

Abstract
The identification of transporters of the HIV integrase inhibitor raltegravir could be a factor in an understanding of the pharmacokinetic-pharmacodynamic relationship and reported drug interactions of raltegravir. Here we determined whether raltegravir was a substrate for ABCB1 or the influx transporters SLCO1A2, SLCO1B1, SLCO1B3, SLC22A1, SLC22A6, SLC10A1, SLC15A1, and SLC15A2. Raltegravir transport by ABCB1 was studied with CEM, CEMVBL100, and Caco-2 cells. Transport by uptake transporters was assessed by using a Xenopus laevis oocyte expression system, peripheral blood mononuclear cells, and primary renal cells. The kinetics of raltegravir transport and competition between raltegravir and tenofovir were also investigated using SLC22A6-expressing oocytes. Raltegravir was confirmed to be an ABCB1 substrate in CEM, CEMVBL100, and Caco-2 cells. Raltegravir was also transported by SLC22A6 and SLC15A1 in oocyte expression systems but not by other transporters studied. The Km and Vmax for SLC22A6 transport were 150 μM and 36 pmol/oocyte/h, respectively. Tenofovir and raltegravir competed for SLC22A6 transport in a concentration-dependent manner. Raltegravir inhibited 1 μM tenofovir with a 50% inhibitory concentration (IC50) of 14.0 μM, and tenofovir inhibited 1 μM raltegravir with an IC50 of 27.3 μM. Raltegravir concentrations were not altered by transporter inhibitors in peripheral blood mononuclear cells or primary renal cells. Raltegravir is a substrate for SLC22A6 and SLC15A1 in the oocyte expression system. However, transport was limited compared to endogenous controls, and these transporters are unlikely to have a great impact on raltegravir pharmacokinetics.
HIV infection and AIDS continue to be a major cause of worldwide mortality in the 21st century. A UNAIDS/WHO report in 2009 estimated that 33.4 million people worldwide were infected with HIV in 2008, with AIDS-related deaths numbering 2 million. Recent attempts to develop a vaccine for HIV have been largely unsuccessful (18). This, combined with increasing drug resistance, has emphasized the need to develop new drugs with unique mechanisms of action.
Raltegravir represents a new class of antiretroviral treatment (8), targeting the HIV-1 integrase enzyme by binding to the active site and preventing viral DNA insertion into the host genome (11). Recent trials have shown raltegravir to have a sustained antiretroviral effect and good tolerability in treatment-experienced HIV-1 patients (33). The primary route of raltegravir metabolism is glucuronidation via UGT1A1, and raltegravir is not a substrate or an inhibitor of the major cytochrome P450 enzymes (19, 24). However, the involvement of human drug transporters in raltegravir absorption, disposition, metabolism, and excretion (ADME) has not been fully investigated. Raltegravir has been described as being an ABCB1 substrate (25), but there are no data yet in the public domain. Raltegravir has shown higher concentrations (1.7-fold) in semen (4) and lower concentrations (0.04- to 0.39-fold) in cerebrospinal fluid (7, 41) than in plasma, which may be facilitated by drug transporters present at membrane barriers.
There are important reasons why raltegravir should be screened for potential transport by known drug transporters. First, by regulating intracellular permeation, drug transporters could be an important factor in an understanding of the lack of a clear pharmacokinetic-pharmacodynamic (PK-PD) relationship of raltegravir, i.e., the similar virological response observed for patients given a wide range of raltegravir doses (29). Second, a knowledge of the mechanisms that control raltegravir disposition may help rationalize or even anticipate drug-drug interactions in the clinic. Since raltegravir represents the first member of a new drug class, possessing a unique chemical structure containing a diketo acid derivative (34), class-specific trends in drug transport may be evident, such as those reported previously for protease inhibitors with ABCB1 (26, 37) or nucleoside reverse transcriptase inhibitors with organic anion and cation transporters (35, 36). Finally, knowledge of which transporters are involved in raltegravir ADME will identify candidate genes for future pharmacogenetic studies.
There have been a number of studies undertaken to evaluate the pharmacokinetic interactions between raltegravir and coadministered drugs. Most studies have shown raltegravir metabolism and disposition to be largely unaffected. For example, etravirine (1), maraviroc (3), darunavir (2), and rifabutin (6) had no or a relatively modest effect on raltegravir plasma concentrations. In addition, despite ritonavir being an inducer of both ABCB1 (9) and UGT1A1 (12), the drug caused only a minimal reduction in the raltegravir plasma concentration (22). Similarly, tipranavir combined with ritonavir had little impact on raltegravir pharmacokinetics (13). However, there are interactions between raltegravir and coadministered drugs that have a more marked effect on raltegravir disposition.
Atazanavir is an inhibitor of UGT1A1 (42), and the coadministration of atazanavir (400 mg once a day [QD]) with raltegravir (100 mg single dose) resulted in increased raltegravir plasma area under the concentration-time curve (AUC), Cmax, and Cmin values of 72%, 53%, and 95%, respectively (20). This interaction was also confirmed in a separate study (43). Efavirenz and rifampin are inducers of UGT1A1 expression (14, 40) and have been shown to decrease raltegravir plasma exposure (22, 39). Other interactions were reported previously for omeprazole (21) and fosamprenavir (28).
One intriguing interaction is that of tenofovir, causing a moderate increase in the raltegravir AUC and Cmax by 49% and 64%, respectively (38). Although the increase in the raltegravir plasma concentration is unlikely to have any important clinical significance (i.e., no increase in toxicity), the mechanism of the interaction is currently unexplained. Interestingly, tenofovir is an anionic compound when charged and is therefore a substrate of the organic anion uptake transporters SLC22A6 and SLC22A8 (36).
The aim of this study was to confirm the transport of raltegravir by ABCB1 (phosphoglycoprotein) and to investigate potential transport by major human drug influx transporters (25), with the exception of CNTs (concentrative nucleoside transporters) and ENTs (equilibrative nucleoside transporters), since these are specific to nucleotides. The influx transporters characterized were SLCO1A2 (OATPA), SLCO1B1 (OATPC), SLCO1B3 (OATP8), SLC22A1 (OCT1), SLC22A6 (OAT1), SLC10A1 (NTCP), SLC15A1 (PEPT1), and SLC15A2 (PEPT2). Since tenofovir is a substrate for SLC22A6, the putative role for this transporter in the interaction between raltegravir and tenofovir was also investigated.
MATERIALS AND METHODS
Chemical reagents used.
CellFix was obtained from Becton Dickinson (Oxford, United Kingdom). UIC2 (anti-ABCB1) antibody was obtained from Immunotech (Marseilles, France). The IgG2a negative control and goat anti-mouse IgG2a-RPE (R-phycoerythrin) were obtained from Serotech Ltd. (Oxford, United Kingdom). CEM and CEMVBL100 cells were donated by Ross Davey, Bill Walsh Cancer Research Laboratories (St. Leonards, Australia). Caco-2 cells were purchased from the European Collection of Cell Cultures (Salisbury, United Kingdom). Primary renal proximal tubule epithelial cells, renal cell basal medium, and renal cell growth kit components were purchased from the American Type Culture Collection. [3H]raltegravir (specific activity = 32.85 Ci/mmol) and nonradiolabeled raltegravir sodium salt were gifts from Merck. [3H]digoxin (specific activity = 40 Ci/mmol) was purchased from Perkin-Elmer. [3H]saquinavir (specific activity = 0.2 Ci/mmol) and [3H]tenofovir (specific activity = 3.4 Ci/mmol) were purchased from Moravek. [3H]estrone-3-sulfate (specific activity = 50 Ci/mmol), [14C]tetraethyl ammonium (specific activity = 55 mCi/mmol), [3H]taurocholic acid (specific activity = 10 Ci/mmol), [3H]glycyl sarcosine (specific activity = 60 Ci/mmol), and [3H]aminohippuric acid (specific activity = 5 Ci/mmol) were purchased from American Radiolabeled Chemicals. Tariquidar was purchased from Xenova (Sloane, United Kingdom). Nonradiolabeled tenofovir was a gift from Gilead. Image clones of SLC22A6 and SLC22A8 were purchased from Geneservice. Ficoll-Paque Plus was purchased from GE Healthcare (Buckinghamshire, United Kingdom). All other reagents were obtained from Sigma (Poole, United Kingdom).
Cell culture.
CEM and CEMVBL100 cells were maintained in cell culture medium (RPMI 1640 medium, 10% [wt/vol] fetal calf serum [FCS]) prior to the experiment (37°C in 5% CO2). CEM cells are a wild-type T-lymphoblastoid cell line. CEMVBL100 cells are CEM cells that have greatly increased ABCB1 expression (selected using vinblastine up to a concentration of 100 ng/ml). Caco-2 cells were maintained in cell culture by passaging at 70% confluence using cell culture medium (Dulbecco's modified Eagle's medium [DMEM], 15% [wt/vol] FCS) prior to the experiment (37°C in 5% CO2). Primary renal proximal tubule epithelial cells were maintained in cell culture by passage at 95% confluence using renal epithelial cell basal medium with essential components (0.5% [wt/vol] FCS, 10 nM triiodothyronine, 10 ng/ml recombinant human epidermal growth factor [rhEGF], 100 ng/ml hydrocortisone hemisuccinate, 5 μg/ml rh insulin, 1 μM epinephrine, 5 μg/ml transferrin, 2.4 mM l-alanyl-l-glutamine) prior to the experiment (37°C in 5% CO2).
Cytotoxicity testing.
CEM and Caco-2 cell lines (100 μl; 2 × 105 cells/ml) were incubated in a 96-well Nunc flat-bottom plate (37°C in 5% CO2 for 120 h) with 0, 0.001, 0.01, 0.1, 1, 10, and 100 μM raltegravir. To validate the study, the cytotoxic control compounds epirubicin and vinblastine were incubated (0.1, 1, 10, and 100 μM) with CEM and Caco-2 cells, respectively. Following incubation, the plates were centrifuged (2,000 rpm for 5 min), and the supernatant was discarded and replaced with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (1 mg/ml MTT, 100 μl Hanks balanced salt solution [HBSS]). A standard MTT assay was performed, the mean absorption was calculated for each raltegravir concentration (n = 6), and results were expressed as the percent absorbance compared to the vehicle control (30).
Confirmation of ABCB1 expression in CEM and CEMVBL100 cells by flow cytometry.
The expression of ABCB1 in CEM and CEMVBL100 cells was determined by using a Coulter Epics XL-MCL flow cytometer as previously described (27). Results are given as relative fluorescence units (mean fluorescence minus that of the isotype control; n = 3) ± standard deviations (SD).
Cellular accumulation of raltegravir in CEM and CEMVBL100 cells.
CEM and CEMVBL100 cells of a constant cell density (1 ml; 2.5 × 106 cells/ml) were incubated (37°C in 5% CO2) for 30 min in RPMI 1640 medium (10% [wt/vol] FCS) containing [3H]raltegravir (1 μM; 0.2 μCi/ml) or the control ABCB1 substrate [3H]saquanavir (1 μM; 0.2 μCi/ml). A separate incubation was undertaken where the cells were preincubated prior to the substrate addition with RPMI 1640 medium (10% [wt/vol] FCS) containing the potent noncompetitive ABCB1 inhibitor tariquidar (300 nM for 30 min), which was also included during the 30 min of substrate incubation. Following incubation, the cells were centrifuged (2,000 rpm at 1°C for 1 min), and 100 μl supernatant aliquots were taken and added to scintillation vials in order to calculate extracellular drug concentrations. The remaining supernatant was discarded, and the cells were washed with ice-cold HBSS and centrifuged (2,000 rpm at 1°C for 1 min). This HBSS wash was repeated a total of 3 times, after which the HBSS was discarded and 100 μl tap water was added to lyse the cells. The incubations were vortexed for 5 min, and 100 μl samples were added to scintillation vials. Four milliliters of scintillation fluid was added to all the scintillation vials, which were then loaded into a liquid scintillation analyzer (Tri-Carb). Using supernatant and intracellular radioactivity readings, cellular accumulation ratios (CARs) (ratio of drug in the cell pellet compared with drug in the supernatant, assuming a 1 pl volume per cell) were calculated for raltegravir and saquinavir in each cell line as described previously (23).
Bidirectional transport of raltegravir using Caco-2 cell monolayers.
Caco-2 monolayer experiments were performed as previously described (17), with slight modifications. When confluent, Caco-2 cells (passage 30) were seeded onto polycarbonate membrane transwells at a density of 5 × 105 cells/cm2. The medium was replaced every 2 days, and plates were used in the experiments after 21 days. Monolayer integrity was checked by using a MillicellERS instrument (Millipore) to determine the transepithelial electrical resistance (TEER) across the monolayer. A TEER of >600 was deemed acceptable. On the day of the experiment, the TEER was assessed, and the medium in each plate was replaced with warm transport buffer (HBSS containing 25 mM HEPES and 0.1% [wt/vol] bovine serum albumin [pH 7.4]) and allowed to equilibrate (37°C for 30 min). For inhibition studies this transport buffer contained tariquidar (300 nM). The transport buffer in the apical (for apical-to-basolateral transport) and basolateral (for basolateral-to-apical transport) sides was replaced with transport buffer containing either the test substrate [3H]raltegravir or the control ABCB1 substrate [3H]digoxin (1 μM; 0.33 μCi/ml) with or without 300 nM tariquidar. Samples were taken from the receiver compartment at 30, 60, 90, 120, and 180 min and analyzed by using a liquid scintillation counter (Tri-Carb). Results were used to determine the apparent permeability (Papp) (cm/s) for each direction and the efflux ratio (ratio of basolateral-to-apical Papp to the apical-to-basolateral Papp). The Papp was calculated by using the following equation, as described previously (10):
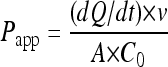 |
where dQ/dt is the change in the drug concentration in the receiver chamber over time (nM/s), v is the volume in the receiver compartment (ml), A is the total surface area of the transwell membrane (cm2), C0 is the initial drug concentration in the donor compartment (nM), and Papp is the apparent permeability (cm/s).
Production of uptake transporter cRNA for Xenopus laevis oocyte injection.
SLCO1A2, SLCO1B1, and SLCO1B3 were cloned from cDNA extracted from Huh-7D12 and A549 cells as described previously (15). IMAGE clones were linearized and used as a template in cRNA production by using an mMessage mMachine RNA transcription kit (Ambion) according to the manufacturer's protocol. SLC10A1, SLC15A1, and SLC15A2 cRNAs were provided by Becton Dickinson (Oxford, United Kingdom).
X. laevis oocyte isolation, collagenase treatment, and microinjection.
Oocytes were harvested from sacrificed adult female X. laevis frogs and treated with modified Barth's solution minus calcium (88 mM NaCl, 1 mM KCl, 15 mM HEPES, 100 U penicillin, 100 μg streptomycin [pH 7.4]) containing collagenase (1 mg/ml at 22°C in a 60-rpm shaker for 1 h). Cells were washed and transferred into Barth's solution containing calcium (88 mM NaCl, 1 mM KCl, 15 mM HEPES, 0.3 mM CaCNO3·6H2O, 41 μM CaCl2·6H2O, 0.82 mM MgSO4·7H2O, 100 U penicillin, 100 μg streptomycin [pH 7.4]) and stored in a cold room at 8°C. Healthy cells were selected and injected with transporter cRNA (50 ng per oocyte; 1 ng/nl) or sterile water (50 nl) and maintained in Barth's solution containing calcium to allow transporter expression (5 days for SLCO1B3-injected oocytes and 3 days for all other conditions; 18°C). Barth's solution was replaced daily, and damaged oocytes were removed.
Drug accumulation in transporter RNA-injected X. laevis oocytes.
Drug accumulation studies using X. laevis oocytes were performed as described previously, with slight modifications (15). Unless otherwise stated, radiolabeled drug was incubated in Hanks balanced salt solution (pH 7.4) with ≥4 oocytes per condition in a 48-well Nunc flat-bottom plate (500 μl; 0.33 μCi/ml; 1 μM; room temperature in a 60-rpm shaker for 1 h). Radiolabeled positive-control drugs were tested alongside [3H]raltegravir to ensure successful transporter expression. Positive-control drugs used were [3H]estrone-3-sulfate for SLCO1A2, SLCO1B1, and SLCO1B3; [3H]aminohippuric acid for SLC22A6; [3H]taurocholic acid for SLC10A1; [3H]glycyl sarcosine for SLC15A1 and SLC15A2; and [14C]tetraethyl ammonium for SLC22A1. All incubations were terminated by transferring the oocytes into cell strainers and washing them in ice-cold HBSS to remove extracellular drug. Each oocyte was placed into a separate scintillation vial followed by treatment with 100 μl 10% SDS. After the disintegration of the oocytes by the SDS treatment, 4 ml scintillation fluid was added to all vials, which were then loaded into a liquid scintillation analyzer (Tri-Carb). Results are expressed as the amount of drug per oocyte (pmol/oocyte), assuming that each oocyte had a volume of 1 μl (15). Results were obtained using oocytes from ≥2 X. laevis frogs.
Determination of raltegravir and tenofovir Km and Vmax using SLC22A6-injected oocytes.
The time-dependent SLC22A6-mediated transport of [3H]raltegravir, [3H]tenofovir, and the control compound [3H]aminohippuric acid was investigated by incubating each drug with SLC22A6- and H2O-injected oocytes at a standard concentration of 1 μM for various lengths of time (1, 2, 5, 10, 15, 30, 60, 120, 180, or 240 min). From these results, an incubation time was chosen that gave a high accumulation rate in SLC22A6-injected oocytes compared to the H2O-injected controls and also allowed enough radiolabeled drug to enter the oocytes for it to be detectable. This time point was then used for kinetic experiments in which [3H]raltegravir and [3H]tenofovir were incubated with SLC22A6- and H2O-injected oocytes (1 h for both drugs) using a range of concentrations (0.1, 0.3, 1, 3, 10, 30, 100, 300, and 1000 μM for raltegravir and 0.3, 1, 3, 10, 30, 100, 200, and 300 μM for tenofovir). Incubations were terminated as described above. The transport of drug by SLC22A6 was determined by subtracting the drug accumulation in H2O-injected oocytes from the drug accumulation in SLC22A6-injected oocytes. The Km and Vmax were calculated by plotting the initial rate of drug transport by SLC22A6 (pmol/oocyte/h) against the drug incubation concentration (μM). The intrinsic clearance (CLint) was calculated by dividing the Vmax by the Km.
Determination of competition between raltegravir and tenofovir for SLC22A6 transport.
[3H]raltegravir (0.33 μCi/ml; 1 μM) was incubated for 1 h with SLC22A6-injected oocytes with various concentrations of nonradiolabeled tenofovir (1, 3, 10, 30, 100, and 300 μM), and the effect on [3H]raltegravir uptake was determined. [3H]raltegravir (0.33 μCi/ml; 1 μM) was also incubated with H2O-injected cells with and without 300 μM tenofovir in order to confirm the role of SLC22A6. This experiment was repeated using [3H]tenofovir as the substrate and nonradiolabeled raltegravir as the competitor drug using the same concentration range and was terminated as described above. Fifty percent inhibitory concentrations (IC50) were generated for both drugs by using Prism 3.0.
Isolation of peripheral blood mononuclear cells from human blood.
Venous blood samples (60 ml) were obtained from healthy volunteers via venipuncture. Blood was layered onto Ficoll-Paque Plus, and peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation according to the manufacturer's protocol. Cells were washed with ice-cold HBSS and centrifuged (800 × g at 1°C for 5 min). This HBSS wash was repeated a total of 3 times, after which the HBSS was discarded and cells were resuspended in RPMI 1640 medium (10% [wt/vol] FCS) for use in experiments.
Cellular accumulation of raltegravir in peripheral blood mononuclear cells.
The accumulation of raltegravir in peripheral blood mononuclear cells was determined by using the same method as that used with CEM and CEMVBL cells, with slight modifications. Briefly, cells of a constant cell density (1 ml; 5 × 106 cells/ml) were incubated (37°C in 5% CO2) for 30 min in RPMI 1640 medium (10% [wt/vol] FCS) containing [3H]raltegravir (1 μM; 0.2 μCi/ml). Separate incubations were undertaken where cells were preincubated prior to substrate addition with RPMI 1640 medium (10% [wt/vol] FCS) containing either the noncompetitive ABCB1 inhibitor tariquidar (300 nM for 30 min), the competitive SLC22A6 inhibitor probenecid (1 mM for 30 min), or the competitive SLC15A1 inhibitor glycyl sarcosine (1 mM for 30 min), which were also included during the 30 min of substrate incubation. Cells were washed and treated for analysis as described above for CEM and CEMVBL cells.
Cellular accumulation of raltegravir in primary renal proximal tubule epithelial cells.
When confluent, renal cells (passage 4) were seeded onto polyester membrane transwells at a density of 5 × 105 cells/cm2. The medium was replaced daily, and plates were used for experiments after 5 days. On the day of the experiment, medium was replaced with buffered HBSS (25 mM HEPES [pH 7.4]) containing either [3H]raltegravir (1 μM; 0.6 μCi/ml), [3H]tenofovir (1 μM; 0.6 μCi/ml), or the control SLC22A6 substrate [3H]aminohippuric acid (1 μM; 0.6 μCi/ml) and incubated (3 h at 37°C in 5% CO2). A separate incubation was undertaken where the cells were preincubated prior to substrate addition with buffered HBSS (25 mM HEPES [pH 7.4]) containing the SLC22A6 inhibitor probenecid (1 mM for 30 min), which was also included during the 3 h of substrate incubation. We also investigated the competition between raltegravir and tenofovir by incubating [3H]raltegravir (1 μM; 0.6 μCi/ml) with 100 μM tenofovir and [3H]tenofovir (1 μM; 0.6 μCi/ml) with 100 μM raltegravir (3 h at 37°C in 5% CO2). Once incubation was complete, an extracellular sample was taken, and incubations were terminated by washing each well with cold HBSS (4°C; 3 ml) three times to remove excess drug. Cells were lysed with 0.5 ml tap water, and contents were analyzed by liquid scintillation as described above for the CEM experiments.
Determination of transporter mRNA expression in primary renal proximal tubule cells and whole kidney.
mRNA from primary renal proximal tubule cells were isolated by using Tri reagent according to the manufacturer's protocol. mRNA was then reverse transcribed by using the TaqMan reverse transcription kit according to the manufacturer's protocol. Real-time PCR using TaqMan array plates was combined with cDNA (40 ng) and used to quantify mRNA expression by standard methodologies. This process was repeated, and mRNA quantification was obtained for whole kidneys purchased from Ambion (United Kingdom), which was created from a pool of three individuals. Transporters were quantified by using the ΔΔCT method and included SLC22A6, SLC22A1, SLC22A2, SLC22A3, ABCC1, ABCC2, ABCC3, ABCC4, and ABCC10. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the housekeeping gene.
Statistical analysis.
Data were analyzed by using SPSS 13.0 for Windows. All data were tested for normality by using the Shapiro-Wilk test. An independent t test was used to determine the significance of the ABCB1 flow cytometry data, Caco-2 transwell data, PBMC accumulation data, and renal cell accumulation data. The Mann-Whitney U test was used for all other data. A two-tailed P value of <0.05 was accepted as being statistically significant.
RESULTS
Toxicity of raltegravir in CEM and Caco-2 cell lines.
Raltegravir was not cytotoxic in CEM or Caco-2 cell lines at the tested concentrations. Cell viability (percent mean viability compared to the drug-free control ± SD; n = 6) was unaffected by concentrations of up to 100 μM in the Caco-2 (104.9% ± 12.7%) and CEM (113.9% ± 13.0%) cell lines. A concentration of 1 μM raltegravir was used in subsequent experiments with these cells to minimize the risk of transporter saturation, as previously recommended (17).
ABCB1 expression levels in CEM and CEMVBL100 cell lines.
The expression of ABCB1 in CEM and CEMVBL100 cell lines was determined by using flow cytometry. CEMVBL100 cells had a significantly higher level of ABCB1 cell surface expression (relative fluorescence = 246.7 ± 9.3; P < 0.001) than in CEM cells (relative fluorescence = 1.5 ± 0.5).
Effect of the ABCB1 inhibitor tariquidar on the accumulation of raltegravir and saquinavir using CEM and CEMVBL100 cell lines.
The cellular accumulation of [3H]raltegravir was determined with CEM and ABCB1-overexpressing CEMVBL100 cells, and the effect of the ABCB1 inhibitor tariquidar on this accumulation was investigated (Fig. 1 A). The level of [3H]raltegravir accumulation was lower in CEMVBL100 cells (CAR = 1.4 ± 0.2) than in CEM cells (CAR = 2.1 ± 0.2; P = 0.02). This difference was reversed when CEMVBL100 cells were treated with tariquidar (CAR = 2.0 ± 0.4). The control ABCB1 substrate [3H]saquinavir had a lower level of accumulation in CEMVBL100 cells (CAR = 19.0 ± 5.6) than in CEM cells (CAR = 37.5 ± 2.1; P = 0.02) (Fig. 1B). This difference was also reversed when CEMVBL100 cells were treated with tariquidar (CAR = 37.8 ± 8.5).
FIG. 1.

(A) [3H]raltegravir (1 μM) accumulation in CEM, CEMVBL100, and CEMVBL100 cells treated with 300 nM tariquidar. (B) [3H]saquinavir (1 μM) accumulation in CEM, CEMVBL100, and CEMVBL100 cells treated with 300 nM tariquidar. Data in A and B are expressed as mean CARs (n = 4 biological replicates; n = 4 experimental replicates per biological replicate) ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Apparent permeability of [3H]raltegravir and [3H]digoxin in the A-to-B (apical-to-basolateral) and B-to-A (basolateral-to-apical) directions across the Caco-2 transwell membrane, with and without the presence of 300 nM tariquidar. Data are expressed as mean apparent permeabilities (×10−6 cm/s; n = 3 experimental replicates) ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Effect of the ABCB1 inhibitor tariquidar on the bidirectional transport of raltegravir and digoxin using a Caco-2 monolayer.
The Papp values obtained for [3H]raltegravir and [3H]digoxin with and without tariquidar are given in Fig. 1C. All Papp and efflux ratio calculations were made by using the samples taken after 120 min of incubation as sink conditions were maintained. [3H]raltegravir showed significantly higher transport in the B→A direction (Papp = 13.4 × 10−6 ± 2.1 × 10−6) compared to the A→B direction (Papp = 7.3 × 10−6 ± 2.2 × 10−6; P = 0.02). The efflux ratio (B→A/A→B) of [3H]raltegravir at 120 min was 1.9. The presence of tariquidar reduced the efflux ratio of [3H]raltegravir to 1.3 (P = 0.30). The control compound [3H]digoxin showed significantly higher levels of transport in the B→A direction (Papp = 12.9 × 10−6 ± 0.6 × 10−6) compared to the A→B direction (Papp = 2.1 × 10−6 ± 0.3 × 10−6; P < 0.001). The efflux ratio of [3H]digoxin at 120 min was 6.3. The presence of tariquidar reduced the efflux ratio of [3H]digoxin to 0.9 (P = 0.58).
Accumulation of raltegravir in oocytes by uptake transporters.
[3H]raltegravir transport by the investigated uptake transporters is given in Table 1. The level of [3H]raltegravir accumulation was significantly higher in SLC22A6-injected oocytes (0.44 ± 0.12 pmol/oocyte; n = 18) than in H2O-injected oocytes (0.20 ± 0.05 pmol/oocyte; n = 19; P < 0.001). [3H]raltegravir accumulation was also significantly higher in SLC15A1-injected oocytes (0.26 ± 0.12 pmol/oocyte; n = 19) than in H2O-injected oocytes (0.17 ± 0.03 pmol/oocyte; n = 19; P = 0.003). The positive-control compounds [3H]estrone-3-sulfate, [3H]aminohippuric acid, [3H]-taurocholic acid, [3H]glycyl sarcosine, and [14C]tetraethyl ammonium all showed significant increases in levels of accumulation in transporter RNA-injected oocytes compared to H2O-injected oocytes.
TABLE 1.
Accumulation of 1 μM raltegravir and various positive-control compounds in oocytesa
| Transporter | Drug | Mean drug concn (pmol/oocyte) ± SD |
RNA/H2O ratio (P value) | |
|---|---|---|---|---|
| RNA-injected oocytes | H2O-injected oocytes | |||
| SLCO1A2 | RAL | 0.39 ± 0.14 | 0.35 ± 0.07 | 1.11 (0.09) |
| E3S | 1.48 ± 0.63 | 0.27 ± 0.09 | 5.48 (<0.01) | |
| SLCO1B1 | RAL | 0.41 ± 0.05 | 0.38 ± 0.05 | 1.08 (0.09) |
| E3S | 2.68 ± 1.55 | 0.34 ± 0.17 | 7.97 (<0.01) | |
| SLCO1B3 | RAL | 0.63 ± 0.19 | 0.65 ± 0.13 | 0.97 (0.09) |
| E3S | 0.56 ± 0.23 | 0.21 ± 0.03 | 2.60 (<0.01) | |
| SLC22A6 | RAL | 0.44 ± 0.12 | 0.20 ± 0.03 | 2.22 (<0.01) |
| AHA | 9.39 ± 2.64 | 0.17 ± 0.04 | 56.9 (<0.01) | |
| SLC10A1 | RAL | 0.20 ± 0.03 | 0.20 ± 0.03 | 1.03 (0.57) |
| TCA | 0.28 ± 0.10 | 0.08 ± 0.05 | 3.51 (<0.01) | |
| SLC15A1 | RAL | 0.26 ± 0.12 | 0.17 ± 0.03 | 1.52 (<0.01) |
| GLY | 0.45 ± 0.13 | 0.11 ± 0.05 | 4.22 (<0.01) | |
| SLC15A2 | RAL | 0.19 ± 0.04 | 0.17 ± 0.03 | 1.08 (0.16) |
| GLY | 0.29 ± 0.19 | 0.11 ± 0.05 | 2.69 (<0.01) | |
| SLC22A1 | RAL | 0.21 ± 0.03 | 0.17 ± 0.04 | 1.21 (0.06) |
| TEA | 0.34 ± 0.06 | 0.17 ± 0.04 | 1.99 (<0.01) | |
Results are expressed as the mean drug concentrations per oocyte (pmol/oocyte) (n ≥ 2 biological replicates; n ≥ 4 experimental replicates per biological replicate) ± standard deviations. Also shown are the ratios of drug accumulation between transporter RNA-injected and H2O-injected oocytes. RAL, raltegravir; E3S, estrone-3-sulfate; AHA, amminohippuric acid; TCA, taurocholic acid; GLY, glycyl sarcosine; TEA, tetraethyl ammonium.
Determination of the time-dependent accumulation of raltegravir, tenofovir, and aminohippuric acid in SLC22A6-injected oocytes.
All drugs tested had a greater accumulation rate in SLC22A6-injected oocytes than in H2O-injected oocytes (Fig. 2 A, B, and C). [3H]raltegravir concentrations continued to increase in SLC22A6-injected oocytes throughout the 4-h incubation period, whereas saturation was reached in H2O-injected oocytes after 2 h. Both [3H]tenofovir and [3H]aminohippuric acid showed virtually no accumulation in H2O-injected oocytes.
FIG. 2.

(A) SLC22A6- and H2O-injected oocyte uptake of [3H]raltegravir over a 4-h incubation period. (B) SLC22A6- and H2O-injected oocyte uptake of [3H]tenofovir over a 4-h incubation period. (C) SLC22A6- and H2O-injected oocyte uptake of [3H]aminohippuric acid over a 4-h incubation period. Data in A, B, and C are expressed as mean drug accumulations (pmol/oocyte) (n = 5 experimental replicates from one biological replicate) ± standard errors (SE). (D) Concentration dependency of the uptake of [3H]raltegravir by SLC22A6. (E) Concentration dependency of the uptake of [3H]tenofovir by SLC22A6. Data in D and E are expressed as mean rates of [3H]tenofovir transport (pmol/oocyte/h) (n = 4 experimental replicates from one biological replicate) ± SE.
Determination of raltegravir and tenofovir Km and Vmax values using SLC22A6-injected oocytes.
An incubation time of 1 h was chosen for subsequent kinetic studies. [3H]raltegravir and [3H]tenofovir kinetics were determined for SLC22A6 in the oocyte expression system (Fig. 2D and E). The raltegravir Km and Vmax were calculated to be 150 μM and 36 pmol/oocyte/h, respectively. The raltegravir CLint (Vmax/Km) was calculated to be 0.2 μl/oocyte/h. The tenofovir Km and Vmax were calculated to be 25 μM and 129 pmol/oocyte/h, respectively. The tenofovir CLint was calculated to be 5.2 μl/oocyte/h.
Competition between raltegravir and tenofovir for SLC22A6 transport.
When incubated at 1 μM for 1 h, [3H]raltegravir showed a significantly higher level of accumulation in SLC22A6-injected oocytes than in H2O-injected oocytes (1.73 ± 0.46 pmol/oocyte versus 0.54 ± 0.06 pmol/oocyte; n = 5; P = 0.014) (Fig. 3 A). The coincubation of [3H]raltegravir with 1, 3, 10, 30, 100, and 300 μM tenofovir resulted in levels of [3H]raltegravir accumulation in SLC22A6-injected oocytes of 1.78 ± 0.41 pmol/oocyte, 1.63 ± 0.62 pmol/oocyte, 1.48 ± 0.58 pmol/oocyte, 1.28 ± 0.27 pmol/oocyte, 0.96 ± 0.28 pmol/oocyte, and 0.83 ± 0.31 pmol/oocyte, respectively (Fig. 3C). There was a statistically significant decrease (P < 0.05) in the level of [3H]raltegravir accumulation when concentrations of 100 μM tenofovir and higher were added to the incubation mixture.
FIG. 3.

(A) Accumulation of 1 μM [3H]raltegravir in SLC22A6- and H2O-injected oocytes with and without the addition of 300 μM tenofovir. (B) Accumulation of 1 μM [3H]tenofovir in SLC22A6- and H2O-injected oocytes with and without the addition of 300 μM raltegravir. Data in A and B are expressed as mean drug concentrations per oocyte (pmol/oocyte) (n = 5 experimental replicates from one biological replicate) ± SE. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Determination of IC50 for inhibition of 1 μM [3H]raltegravir SLC22A6 transport by tenofovir. Data are expressed as mean [3H]raltegravir oocyte concentrations (pmol/oocyte) (n = 5 experimental replicates from one biological replicate) ± SE. (D) Determination of IC50 for inhibition of 1 μM [3H]tenofovir SLC22A6 transport by raltegravir. Data are expressed as mean [3H]tenofovir oocyte concentrations (pmol/oocyte) (n = 5 experimental replicates from one biological replicate) ± SE.
Similar results were seen when [3H]tenofovir accumulation was investigated in the presence of various concentrations of raltegravir. When incubated at 1 μM for 1 h, [3H]tenofovir showed a significantly higher level of accumulation in SLC22A6-injected oocytes than in H2O-injected oocytes (2.11 ± 0.37 pmol/oocyte versus 0.03 ± 0.01 pmol/oocyte; n = 5; P < 0.009) (Fig. 3B). The coincubation of [3H]tenofovir with 1, 3, 10, 30, 100, and 300 μM raltegravir resulted in levels of [3H]tenofovir accumulation in SLC22A6-injected oocytes of 2.11 ± 0.14 pmol/oocyte, 2.07 ± 0.38 pmol/oocyte, 1.47 ± 0.43 pmol/oocyte, 0.78 ± 0.50 pmol/oocyte, 0.49 ± 0.06 pmol/oocyte, and 0.28 ± 0.08 pmol/oocyte, respectively (Fig. 3D). There was a statistically significant decrease (P < 0.05) in levels of [3H]tenofovir accumulation when concentrations of 10 μM raltegravir and higher were added to the incubation mixtures.
Effect of transporter inhibitors on the accumulation of raltegravir in peripheral blood mononuclear cells.
The cellular accumulation of [3H]raltegravir in peripheral blood mononuclear cells was determined (CAR = 3.02 ± 0.67) (Fig. 4 A). Cellular accumulation was not significantly altered by coincubation with tariquidar (CAR = 3.78 ± 1.56; P = 0.77), probenecid (CAR = 3.26 ± 0.98; P = 0.77), or glycyl sarcosine (CAR = 3.23 ± 0.83; P = 0.56).
FIG. 4.

(A) [3H]raltegravir (1 μM) accumulation in peripheral blood mononuclear cells alone or treated with 300 nM tariquidar, 1 mM probenecid, or 1 mM glycyl sarcosine. (B) [3H]raltegravir (1 μM) accumulation in primary renal proximal tubule cells alone or treated with 1 mM probenecid or 100 μM tenofovir. (C) [3H]tenofovir (1 μM) accumulation in primary renal proximal tubule cells alone or treated with 1 mM probenecid or 100 μM raltegravir. (D) [3H]aminohippuric acid (1 μM) accumulation in primary renal proximal tubule cells alone or treated with 1 mM probenecid. Data in A, B, C, and D are expressed as mean CARs (n = 3 experimental replicates) ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) Relative abundance of transporter RNA in cultured renal proximal tubule cells compared to transporter RNA in whole kidney (log percent) (n = 1 experimental replicate).
Interactions in primary renal proximal tubule epithelial cells.
The levels of cellular accumulation of [3H]raltegravir (CAR = 2.01 ± 0.20) (Fig. 4B), [3H]tenofovir (CAR = 3.45 ± 0.76) (Fig. 4C), and [3H]aminohippuric acid (CAR = 0.50 ± 0.02) (Fig. 4D) in renal proximal tubule epithelial cells were determined. [3H]raltegravir cellular accumulation was not significantly altered by treatment with 1 mM probenecid (CAR = 2.19 ± 0.45; P = 0.56) or 100 μM tenofovir (CAR = 2.07 ± 0.08; P = 0.66). [3H]tenofovir showed a high level of cellular accumulation, which was not significantly altered by treatment with 1 mM probenecid (CAR = 2.59 ± 0.56; P = 0.19) or 100 μM raltegravir (CAR = 3.15 ± 0.74; P = 0.65). For [3H]aminohippuric acid there was a trend toward a lower level of cellular accumulation when incubated with 1 mM probenecid (CAR = 0.44 ± 0.03; P = 0.08).
Transporter expression in primary renal proximal tubule cells versus whole kidneys.
All transporters tested showed lower or undetectable levels of expression in primary renal proximal tubule cells compared to whole kidney (Fig. 4E). Importantly, SLC22A6 was undetectable in primary renal cells.
DISCUSSION
The results from CEM/CEMVBL accumulation and Caco-2 bidirectional transport experiments confirm that raltegravir is transported by ABCB1. The extent of raltegravir transport by ABCB1 was small compared to the transport of the positive controls saquinavir and digoxin. Indeed, FDA guidelines recommend that a drug should achieve an efflux ratio of at least 2 in Caco-2 cell monolayers and show greater than a 50% reduction in the efflux ratio when an ABCB1 inhibitor is used in order for ABCB1 transport to be considered relevant in vivo (16). In our Caco-2 experiment raltegravir achieved an efflux ratio of only 1.9 and a reduction of 32% when tariquidar was used to inhibit ABCB1. The low rate of raltegravir transport by ABCB1 may explain the absence of major drug interactions with known potent ABCB1 inhibitors. This is consistent with a previous report that the coadministration of low-dose ritonavir had no major effect on raltegravir pharmacokinetics, and no dose adjustment is required for patients (22).
Raltegravir showed significantly increased levels of accumulation in SLC15A1- and SLC22A6-injected oocytes compared to H2O-injected controls, but accumulation was not higher in oocytes expressing SLCO1A2, SLCO1B1, SLCO1B3, SLC15A2, and SLC10A1. In SLC22A6-injected oocytes, both raltegravir and tenofovir inhibited the accumulation of the other in a concentration-dependent manner (Fig. 3C and D). No competition was observed for H2O-injected oocytes, which supports the hypothesis that raltegravir and tenofovir are competing for SLC22A6 transport and are not having nonspecific effects on oocyte membrane permeability. IC50 values of 27.3 μM and 14.0 μM were determined for tenofovir and raltegravir, respectively. The IC50 obtained for raltegravir was much lower than the observed Km for SLC22A6 (IC50 of 14.0 μM versus a Km of 147 μM). These results suggest that raltegravir is a more efficient SLC22A6 inhibitor than it is a substrate. Previous studies indicated that SLC22A6 and SLC15A1 are absent from PBMCs (5), and so these transporters are unlikely to explain the unusual PK-PD relationship for raltegravir. Indeed, our studies of PBMCs with known inhibitors of ABCB1, SLC22A6, and SLC15A1 revealed no significant interaction with raltegravir.
Wenning et al. (38) previously studied the interaction of raltegravir (400 mg twice daily) and tenofovir (300 mg once daily). The study showed increased raltegravir AUC (49%) and Cmax (64%) values but no effect on the raltegravir Cmin and a decrease in the tenofovir AUC (10%), Cmax (23%), and Cmin (13%) (38). SLC22A6 is expressed predominantly in the proximal tubule of the kidney on the basolateral (blood-facing) surface, thereby facilitating the removal of drugs out of the blood and into the urine (31). Therefore, a possible mechanism of interaction is the inhibition of SLC22A6-mediated raltegravir transport at the kidney-proximal tubule by tenofovir, resulting in increased raltegravir plasma concentrations. In order to investigate interactions at the level of the kidney, we conducted a number of experiments with primary renal proximal cells. No interaction between tenofovir and raltegravir was observed for these cells, but neither was an interaction with the positive-control substrate and inhibitor. Subsequent analyses revealed the expression of SLC22A6 to be undetectable, unlike kidney tissue. Furthermore, all transporters that were assessed were at lower levels of expression than in kidney tissue, and the absence of a robust primary cell model for these studies is a limitation.
Since only a small percentage (around 30%) of raltegravir excreted via the kidney is in the parent form, with the remaining 70% being the glucuronide metabolite (24), it is important to determine if the raltegravir glucuronide is also transported by SLC22A6 and inhibited by tenofovir. It would also be interesting to investigate the transport and inhibitory potential of tenofovir diphosphate for SLC22A6 and whether this affects raltegravir transport to the same degree.
The Xenopus laevis oocyte expression system has several advantages when investigating drug transport. The large size and high level of protein production of oocytes provide robust and reliable data. Also, the level of expression of endogenous primary and secondary active xenobiotic transporters in oocytes is low (32). However, there are also disadvantages. The temperature must be maintained at 18°C during protein expression and at around room temperature during any accumulation experiments to avoid degradation, and this may impact transporter kinetics. Also, as in other models, the expression of the investigated transporters is superphysiological. Therefore, although they allow an investigation of low-affinity or high-permeability substrates, this means that caution should be taken when extrapolating the data to in vivo observations.
In summary, our studies have shown raltegravir to have minimal interactions with known drug transporters. Raltegravir is transported by ABCB1 in vitro, although the rate of transport is low and the potential for interactions is expected to be small. Raltegravir is a substrate for SLC22A6 and SLC15A1 in X. Laevis oocyte expression systems and competes with tenofovir for SLC22A6 transport. Polymorphisms in SLC22A6 have previously been described and now warrant study in the context of raltegravir.
Acknowledgments
This work was supported in part by a research grant from the Investigator-Initiated Studies Program of Merck Sharp & Dohme Corp. This work was funded by Merck & Co. Inc., (Whitehouse Station, NJ).
The opinions expressed in this paper are those of the authors and do not necessarily represent those of Merck Sharp & Dohme Corp.
Footnotes
Published ahead of print on 15 November 2010.
REFERENCES
- 1.Anderson, M. S., T. N. Kakuda, W. Hanley, J. Miller, J. T. Kost, R. Stoltz, L. A. Wenning, J. A. Stone, R. M. Hoetelmans, J. A. Wagner, and M. Iwamoto. 2008. Minimal pharmacokinetic interaction between the human immunodeficiency virus nonnucleoside reverse transcriptase inhibitor etravirine and the integrase inhibitor raltegravir in healthy subjects. Antimicrob. Agents Chemother. 52:4228-4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, M. S., V. Sekar, F. Tomaka, J. Mabalot, R. Mack, L. Lionti, S. Zajic, L. Wenning, C. Vanden Abeele, M. Zinny, N. M. Lunde, B. Jin, J. A. Wagner, and M. Iwamoto. 2008. Pharmacokinetic (PK) evaluation of darunavir/ritonavir (DRV/r) and raltegravir (RAL) in healthy subjects, abstr. A-962. Abstr. 48th Annu. Intersci. Conf. Antimicrob. Agents Chemother. (ICAAC)-Infect. Dis. Soc. Am. (IDSA) 46th Annu. Meet. American Society for Microbiology and Infectious Diseases Society of America, Washington, DC.
- 3.Andrews, E., P. Glue, J. Fang, P. Crownover, R. Tressler, and B. Damle. 2008. A pharmacokinetic study to evaluate an interaction between maraviroc and raltegravir in healthy adults, abstr. H-4055. Abstr. 48th Annu. Intersci. Conf. Antimicrob. Agents Chemother. (ICAAC)-Infect. Dis. Soc. Am. (IDSA) 46th Annu. Meet. American Society for Microbiology and Infectious Diseases Society of America, Washington, DC.
- 4.Barau, C., C. Delaugerre, J. Braun, N. de Castro, V. Furlan, I. Charreau, L. Gerard, C. Lascoux-Combe, J. M. Molina, and A. M. Taburet. 2010. High concentration of raltegravir in semen of HIV-infected men: results from a substudy of the EASIER-ANRS 138 trial. Antimicrob. Agents Chemother. 54:937-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bleasby, K., J. C. Castle, C. J. Roberts, C. Cheng, W. J. Bailey, J. F. Sina, A. V. Kulkarni, M. J. Hafey, R. Evers, J. M. Johnson, R. G. Ulrich, and J. G. Slatter. 2006. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: a resource for investigations into drug disposition. Xenobiotica 36:963-988. [DOI] [PubMed] [Google Scholar]
- 6.Brainard, L. M., A. S. Petry, L. Fang, C. Liu, S. A. Breidinger, E. P. DeNoia, J. A. Stone, J. A. Chodakewitz, L. A. Wenning, J. A. Wagner, and M. Iwamoto. 2009. Lack of a clinically important effect of rifabutin (RFB) on raltegravir (RAL) pharmacokinetics, abstr. A1-1296. Abstr. 49th Annu. Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC.
- 7.Calcagno, A., S. Bonora, R. Bertucci, A. Lucchini, A. D'Avolio, and G. Di Perri. 2010. Raltegravir penetration in the cerebrospinal fluid of HIV-positive patients. AIDS (London) 24:931-932. [DOI] [PubMed] [Google Scholar]
- 8.Chirch, L. M., S. Morrison, and R. T. Steigbigel. 2009. Treatment of HIV infection with raltegravir. Expert Opin. Pharmacother. 10:1203-1211. [DOI] [PubMed] [Google Scholar]
- 9.Dixit, V., N. Hariparsad, F. Li, P. Desai, K. E. Thummel, and J. D. Unadkat. 2007. Cytochrome P450 enzymes and transporters induced by anti-human immunodeficiency virus protease inhibitors in human hepatocytes: implications for predicting clinical drug interactions. Drug Metab. Dispos. 35:1853-1859. [DOI] [PubMed] [Google Scholar]
- 10.Elsby, R., D. D. Surry, V. N. Smith, and A. J. Gray. 2008. Validation and application of Caco-2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P-glycoprotein to support regulatory submissions. Xenobiotica 38:1140-1164. [DOI] [PubMed] [Google Scholar]
- 11.Espeseth, A. S., P. Felock, A. Wolfe, M. Witmer, J. Grobler, N. Anthony, M. Egbertson, J. Y. Melamed, S. Young, T. Hamill, J. L. Cole, and D. J. Hazuda. 2000. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc. Natl. Acad. Sci. U. S. A. 97:11244-11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foisy, M. M., E. M. Yakiwchuk, and C. A. Hughes. 2008. Induction effects of ritonavir: implications for drug interactions. Ann. Pharmacother. 42:1048-1059. [DOI] [PubMed] [Google Scholar]
- 13.Hanley, W. D., L. A. Wenning, A. Moreau, J. T. Kost, E. Mangin, T. Shamp, J. A. Stone, K. M. Gottesdiener, J. A. Wagner, and M. Iwamoto. 2009. Effect of tipranavir-ritonavir on pharmacokinetics of raltegravir. Antimicrob. Agents Chemother. 53:2752-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hariparsad, N., S. C. Nallani, R. S. Sane, D. J. Buckley, A. R. Buckley, and P. B. Desai. 2004. Induction of CYP3A4 by efavirenz in primary human hepatocytes: comparison with rifampin and phenobarbital. J. Clin. Pharmacol. 44:1273-1281. [DOI] [PubMed] [Google Scholar]
- 15.Hartkoorn, R. C., W. S. Kwan, V. Shallcross, A. Chaikan, N. Liptrott, D. Egan, E. S. Sora, C. E. James, S. Gibbons, P. G. Bray, D. J. Back, S. H. Khoo, and A. Owen. 2010. HIV protease inhibitors are substrates for OATP1A2, OATP1B1 and OATP1B3 and lopinavir plasma concentrations are influenced by SLCO1B1 polymorphisms. Pharmacogenet. Genomics 20:112-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang, S. M., J. M. Strong, L. Zhang, K. S. Reynolds, S. Nallani, R. Temple, S. Abraham, S. A. Habet, R. K. Baweja, G. J. Burckart, S. Chung, P. Colangelo, D. Frucht, M. D. Green, P. Hepp, E. Karnaukhova, H. S. Ko, J. I. Lee, P. J. Marroum, J. M. Norden, W. Qiu, A. Rahman, S. Sobel, T. Stifano, K. Thummel, X. X. Wei, S. Yasuda, J. H. Zheng, H. Zhao, and L. J. Lesko. 2008. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 48:662-670. [DOI] [PubMed] [Google Scholar]
- 17.Hubatsch, I., E. G. Ragnarsson, and P. Artursson. 2007. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2:2111-2119. [DOI] [PubMed] [Google Scholar]
- 18.Iaccino, E., M. Schiavone, G. Fiume, I. Quinto, and G. Scala. 2008. The aftermath of the Merck's HIV vaccine trial. Retrovirology 5:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwamoto, M., K. Kassahun, M. D. Troyer, W. D. Hanley, P. Lu, A. Rhoton, A. S. Petry, K. Ghosh, E. Mangin, E. P. DeNoia, L. A. Wenning, J. A. Stone, K. M. Gottesdiener, and J. A. Wagner. 2008. Lack of a pharmacokinetic effect of raltegravir on midazolam: in vitro/in vivo correlation. J. Clin. Pharmacol. 48:209-214. [DOI] [PubMed] [Google Scholar]
- 20.Iwamoto, M., L. A. Wenning, G. C. Mistry, A. S. Petry, S. Y. Liou, K. Ghosh, S. Breidinger, N. Azrolan, M. J. Gutierrez, W. E. Bridson, J. A. Stone, K. M. Gottesdiener, and J. A. Wagner. 2008. Atazanavir modestly increases plasma levels of raltegravir in healthy subjects. Clin. Infect. Dis. 47:137-140. [DOI] [PubMed] [Google Scholar]
- 21.Iwamoto, M., L. A. Wenning, B. Y. Nguyen, H. Teppler, A. R. Moreau, R. R. Rhodes, W. D. Hanley, B. Jin, C. M. Harvey, S. A. Breidinger, N. Azrolan, H. F. Farmer, Jr., R. D. Isaacs, J. A. Chodakewitz, J. A. Stone, and J. A. Wagner. 2009. Effects of omeprazole on plasma levels of raltegravir. Clin. Infect. Dis. 48:489-492. [DOI] [PubMed] [Google Scholar]
- 22.Iwamoto, M., L. A. Wenning, A. S. Petry, M. Laethem, M. De Smet, J. T. Kost, S. A. Breidinger, E. C. Mangin, N. Azrolan, H. E. Greenberg, W. Haazen, J. A. Stone, K. M. Gottesdiener, and J. A. Wagner. 2008. Minimal effects of ritonavir and efavirenz on the pharmacokinetics of raltegravir. Antimicrob. Agents Chemother. 52:4338-4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janneh, O., B. Chandler, R. Hartkoorn, W. S. Kwan, C. Jenkinson, S. Evans, D. J. Back, A. Owen, and S. H. Khoo. 2009. Intracellular accumulation of efavirenz and nevirapine is independent of P-glycoprotein activity in cultured CD4 T cells and primary human lymphocytes. J. Antimicrob. Chemother. 64:1002-1007. [DOI] [PubMed] [Google Scholar]
- 24.Kassahun, K., I. McIntosh, D. Cui, D. Hreniuk, S. Merschman, K. Lasseter, N. Azrolan, M. Iwamoto, J. A. Wagner, and L. A. Wenning. 2007. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab. Dispos. 35:1657-1663. [DOI] [PubMed] [Google Scholar]
- 25.Kis, O., K. Robillard, G. N. Chan, and R. Bendayan. 2010. The complexities of antiretroviral drug-drug interactions: role of ABC and SLC transporters. Trends Pharmacol. Sci. 31:22-35. [DOI] [PubMed] [Google Scholar]
- 26.Lee, C. G., M. M. Gottesman, C. O. Cardarelli, M. Ramachandra, K. T. Jeang, S. V. Ambudkar, I. Pastan, and S. Dey. 1998. HIV-1 protease inhibitors are substrates for the MDR1 multidrug transporter. Biochemistry 37:3594-3601. [DOI] [PubMed] [Google Scholar]
- 27.Liptrott, N. J., M. Penny, P. G. Bray, J. Sathish, S. H. Khoo, D. J. Back, and A. Owen. 2009. The impact of cytokines on the expression of drug transporters, cytochrome P450 enzymes and chemokine receptors in human PBMC. Br. J. Pharmacol. 156:497-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luber, A., P. D. Slowinski, and E. Acosta. 2009. Steady-state pharmacokinetics of fosamprenavir and raltegravir alone and combined with unboosted and ritonavir-boosted fosamprenavir, abstr. A1-1297. Abstr. 49th Annu. Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC.
- 29.Markowitz, M., J. O. Morales-Ramirez, B. Y. Nguyen, C. M. Kovacs, R. T. Steigbigel, D. A. Cooper, R. Liporace, R. Schwartz, R. Isaacs, L. R. Gilde, L. Wenning, J. Zhao, and H. Teppler. 2006. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naive HIV-1-infected individuals. J. Acquir. Immune Defic. Syndr. 43:509-515. [DOI] [PubMed] [Google Scholar]
- 30.Mosmann, T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55-63. [DOI] [PubMed] [Google Scholar]
- 31.Rizwan, A. N., and G. Burckhardt. 2007. Organic anion transporters of the SLC22 family: biopharmaceutical, physiological, and pathological roles. Pharm. Res. 24:450-470. [DOI] [PubMed] [Google Scholar]
- 32.Sobczak, K., N. Bangel-Ruland, G. Leier, and W. M. Weber. 2010. Endogenous transport systems in the Xenopus laevis oocyte plasma membrane. Methods 51:183-189. [DOI] [PubMed] [Google Scholar]
- 33.Steigbigel, R. T., D. A. Cooper, H. Teppler, J. J. Eron, J. M. Gatell, P. N. Kumar, J. K. Rockstroh, M. Schechter, C. Katlama, M. Markowitz, P. Yeni, M. R. Loutfy, A. Lazzarin, J. L. Lennox, B. Clotet, J. Zhao, H. Wan, R. R. Rhodes, K. M. Strohmaier, R. J. Barnard, R. D. Isaacs, and B. Y. Nguyen. 2010. Long-term efficacy and safety of raltegravir combined with optimized background therapy in treatment-experienced patients with drug-resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 phase III trials. Clin. Infect. Dis. 50:605-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Summa, V., A. Petrocchi, F. Bonelli, B. Crescenzi, M. Donghi, M. Ferrara, F. Fiore, C. Gardelli, O. Gonzalez Paz, D. J. Hazuda, P. Jones, O. Kinzel, R. Laufer, E. Monteagudo, E. Muraglia, E. Nizi, F. Orvieto, P. Pace, G. Pescatore, R. Scarpelli, K. Stillmock, M. V. Witmer, and M. Rowley. 2008. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 51:5843-5855. [DOI] [PubMed] [Google Scholar]
- 35.Takeda, M., S. Khamdang, S. Narikawa, H. Kimura, Y. Kobayashi, T. Yamamoto, S. H. Cha, T. Sekine, and H. Endou. 2002. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J. Pharmacol. Exp. Ther. 300:918-924. [DOI] [PubMed] [Google Scholar]
- 36.Uwai, Y., H. Ida, Y. Tsuji, T. Katsura, and K. Inui. 2007. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 24:811-815. [DOI] [PubMed] [Google Scholar]
- 37.van der Sandt, I. C., C. M. Vos, L. Nabulsi, M. C. Blom-Roosemalen, H. H. Voorwinden, A. G. de Boer, and D. D. Breimer. 2001. Assessment of active transport of HIV protease inhibitors in various cell lines and the in vitro blood-brain barrier. AIDS (London) 15:483-491. [DOI] [PubMed] [Google Scholar]
- 38.Wenning, L. A., E. J. Friedman, J. T. Kost, S. A. Breidinger, J. E. Stek, K. C. Lasseter, K. M. Gottesdiener, J. Chen, H. Teppler, J. A. Wagner, J. A. Stone, and M. Iwamoto. 2008. Lack of a significant drug interaction between raltegravir and tenofovir. Antimicrob. Agents Chemother. 52:3253-3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wenning, L. A., W. D. Hanley, D. M. Brainard, A. S. Petry, K. Ghosh, B. Jin, E. Mangin, T. C. Marbury, J. K. Berg, J. A. Chodakewitz, J. A. Stone, K. M. Gottesdiener, J. A. Wagner, and M. Iwamoto. 2009. Effect of rifampin, a potent inducer of drug-metabolizing enzymes, on the pharmacokinetics of raltegravir. Antimicrob. Agents Chemother. 53:2852-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie, W., M. F. Yeuh, A. Radominska-Pandya, S. P. Saini, Y. Negishi, B. S. Bottroff, G. Y. Cabrera, R. H. Tukey, and R. M. Evans. 2003. Control of steroid, heme, and carcinogen metabolism by nuclear pregnane X receptor and constitutive androstane receptor. Proc. Natl. Acad. Sci. U. S. A. 100:4150-4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yilmaz, A., M. Gisslen, S. Spudich, E. Lee, A. Jayewardene, F. Aweeka, and R. W. Price. 2009. Raltegravir cerebrospinal fluid concentrations in HIV-1 infection. PLoS One 4:e6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang, D., T. J. Chando, D. W. Everett, C. J. Patten, S. S. Dehal, and W. G. Humphreys. 2005. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation. Drug Metab. Dispos. 33:1729-1739. [DOI] [PubMed] [Google Scholar]
- 43.Zhu, L., L. Mahnke, J. Butterton, A. Persson, M. Stonier, W. Comisar, D. Panebianco, S. Breidinger, J. Zhang, and R. Bertz. 2009. Pharmacokinetics and safety of twice daily atazanavir 300 mg and raltegravir 400 mg in healthy subjects, abstr. 693. Abstr. 16th Conf. Retroviruses Opportun. Infect., Montreal, Canada.