | GCCATGTGGCTAAAACTGGCC |
| P2 | CCCAAGCTTCCAAACTTTTATGTAATTATTTC |
| P3 | TGTTGCTAAAACTGGTTCAGATGAAGGGAAAGGA |
| P4 | TGATATTCCATTTTTTTATCTCCTTCTTGTCGAC |
The noncapitalized nucleotides represent degenerated or mutated nucleotides. The boldface nucleotides indicate differences among the RSD-PCR primers D1, D2, D3, and D4. The primers P1 and P2 were used for subcloning of the xylC gene, and the two oligonucleotides P3 and P4 were used as primers for the mutation. The underlining indicates the restriction site; the boldface indicates mutation sites.
To raise the expression level of xylC, site-directed mutagenesis was designed based on the analysis of a predicted secondary structure of the mRNA. The Mfold analysis was arbitrarily defined for 70 nucleotides (nt) starting at the 35th nucleotide upstream from xylC in pHsh-xylC and conducted under default conditions with a change of temperature from 37 to 42°C. According to the Mfold analysis, two mutagenic oligonucleotides, P3 and P4, were synthesized as primers for the mutation to reduce the secondary structure of mRNA in the translation initiation region (TIR) (Table 1).
Enzyme activity assay and analysis of hydrolysis products.
Xylosidase activities are reported in international units (U), the amount of enzyme which releases 1 μmol product per minute. Specific activity is defined as the number of international units per milligram of protein. Protein concentrations were determined according to the work of Bradford (2).
The β-xylosidase activity was determined by assaying the amount of p-nitrophenol released from the substrate p-nitrophenyl-β-d-xylopyranoside (pNP-xyloside) (Sigma). The reaction mixture contained 10 μl of suitably diluted enzyme, 180 μl of 0.1 M potassium phthalate buffer (pH 6.0), and 10 μl of substrate solution (40 mM p-nitrophenyl xylopyranoside) as described elsewhere (26). After incubation at 65°C for 2 to 5 min, the reaction was stopped by adding 0.6 ml of 1 M Na2CO3, and the A405 was read. A standard curve was prepared by using p-nitrophenol.
For determination of the kinetic parameters, the purified enzymes (0.15 μg) were assayed with the substrate pNP-xyloside at five concentrations in the range of 1.8 to 58.5 mM. Kinetic parameters, Km and Vmax, were determined from Lineweaver-Burk plots. To verify xylosidase activity, the assay was performed similarly to the above-described assay but with 5 mM concentrations of the true substrates xylobiose and xylotriose as well as 1% (wt/vol) birch xylan.
Transglycosylation activity of XylC was demonstrated by using alcohols as the acceptors of glycosyl moieties. The action of XylC on 5 mM pNP-xyloside was determined in the presence of optimal concentrations of various alcohols and 50 mM potassium phthalate buffer (pH 6.0). After incubation at 60°C for 1.5 h, the reaction mixtures were concentrated and spotted on silica gel 60 F254 (Merck), and different alkyl xylosides were partitioned by thin-layer chromatography with acetonitrile and water (85:15, vol/vol) as the elution system.
Possible pectinase activity was analyzed in the pH range of 4.2 to 8.2 and between 40 and 80°C, using 2 μg purified enzyme in a 200-μl reaction mixture containing 10 mM citrus peel pectin (Sigma Chemical, St. Louis, MO).
Stereochemical analysis of the catalytic reaction was performed with purified XylC. The enzyme was incubated with 15 mM pNP-xyloside in 50 mM potassium phthalate buffer (pH 6.0) at 40°C for 1, 2, 5, and 10 min, and the reaction mixture was examined by high-pressure liquid chromatography (HPLC) for inversion of anomeric configuration. The HPLC analysis was performed on a column of TSKgel Amide-80 (1.5 by 80 mm) (Waters) with 70% acetronitrile as the flow phase at a flow rate of 1 ml/min. A standard of equilibrium states was prepared by using xylose (Sigma).
Mixtures of xylo-oligosaccharides containing xylobiose and xylotriose were obtained by hydrolyzing xylan with purified recombinant xylanase of Thermomyces lanuginosus. Hydrolysis products were analyzed by thin-layer chromatography using silica gel 60 F254 (Merck) and n-butanol/acetic acid/water (2:1:1, vol/vol/vol) as the mobile phase. After being partitioned, the sugar spots on the silica plates were stained by spraying the air-dried plate with a solution containing H2SO4-methanol (9:1) and heating it at 100°C for a few minutes.
Nucleotide sequence accession number.
The nucleotide sequence of the β-xylosidase gene xylC from T. saccharolyticum JW/SL-YS485 was deposited in the GenBank database under accession number EF193646.
RESULTS
Purification and protein properties of xylosidases.
Two xylosidases were purified from crude extract of the parental strain T. saccharolyticum JW/SL-YS485 using standard purification procedures as described in Materials and Methods. Two xylosidase activity peaks were obtained during hydrophobic chromatography on phenyl 650 M (Table 2). The corresponding xylosidase eluted first was named later XylA, and the other was named XylC. A XylB xylosidase was previously characterized (13) as a recombinant enzyme but was not observed as a corresponding activity during chromatographic analysis of crude extracts from the parental strain. After subsequent gel filtration chromatography, both XylA and XylB gave single electrophoretic bands on both native gradient gels and SDS-polyacrylamide gels. Protein properties confirm that XylA and XylC are different xylosidases: XylA and XylC have molecular masses of 122 and 210 kDa, respectively, determined via gel filtration chromatography; 112 and 265 kDa, determined by native gradient gel electrophoresis (Fig. 1); and 86 and 78 kDa for the subunits by SDS gel electrophoresis, respectively. XylA has a pI at pH 4.44 and XylC has a pI at pH 4.45, as determined by isoelectric focusing. XylC contained 6% (wt/wt) carbohydrates while no carbohydrates were observed in XylA, the enzyme with the higher molecular mass of the subunit (SDS gel), suggesting that XylA is not XylC without glycosylation.
TABLE 2.
Purification of XylC from T. saccharolyticum strain JW/SL-YS485
| Purification step | Enzyme | Total protein (mg) | Total activity (U) | Sp act (U/mg)a | Purification (fold) | Recovery (%) |
|---|---|---|---|---|---|---|
| Crude extract | Mixture | 1,069 | 1,847 | 1.73 | 1.00 | 100 |
| DEAE-Sepharose | Mixture | 121 | 1,749 | 14.5 | 8.35 | 95 |
| Phenyl- | XylA | 51.0 | 894 | 17.5 | 12.2 | 56 |
| Sepharose | XylC | 39.0 | 140 | 3.59 | ||
| DEAE- | XylA | 11.6 | 334 | 28.9 | 22.5 | 21 |
| Sepharose | XylC | 5.88 | 59.6 | 10.1 | ||
| Gel filtration | XylA | 5.06 | 172 | 34.1 | 46.0 | 12 |
| XylC | 1.17 | 53.8 | 45.8 |
Activity was determined with p-nitrophenyl-β-d-xylopyranoside as substrate (1 mM) in 50 mM potassium hydrogen phthalate-imidazole buffer, pH 6.0, at 65°C.
FIG. 1.
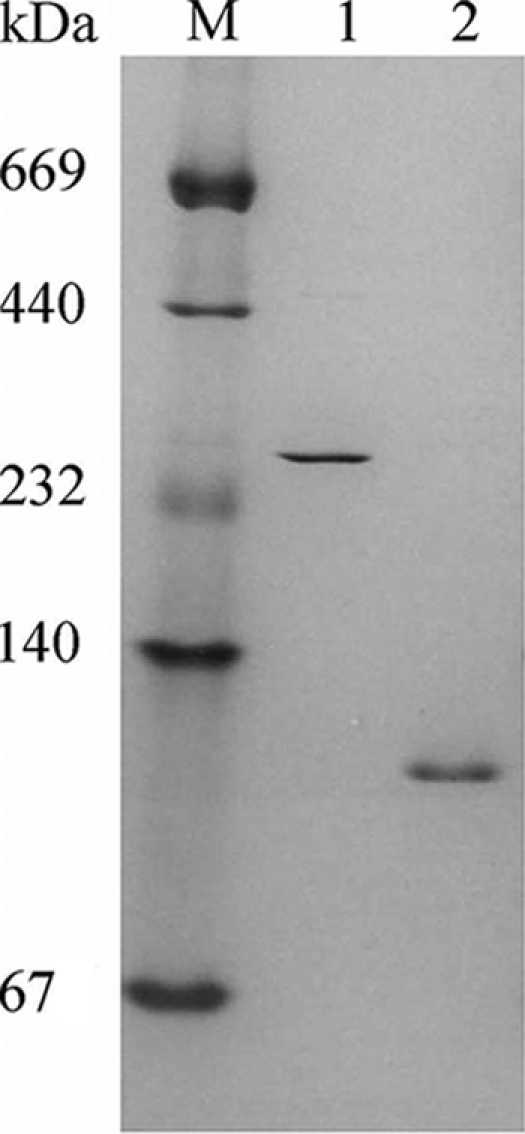
Nondenaturing gradient (4 to 30%) polyacrylamide gel analysis of purified native XylA and C. Lanes: M, molecular mass markers (in kDa); 1, XylC (5 μg); 2, XylA (2 μg). Enzymes were purified by Sepharose column chromatography and gel filtration as presented in Table 2. XylA and XylC were separated during phenyl-Sepharose chromatography.
Sequencing and cloning of XylC.
The NH2-terminal amino acid sequence of XylC was MEYHVAKTGS. Based on the amino acid residues of MEYHVA, the xylC gene was obtained by RSD-PCR when primer D4 (Table 1) was paired with RSD primers. Chromosome walking confirmed that the complete open reading frame (ORF) for xylC was 1,914 kb, encoding a protein with the same NH2-terminal sequence as that obtained from the purified parental enzyme. The gene encoded a 638-amino-acid protein with a theoretical molecular mass of 73 kDa, compared to the 78-kDa protein from SDS gel electrophoresis. The expression plasmid pHsh-xylC was generated by ligating this gene to the 2,442-bp plasmid pHsh (34). The nucleotide sequence of the β-xylosidase gene xylC from T. saccharolyticum JW/SL-YS485 was deposited in the GenBank database under accession number EF193646.
Potential secondary structure of mRNA, mutagenesis, and overexpression of xylCm.
The secondary structure of the first 70 nucleotides (nt) of the mRNA transcribed from pHsh-xylC was predicted as shown in Fig. 2a. In this predicted structure, the Shine-Dalgarno (SD) region and the start codon ATG were hidden in the loops formed at 42°C, the temperature for gene expression. Based on the premise of ensuring that the SD region and the start codon ATG were as exposed as possible, a mutation was designed to change 8 nt (TACCC→AAAAA; GGC→ATA and a G-to-T change) as indicated by the oversized italic bases in Fig. 3a to form a new predicted secondary structure for the first 70 nt (Fig. 2b). The calculations using the Mfold algorithm online indicate that the free energy (ΔG) changed from −38.5 to −26.3 kJ/mol, implying that the energy for forming the secondary structure of mRNA in TIR can be significantly reduced and thus that the translation efficiency can be increased by site-directed mutagenesis. In contrast to the mesophilic temperature required for using E. coli as the expression vector, at the optimal growth temperature of 65 to 68°C for T. saccharolyticum, the loop is much less stable and thus probably does not cause much reduction in the xylC expression.
FIG. 2.

Secondary structure of the translation initiation region (TIR) of the target gene. Predicted secondary structure of the 70 nt of TIR of pHsh-xylC (parental gene structure) (a) and pHsh-xylCm (modified gene structure) (b) calculated with the Mfold algorithm of Zuker (38). The elimination of the loop in pHsh-xylCm did not lead to a change of the amino acid sequence in the heterologously expressed (recombinant) enzyme XylCrec.
FIG. 3.

Gene sequence for the xylCm insert in plasmid pHsh-xylCm (a) and SDS-PAGE analysis of total soluble proteins in recombinant E. coli JM109 cells harboring pHsh-xylC or pHsh-xylCm (b). (a) Restriction sites, including the 3′ HindIII cloning sites, Hsh promoter sites (−10 and −35 regions), translation initiation region (70 nt), and transcription terminator sites, are illustrated. Oversized italic bases represent mutant nucleotides. rbs, ribosome binding site. (b) Lane M, low-molecular-weight marker; lane 1, E. coli JM109 containing the plasmid pHsh; lane 2, E. coli JM109 containing the plasmid pHsh-xylC; lane 3, E. coli JM109 containing the plasmid pHsh-xylCm; lane 4, purified recombinant XylCrec.
Site-directed mutagenesis was carried out according to the above-described design, and DNA sequencing confirmed that the 9 nt between the SD sequence and the N-terminal region of xylC were mutated (Fig. 3a). This mutated plasmid was designated pHsh-xylCm. SDS-PAGE analysis of a pHsh-xylCm cell-free lysate revealed a prominent protein band migrating at ∼72 kDa (Fig. 3b, lane 2); the recombinant form had an apparent molecular size which was slightly smaller than that of the native form. The difference in relative mobilities between the two forms is likely due to a lack of, or decrease in, glycosylation in the recombinant enzyme. The final cell densities of E. coli harboring different recombinant plasmids were about the same (optical density at 600 nm [OD600] around 3.2), but on the basis of densitometer scanning (Gel Doc 2000; Vilber Lourmat) of the SDS-PAGE gel (Fig. 3b, lane 4), the expressed level of XylCrec was significantly higher in pHsh-xylCm than in pHsh-xylC cultures (Fig. 3b, lanes 2 and 3). Furthermore, the activity in pHsh-xylCm cell extracts (∼5.5 U/ml) was 2.9-fold greater than that in pHsh-xylC cell extracts (∼1.4 U/ml). The high expression of xylCm was reproducible and was obtained every time. Recombinant XylC (XylCrec) was purified as described in Materials and Methods to gel electrophoretic homogeneity for further characterization (Fig. 3b, lane 4).
Kinetics and stability of XylC and substrate specificity and reaction products of XylC.
Using the artificial substrate pNP-xyloside, analysis of XylCrec activity demonstrated that the purified recombinant form exhibited high levels of thermostable xylosidase activity as well as temperature and pH optima that were essentially identical to values previously determined for XylC purified from T. saccharolyticum JW/SL-YS485. XylC and XylCrec hydrolyzed xylobiose and xylotriose (other oligomers not tested) and produced xylose from pNP-xyloside and xylo-oligosaccharides as shown by thin-layer chromatography. No significant activity was observed with pNP-α-l- arabinofuranoside (pNPAF), pNP-α-d-xylopyranoside, pNP-α-d-glucopyranoside, and pNP-β-d-glucopyranoside, nor with oat spelt xylan, birch wood xylan, or carboxymethyl cellulose (CMC) (detailed data not shown).
The optimal reaction conditions were pH 6.0 at 65°C for XylC as well as for the recombinant XylCrec in 2-min assays using pNP-xyloside as a substrate (Fig. 4a and b). Purified XylC was stable over a pH range of 6.0 to 8.0 and most stable at pH 7.0 (Fig. 4c). The enzyme was relatively stable under the optimal reaction conditions (pH 6.0, 65°C) (Fig. 4d) and exhibited a 1-h half-life at 67°C (Fig. 4e). Enzyme activity was not inhibited by EDTA at concentrations up to 10 mM (data not shown).
FIG. 4.

(a and b) Effects of pH (a) and temperature (b) on activity of purified recombinant XylCrec. The optimum pH for xylosidase activity was determined by incubation at 65°C for 2 min in the pH65°C range from 4.5 to 7.5 (pH was measured at 65°C; see the work of Wiegel [32] for explanation and calibration) in 100 mM potassium hydrogen phthalate-imidazole buffer (▴).The optimum temperature for enzyme activity was assayed in 100 mM potassium hydrogen phthalate-imidazole buffer at pH65°C 6.0 at 45 to 75°C for 5 min (⧫). Each assay was performed with 1.3 μg of protein per ml. (c) Effects of pH on the purified recombinant XylCm activity. Residual activities were assayed in 100 mM potassium hydrogen phthalate-imidazole buffer (pH 6.0) at 65°C for 5 min after preincubation of the enzyme in 50 mM potassium hydrogen phthalate-imidazole buffer at pH65°C 4.5 to 8 at 67°C for 2 h (▪) or 65°C for 1 h (⧫). (d and e) Thermostability of purified recombinant XylCm. Residual activities were assayed in 100 mM potassium hydrogen phthalate-imidazole buffer (pH65°C 6.0) at 65°C for 5 min after preincubation of the enzyme in 50 mM potassium hydrogen phthalate-imidazole buffer (pH65°C 6.0) at 50°C (▴), 65°C (▪), and 70°C (⧫) for varied times (d) and at 66°C, 67°C, 68°C, and 69°C for 1 h (e).
The dependence of the enzyme reaction rate on the pNP-xyloside concentration at pH 6.0 and 65°C followed Michaelis-Menten kinetics. XylCrec exhibited Km and Vmax values of 28 mM and 276 U per mg of protein, respectively. The kinetic properties of XylC and XylCrec were significantly different from those of the other xylosidases, e.g., XylA and the heterologously expressed XylB (13). For example, the isolated XylA and XylC had Km values of 0.68 versus 28.0 mM and a kcat of 5,075 versus 189,292, respectively (Fig. 5). XylC and XylCrec had an unusually high tolerance to inhibition by the XylC product, xylose; up to 70 mM xylose did not affect XylC activity at all, and 70% of its activity was retained in the presence of 200 mM xylose (detailed data not shown).
FIG. 5.
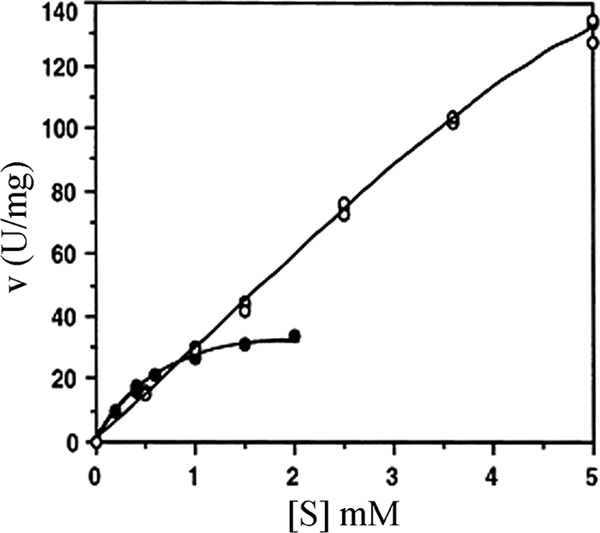
Effects of the substrate pNP-xyloside concentration on the activity of XylC (○) and XylA (•), both purified from T. saccharolyticum JW/SL-YS485. The variations in the duplicate assays are smaller than the symbols.
Under the conditions employed in this work, the standard xylose contained α- and β-xylose at a ratio of 1:1.87 as quantified from the two peaks during HPLC analysis. The retention times were 6.58 and 6.88 min for α- and β-xylose, respectively. During the hydrolysis of pNP-xyloside by XylC, the ratios of α- and β-xylose produced in the reaction mixture were 1:3.66, 1:2.7, 1:2.05, and 1:1.87 after the reaction times of 1, 2, 5, and 10 min, respectively (Fig. 6a), i.e., after 10 min the ratio of α- and β-xylose had reached the equilibrium values of the standard xylose. These results indicated that the enzyme produced β-xylose from β-xylopyranosides while retaining the anomeric configuration of the xylose in the substrate.
FIG. 6.

(a) Stereochemical analysis of reaction mixtures containing pNP-xyloside and XylC from T. saccharolyticum JW/SL-YS485. The enzymatic reaction was carried out at 40°C for 1, 2, 5, and 10 min. The migration times of β-xylose and β-xylopyranoside were 6.58 and 6.88 min, respectively. (b) Thin-layer chromatography of the transglycosylation products from pNP-xyloside (5 mM) and various alcohols in 50 mM morpholineethanesulfonic acid (MES) buffer (pH65°C 6.0). Lane M, pNP-xyloside; lane 0, control (reaction mixture without alcohol); lane 1, plus methanol; lane 2, plus ethanol; lane 3, plus 1-propanol; lane X, standard xylose.
Transglycosylation activities of XylC were detected by using pNP-xyloside as the substrate and various alcohols as receptors (Fig. 6b). XylC produced xylose from pNP-xyloside in the absence of alcohol (Fig. 6b, lanes M and 0) and produced a series of alkyl xylosides (Fig. 6b, lanes 1 to 3) by transferring xyloside from pNP-xyloside to alkyl alcohols.
Assignment to a novel glycosyl hydrolase family.
BLAST searches of the XylC amino acid and nucleotide sequence of xylC against sequences from both protein and nucleic acid databases failed to identify any significantly similar sequences of glycosyl hydrolases. Furthermore, comparison of the predicted amino acid sequence to existing sequences and assignment into one of the glycosidic families suggested that the xylosidases do not fit into any of the xylosidase families (4, 8). The InterProScan analysis (www.ebi.ac.uk/Tools/InterProScan) suggested two domains. The first domain (region 1 to 507) contained a pectin lyase (PL) fold which also included a conserved Zn binding domain (region 351 to 365) typical for alcohol dehydrogenases. The domain also contained five short parallel beta-helix repeats (region 246 to 436). Performance of sequence alignments with other PL-related sequences in the CAZy database (http://www.cazy.org/Glycoside-Hydrolases.html, accessed April 2010) indicated that this sequence seems to be closest to PL6 sequences. However, no pectin lyase activity was detected with the purified enzyme under various conditions (see Materials and Methods). The second domain (508 to 638) had no homology to any known domain. The C-terminal 120 amino acids have sequence similarity to many predicted outer membrane proteins. However, no predicted membrane-associated domain or transmembrane domain was detected.
DISCUSSION
Three xylosidases in T. saccharolyticum JW/SL-YS485.
Previously the β-xylosidase gene xylB from the xylanolytic T. saccharolyticum JW/SL-YS485 was cloned and expressed in E. coli (13). XylB is a homolog of the xynB gene from T. saccharolyticum B6A-RI. In contrast to XylB, which was never encountered as an active enzyme and characterized only as a recombinant enzyme, both XylA and XylC were purified as active enzymes from the parental thermophile. They migrate as much larger proteins on SDS-PAGE gels than did the gene product of xylB (13). The differences in protein and kinetic properties between XylA, XylB, and XylC suggest that they are not the same enzyme. Due to its unusual properties, XylC, encoded by xylC, was further characterized in detail (the gene encoding XylA has yet to be cloned). However, based on the protein purification and genetic analysis results from these studies, it is concluded that T. saccharolyticum JW/SL-YS48 contains at least these three distinct xylosidases. To our knowledge, such a combination of xylosidases has not been observed thus far in any other thermophilic anaerobe and especially not in the type strain B6A1 of T. saccharolyticum, for which only one xylosidase has been published (11). The only other bacterium which contains three xylosidases is the aerobic Bacillus stearothermophilus T-6; the three xylosidases belong to three different enzyme families (reference 3 and literature cited therein), but none of them exhibit properties similar to those of XylC. Based on the characterization reported here, it appears that XylA and XylC function under different substrate conditions and thus are of physiological importance for the bacterium when exposed to differing environments and substrate concentrations.
The XylC gene encodes a novel type of xylosidase.
Based on sequence and size identities between native and recombinant proteins, xylC is the gene encoding XylC. Analysis of xylosidase activity using the substrate pNP-xyloside demonstrated that the purified recombinant XylCrec and the XylC from the parental strain were essentially identical. They have the highest xylosidase activity at pH 6.0 and 65°C. Based on the data from gel filtration (210 kDa) and native and SDS gel electrophoresis (265 kDa and 78 kDa, respectively) and on the calculated molecular mass of 73 kDa, the native XylC is probably a trimer, an unusual quaternary structure for xylosidases. Beside XylC, a trimeric structure has been proposed for another thermostable xylosidase from Geobacillus pallidus. It has a thermal denaturation midpoint of 80°C and belongs to the GH 52 family (20). The majority of fungal enzymes are monomeric; a few are dimeric.
There are two basic types of glycosyl hydrolases: “retaining” and “inverting.” Importantly, retaining enzymes usually can perform both hydrolysis and transglycosylation reactions, whereas inverting enzymes can perform only hydrolysis; Smaali et al. (29) reported that the β-xylosidases from Bacillus halodurans C-125 (belonging to GH 39) functioned via a retaining mechanism exhibiting transglycosylation using either p-nitrophenyl-β-d-xyloside or xylotriose. Similarly, the XylC from T. saccharolyticum JW/SL-YS485 in this study also exhibited retention of the anomeric configuration as well as transglycosylation activity with primary alcohols of straight carbon chains.
Both XylC and recombinant XylCrec exhibited unusually high values for the apparent Km (28 mM pNP-xyloside) and Vmax (276 U/mg). Reported Km values of other β-xylosidases range from 0.08 (19) to 5.8 mM (17) for fungal enzymes and from 0.018 (26) to 10 mM (9) for bacterial enzymes. To our knowledge, XylC from T. saccharolyticum JW/SL-YS485 exhibits the highest Km (i.e., lowest affinity) for the artificial substrate pNP-xyloside. An important unusual feature is the absence of a significant substrate or product inhibition. Most xylosidases are inhibited by high concentrations of pNP-xyloside. In contrast, the catalytic activity of the here-described parental xylosidase XylC and the recombinant XylCrec from T. saccharolyticum JW/SL-YS485 was not reduced in the presence of high pNP-xyloside (absence of substrate inhibition). Furthermore, most importantly, there was no measurable inhibition at 70 mM xylose and only a 30% inhibition in the presence of 200 mM xylose (slight product inhibition). These properties suggest that XylC has its physiological role when such elevated concentrations of xylo-oligomers and xylose are generated and accumulated. The other enzymes known for exhibiting an absence of strong product inhibition are (i) the fungal xylosidase from Scytalidium thermophilum, which is insensitive to inhibition by up to 200 mM xylose (highest value tested) (37); (ii) the enzyme from Humicola grisea var. thermoidea, which is not inhibited by 10 mM xylose (highest concentration tested) (6); and (iii) the enzyme from Aspergillus nidulans, which is inhibited to 44% by 25 mM xylose. Furthermore, a Ki value for xylose of 139 mM was reported for the xylosidase from Paecilomyces thermophila (36). If the affinity was determined, most xylosidases, especially those from bacteria, have Ki values for xylose of between 2 and 10 mM. Additionally, many xylosidases exhibit high activity against pNP-xyloside as well as some activity against p-nitrophenyl-α-l-arabinofuranoside (pNPAF) (26). However, XylC from T. saccharolyticum JW/SL-YS485 did not have any arabinosidase activity.
Beside its unusual kinetic properties, XylC has an unusual amino acid sequence. FASTA and BLAST searches and analysis for known domains of the deduced protein sequence failed to detect any similar xylosidase sequences. The absence of any pectin lyase activity of the purified enzymes makes the observed sequence similarity to pectin lyase interesting for speculations regarding the origin of this unusual xylosidase.
No similarities were observed to the xylosidase-containing glycoside hydrolase families GH 3, 39, 52, and 54; these families contain xylosidases which—like the xylosidase presented here—retain the anomeric configuration during cleavage. Nor was similarity observed to the family GH 43, comprised of xylosidases with an inverting anomeric configuration mechanism (30) (www.cazy.org/Glycoside-Hydrolases.html). Thus, it is proposed that this XylC represents the first enzyme of a novel glycosidase family (B. Henrissat, personal communication).
Overexpression of recombinant protein.
The predicted secondary structures of the mRNAs in TIR suggested that eliminating the loop in the secondary structure of the mRNA in TIR can significantly increase the translation efficiency (18, 33). Because the secondary structure of the mRNA is an important determinant of translation efficiency, replacement of the G or C nucleotides between the SD sequence and the start codon, and replacement of the third nucleotide of codons in the N-terminal region with A or T, markedly increased the expression level of xylCrec compared to that of the parental xylC. The results presented here demonstrate that this assumption was correct and substantiate the idea that mutating nucleotides between the SD sequence and the 5′ flanking region of xylC by reducing the mRNA secondary structure in TIR is a useful approach to control and increase the expression level of heterologous proteins in E. coli cells. The increased expression of xylCm is due only to the removal of the stem-loop structure in xylCm, with the result that the start codon is in a less-structured region. The change in the nucleotide sequence in xylCm does not change the amino acid sequence of XylCrec (Fig. 2). The employed novel expression vector pHsh and the mutated mRNA in TIR led to a high level of the recombinant XylCrec, a requirement for enzymes to be considered for potential applications in biotechnology and biochemical processes (28, 34).
In summary, the xylosidase XylCrec was expressed heterologously at a high expression level in E. coli after a loop within the translational initiation region in the mRNA secondary structure was removed by site-directed mutagenesis, yielding xylCm, which encoded the unusual β-xylosidase XylC, and using the novel expression vector pHsh (yielding pHsh-xylCm). We suggest that the removal of high-expression-hindering loops in the translational initiation region could be a more general approach to high heterologous expression.
XylC is the first member of a novel GH family retaining the anomeric configuration during hydrolysis and exhibiting transglycosylation. Several properties of XylC, including insensitivity to product inhibition and a relatively high turnover rate, suggest that this novel enzyme has several potential industrial applications.
ADDENDUM IN PROOF
Preliminary X-ray crystallographic analysis indicates that XylC is a tetromer in its crystal form (Rey-Ting Guo, personal communication).
Acknowledgments
This work was supported by the National Science Foundation of China (grant no. BK30770061).
We thank Karen Bowers for proofreading the original version of the manuscript.
Footnotes
Published ahead of print on 3 December 2010.
REFERENCES
- 1.Biely, P. 1985. Microbial xylanolytic systems. Trends Biotechnol. 3:286-290. [Google Scholar]
- 2.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 3.Brux, C., et al. 2006. The structure of an inverting GH43 beta-xylosidase from Geobacillus stearothermophilus with its substrate reveals the role of the three catalytic residues. J. Mol. Biol. 359:97-109. [DOI] [PubMed] [Google Scholar]
- 4.Cantarel, B. L., P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard, and B. Henrissat. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. J. Nucleic Acids Res. 37(database issue):D233-D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cournoyer, B., and D. Faure. 2003. Radiation and functional specialization of the family-3 glycoside hydrolases. J. Mol. Microbiol. Biotechnol. 5:190-198. [DOI] [PubMed] [Google Scholar]
- 6.de Almeida, E. M., M. L. T. M. Polizeli, H. F. Terenzi, and J. A. Jorge. 1995. Purification and biochemical characterization of β-xylosidase from Humicola grisea var. thermoidea. FEMS Microbiol. Lett. 130:171-175. [Google Scholar]
- 7.DuBois, M., K. A. Gilles, J. K. Hamilton, P. A. Rebers, and F. Smith. 1956. Colorimetric method for determination of sugars and related substances. Anal. Chem. 28:350-356. [Google Scholar]
- 8.Henrissat, B., and A. Bairoch. 1993. New families in the classification of glycosyl hydrolases on amino acid sequence similarities. Biochem. J. 293:781-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudson, R. C., L. R. Schofield, T. Coolbear, R. M. Daniel, and H. W. Morgan. 1991. Purification and properties of an aryl β-xylosidase from a cellulolytic extreme thermophile expressed in Escherichia coli. Biochem. J. 273:645-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang, Y., J. Pei, X. Song, and W. Shao. 2007. Restriction site-dependent PCR: an efficient technique for cloning of new genes from genomic DNA. DNA Res. 14:283-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee, Y. E., and J. G. Zeikus. 1993. Genetic organization, sequence and biochemical characterization of recombinant β-xylosidase from Thermoanaerobacterium saccharolyticum strain B6A-RI. J. Gen. Microbiol. 139:1235-1243. [DOI] [PubMed] [Google Scholar]
- 12.Liu, S.-Y., F. C. Gherardini, M. Matuschek, H. Bahl, and J. Wiegel. 1996. Cloning, sequencing, and expression of the gene encoding a large S-layer associated endoxylanase from Thermoanaerobacterium saccharolyticum strain JW/SL-YS 485 in Escherichia coli. J. Bacteriol. 178:1539-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorenz, W., and J. Wiegel. 1997. Isolation, analysis, and expression of two genes from Thermoanaerobacterium saccharolyticum strain JW/SL-YS485: a β-xylosidase and a novel acetyl xylan esterase with cephalosporin C deacetylase activity. J. Bacteriol. 179:5436-5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luthi, E., and P. L. Bergquist. 1990. A beta-d-xylosidase from the thermophile Caldocellum saccharolyticum expressed in Escherichia coli. FEMS Microbiol. Lett. 9:291-294. [Google Scholar]
- 15.Mai, V., J. Wiegel, and W. Lorenz. 2000. Cloning, sequencing, and characterization of the bifunctional xylose-arabinosidase from the anaerobic thermophile Thermoanaerobacter ethanolicus. Gene 247:137-143. [DOI] [PubMed] [Google Scholar]
- 16.Manelius, Å., L. Dahlberg, and O. Holst. 1994. Some properties of a thermostable β-xylosidase from Rhodothermus marinus. Appl. Biochem. Biotechnol. 44:39-48. [Google Scholar]
- 17.Matsuo, M., and T. Yasui. 1984. Purification and some properties of β-xylosidase from Trichoderma viride. Agric. Biol. Chem. 48:1845-1852. [Google Scholar]
- 18.Pei, J., and W. Shao. 2008. Purification and characterization of an extracellular α-l-arabinosidase from a novel isolate Bacillus pumilus ARA and its over-expression in Escherichia coli. Appl. Microbiol. Biotechnol. 78:115-121. [DOI] [PubMed] [Google Scholar]
- 19.Poutanen, K., and J. Puls. 1988. Characteristics of Trichoderma reesei β-xylosidase and its use in the hydrolysis of solubilized xylans. Appl. Microbiol. Biotechnol. 28:425-432. [Google Scholar]
- 20.Quintero, D., Z. Velasco, E. Hurtado-Gomez, J. L. Neira, and L. M. Contreras. 2007. Isolation and characterization of a thermostable β-xylosidase in the thermophilic bacterium Geobacillus pallidus. Biochim. Biophys. Acta 1774:510-518. [DOI] [PubMed] [Google Scholar]
- 21.Rohman, A., et al. 2007. Purification, crystallization and preliminary X-ray analysis of a thermostable glycoside hydrolase family 43 β-xylosidase from Geobacillus thermoleovorans IT-08. Acta Crystallogr. F 63:932-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruttersmith, L. D., and R. M. Daniel. 1993. Thermostable α-glucosidase and β-xylosidase from Thermotoga sp. strain FjSS3-B.1. Biochim. Biophys. Acta 1156:167-172. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 24.Shao, W., S. Deblois, and J. Wiegel. 1995. A high-molecular-weight, cell associated xylanase isolated from exponentially growing Thermoanaerobacterium saccharolyticum strain JW/SL-YS 485. Appl. Environ. Microbiol. 61:937-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao, W., S. K. C. Obi, J. Puls, and J. Wiegel. 1995. Purification and characterization of the α-glucuronidase from Thermoanaerobacterium sp. strain JW/SL-YS485, an important enzyme for the utilization of substituted xylans. Appl. Environ. Microbiol. 61:1077-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao, W., and J. Wiegel. 1992. Purification and characterization of a thermostable β-xylosidase from Thermoanaerobacter ethanolicus. J. Bacteriol. 174:5848-5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao, W., and J. Wiegel. 1995. Purification and characterization of two thermostable acetyl xylan esterases from Thermoanaerobacterium sp. strain JW/SL-YS485. Appl. Environ. Microbiol. 61:729-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao, W., H. Wu, and J. Pei. October 2010. Novel expression vector system regulated by sigma 32 and methods for using it to produce recombinant protein. U.S. patent 7,807,460.
- 29.Smaali, M. I., C. Rémond, and M. J. O'Donohue. 2006. Expression in Escherichia coli and characterization of β-xylosidases GH39 and GH-43 from Bacillus halodurans C-125. Appl. Microbiol. Biotechnol. 73:582-590. [DOI] [PubMed] [Google Scholar]
- 30.Stam, M. R., E. Blanc, P. M. Coutinho, and B. Henrissat. 2005. Evolutionary and mechanistic relationships between glycosidases acting on α- and β-bonds. Carbohydr. Res. 340:2728-2734. [DOI] [PubMed] [Google Scholar]
- 31.Sunna, A., J. Puls, and G. Antranikian. 1997. Characterization of the xylanolytic enzyme system of the extreme thermophilic anaerobic bacteria Thermotoga maritima, T. neapolitana, and T. thermarum. Comp. Biochem. Physiol. A Physiol. 118:453-461. [Google Scholar]
- 32.Wiegel, J. 1998. Anaerobic alkalithermophiles, a novel group of extremophiles. Extremophiles 2:257-267. [DOI] [PubMed] [Google Scholar]
- 33.Wu, H., J. Pei, G. Wu, and W. Shao. 2008. Overexpression of GH10 endoxylanase XynB from T. maritima in E. coli by a novel vector with potential for industrial application. Enzyme Microb. Technol. 42:230-234. [Google Scholar]
- 34.Wu, H., J. Pei, Y. Jiang, X. Song, and W. Shao. 14 February 2010. pHsh vectors, a novel expression system of Escherichia coli for the large-scale production of recombinant enzymes. Biotechnol. Lett., in press. doi: 10.1007/s10529-010-0223-y. [DOI] [PubMed]
- 35.Xue, Y. M., and W. L. Shao. 2004. Expression and characterization of a thermostable β-xylosidase from hyperthermophile Thermotoga maritima. Biotechnol. Lett. 26:1511-1515. [DOI] [PubMed] [Google Scholar]
- 36.Yan, Q. J., et al. 2008. A xylose-tolerant β-xylosidase from Paecilomyces thermophila: characterization and its co-action with the endogenous xylanase. Bioresour. Technol. 99:5402-5410. [DOI] [PubMed] [Google Scholar]
- 37.Zanoelo, F. F., M. L. T. M. Polizeli, H. F. Terenzi, and J. A. Jorge. 2004. Purification and biochemical properties of a thermostable xylose-tolerant β-d-xylosidase from Scytalidium thermophilum. J. Ind. Microbiol. Biotechnol. 31:170-176. [DOI] [PubMed] [Google Scholar]
- 38.Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]