

Abstract
The role of mouse peptidoglycan recognition protein PGLYRP-1 in innate immunity against Listeria monocytogenes infection was studied. The recombinant mouse PGLYRP-1 and a polyclonal antibody specific to PGLYRP-1 were prepared. The mouse PGLYRP-1 showed antibacterial activities against L. monocytogenes and other Gram-positive bacteria. PGLYRP-1 mRNA expression was induced in the spleens and livers of mice infected with L. monocytogenes. The viable bacterial number increased, and the production of cytokines such as gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) was reduced in mice when mice had been injected with anti-PGLYRP-1 antibody before infection. The levels of IFN-γ and TNF-α titers in the organs were higher and the viable bacterial number was reduced in mice injected with recombinant mouse PGLYRP-1 (rmPGLYRP-1) before infection. PGLYRP-1 could directly induce these cytokines in spleen cell cultures. The elimination of intracellular bacteria was upregulated in NMuLi hepatocyte cells overexpressing PGLYRP-1. The enhancement of the elimination of L. monocytogenes from the organs was observed in IFN-γ−/− mice by rmPGLYRP-1 administration but not in TNF-α−/− mice. These results suggest that PGLYRP-1 plays a role in innate immunity against L. monocytogenes infection by inducing TNF-α.
Innate immunity is the frontier of host defense against microbial infections. It is directed to components of microorganisms and recognizes them through a series of pattern recognition receptors, which are conserved through species from insects to mammals (3, 10, 12). Peptidoglycan recognition proteins (PGRPs) also are a family of these pattern recognition proteins and are conserved through species (8, 9, 20, 23, 33, 47).
PGRP was first discovered in the silkworm Bombyx mori in 1996 as a 19-kDa protein that could recognize peptidoglycans (PGNs) (61). The genome sequencing of Drosophila melanogaster revealed 17 homologues of PGRPs in this fly (8, 56). In these PGRPs, seven short PGRPs have signal peptides and can be secreted. Ten long PGRPs have predicted transmembrane domains and can be transmembrane proteins. Drosophila PGRPs are expressed in immunocompetent cells such as hemocytes and are upregulated by PGNs (23). Therefore, it is likely that Drosophila PGRPs play a role in insect innate immunity. Recent studies revealed that PGRP-SA, -SD, -LC, and -LE can activate two different pathways, Toll and Imd pathways, that lead to the production of antibacterial peptides (1, 6, 14, 30, 39, 51, 57, 62). In addition, PGRP-LE is reported to be crucial for the induction of autophagy by L. monocytogenes in D. melanogaster (59). Other proteins, PGRP-SC1b, -LB, and -SA, are known to have PGN-degrading activities (25, 36, 37).
Mammals have four homologues of PGRPs: PGLYRP-1, -2, -3, and -4 (initially named PGRP-S, -L, -Iα, and -Iβ, respectively) in humans (23, 33), mice (24, 32, 35), rats (43), and cattle (53, 54). Mammalian PGLYRP-2 is an N-acetyl-muramoyl-l-alanine amidase (9, 13, 34, 55, 62). It can cleave the peptide from the glycan chain of PGNs and is expressed in liver, colon, and skin cells (31, 33, 34, 62), but its function in vivo still is unclear (56, 58). Human PGLYRP-1, -3, and -4 show antibacterial activities (34). Molecular and structural mechanisms of mammalian PGRPs for PGN binding and antibacterial activities have been studied (5, 16-19, 26, 48), as have been insect PGRPs (4, 24, 29, 44). Mammalian PGLYRP-1 is a secretory protein (32). This protein constructs dimer formation (32-34), and the metal ions such as calcium and zinc ions enhance the antimicrobial activities (34). Mammalian PGLYRP-1 is considered a pattern recognition receptor (8, 33). Human PGLYRP-1 is localized in neutrophils and is likely to kill phagocytized bacteria (7, 32, 53). Moreover, the immunomodulatory activity of PGLYRP-1 in PGN-induced arthritis has been reported recently for mice (48). However, a mechanism of pattern recognition and ensuing innate immunity in bacterial infection still is unclear.
Listeria monocytogenes is a Gram-positive intracellular bacterium that is important as an opportunistic pathogen in humans such as immunocompromised hosts, pregnant women, and their fetuses. Macrophages contribute to innate immunity against L. monocytogenes infection, although recent reports showed that granulocytes, including neutrophils, play a role in host resistance against the early stage of L. monocytogenes infection as well as macrophages (15, 60). Tumor necrosis factor-alpha (TNF-α) and gamma interferon (IFN-γ) are known to be important in host resistance against L. monocytogenes infection (2, 11, 21, 22, 40-42, 44). The production of these cytokines is induced by L. monocytogenes infection via several pattern recognition receptors and their downstream components (3). However, the role of PGLYRP-1 in the protection against L. monocytogenes infection is not clear.
In this study, we investigated the immunomodulatory activities of mouse PGLYRP-1 in innate immune systems as well as antibacterial activities. We demonstrated that mouse PGLYRP-1 plays an important role in host resistance against L. monocytogenes infection.
MATERIALS AND METHODS
Mice.
C57BL/6 mice were purchased from Clea Japan Inc., Tokyo, Japan. IFN-γ-deficient (IFN-γ−/−) and TNF-α-deficient (TNF-α−/−) mice (C57BL/6 background) were developed as previously reported (50, 52). Mice were cared for under specific-pathogen-free conditions in the Institute for Animal Experimentation, Hirosaki University Graduate School of Medicine. All animal experiments in this study were performed by following the guidelines for animal experimentation of Hirosaki University.
Infection.
L. monocytogenes strain 1b 1684 (41) was used in this study. Bacteria grown in tryptic soy broth (BD Bioscience, Sparks, MD) were dispersed and stored at −80°C until use. C57BL/6, IFN-γ−/−, and TNF-α−/− mice were infected intravenously with sublethal doses of 5 × 105 CFU (for C57BL/6 mice) and 1 × 105 CFU (for IFN-γ−/− and TNF-α−/− mice) of L. monocytogenes in phosphate-buffered saline (PBS). When the effect of PGLYRP-1 administration was tested, 5 × 106 CFU of L. monocytogenes in PBS was infected.
Spleen cells and macrophages.
Spleens from C57BL/6 mice were minced and filtrated through stainless mesh (size, 100). Erythrocytes were lysed with 0.83% (wt/vol) ammonium chloride and then washed three times with Dulbecco's modified Eagle medium (DMEM; Nissui Pharmaceutical Co., Tokyo, Japan). Splenic macrophages were prepared as follows: spleen cells resuspended in DMEM supplemented with 10% (vol/vol) fetal calf serum (FCS; Nichirei Biosciences Inc., Tokyo, Japan) and 0.03% l-glutamine (Wako Pure Chemical Co., Osaka, Japan) were plated on 25-cm2 culture flasks (Asahi Glass Co., LTD., Tokyo, Japan) for 2 h in a 5% CO2 incubator. The adherent cells were isolated by a cell scraper and counted. Spleen cells and splenic macrophages were cultured in the media in a 5% CO2 incubator. Mouse hepatocyte cell line NMuLi cells (DS Pharma Biomedical, Osaka, Japan) were maintained in DMEM with 10% (vol/vol) FCS.
Expression of mouse PGLYRP-1.
RNA was isolated from the spleens of C57BL/6 mice infected with L. monocytogenes 0, 0.5, 1, 2, 4, 6, and 12 h after infection using TRIzol reagents (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. RNA from three mice was pooled and used for real-time reverse transcription-PCR (RT-PCR). Primers for the amplification of tag7 genes, coding for PGLYRP-1 (25), were p-132 (5′-CAT ATG TGC AGT TTC ATC GTG CCC CGC AG-3′) and p-133 (GGA TCC TCA CTC TCG GTA GTG TTC CCA GC). Primers for the amplification of mouse glyceraldehyde-3-phosphate dehydrogenase (mGAPDH) genes were mGAPDH-F (5′-TGA AGG TCG GTG TGA ACG GAT TTG G-3′) and mGAPDH-R 5′-ACG ACA TAC TCA GCA CCG GCC TCA C-3′). PCR conditions consisted of preheating at 94°C for 10 min, 40 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 45 s, and a final elongation at 72°C for 10 min. PCR products were observed in the agarose gel after electrophoresis. The quantitative analysis of the expression of PGLYRP-1 was performed by an i-Cycler system (Bio-Rad Japan, Tokyo, Japan) using iQ SYBR green Supermix (Bio-Rad Japan) as the chromogen. The expression was analyzed as the relative expression compared to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
rmPGLYRP-1.
RNA samples were obtained from spleens of C57BL/6 mice 2 h after L. monocytogenes infection. Primers for cloning were the same as those for amplification (described above) and were designed for subcloning to the pET15b expression vector (Merck Chemical KgaA, Darmstadt, Germany). After PCR, PCR products were purified and ligated to pCR II TA cloning vector. After the transformation and sequencing of the insert, it was subcloned into pET15b by the NdeI-BamHI restriction site. The expression host, Rosetta (DE3) pLysS, was transformed by this plasmid. After the cultivation of these bacteria in 500 ml LB broth (BD Biosciences) with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol for 2 h, recombinant protein production was induced by the addition of 1 mM isopropylthiogalactopyranoside, and the bacteria were cultured for 2 h. The cultivated bacteria were disrupted by sonication, and recombinant proteins were precipitated in inclusion bodies. Purified inclusion bodies were solubilized by 6 M guanidine-HCl in the washing buffer (50 mM sodium phosphate buffer, pH 7.0, 300 mM NaCl) and immediately reprecipitated by diluting the guanidine concentration to 0.6 M. After centrifugation, the resulting pellets were resolubilized by 6 M guanidine in the washing buffer supplemented with 0.3% N-laurolyl sarcosinate and 1 mM dithiothreitol, and the concentration of guanidine was diluted to 2 M by slowly adding the washing buffer with supplements. Solubilized recombinant mouse PGLYRP-1 (rmPGLYRP-1) then was charged to a Talon 6× histidine affinity column (Takara Bio Inc., Ohtsu, Japan). The column was washed with the washing buffer containing a slowly lowered concentration of guanidine from 2 to 0 M using an Econo gradient pump system (Bio-Rad Japan). The charged rmPGLYRP-1 then was eluted by 150 mM imidazole in the washing buffer. The eluted proteins were concentrated and exchanged with the washing buffer using Vivaspin filter units (Sartorius AG, Goettingen, Germany). Digestion by thrombin and the subsequent removal of thrombin were performed using a thrombin cleavage capture kit (Novagen, Madison, WI), and histidine tag-cleaved rmPGLYRP-1 was stored at −20°C before use. When used for L. monocytogenes infection, 100 μg of rmPGLYRP-1 was administered to mice 6 h before infection.
Tests for endotoxin contamination.
To assess the contamination of lipopolysaccharide (LPS) derived from host Escherichia coli, Limulus amebocyte lysate (LAL) tests were performed using PYROTELL kits (Seikagaku Corp., Tokyo, Japan). Endotoxin was below 3 pg/100 μg rmPGLYRP-1. In some experiments, rmPGLYRP-1 was inactivated by heating the sample for 30 min at 95°C. LPS from E. coli was purchased from Sigma-Aldrich Japan, Tokyo, Japan.
Rabbit antibodies against rmPGLYRP-1.
The rabbits were immunized with 1 mg of rmPGLYRP-1 plus 1 ml of Imject alum (Pierce Biotechnology Inc., Rockford, IL) per rabbit three times at 2-week intervals. The sera were obtained from the rabbits 1 week after final immunization. Immunoglobulin was purified using DEAE-sephacel resin (GE Healthcare, Piscataway, NJ) according to the standard protocol and stored at −80°C before use. The titer of antibodies was 1:102,400, as determined by enzyme-linked immunosorbent assay (ELISA). Mice were administered 1 mg of the antibody 24 h before L. monocytogenes infection. Normal rabbit globulin (NRG) was injected as a negative control. NRG was prepared from normal rabbit sera using the same protocol as that for the antibody.
Assay of antibacterial activities.
Salmonella enterica serovar Typhimurium χ3306, Escherichia coli strain IFO3806, and clinical isolates of Staphylococcus aureus, S. epidermidis, Pseudomonas aeruginosa, and Serratia marcescens were used for assays of antibacterial activities in addition to L. monocytogenes. Bacteria were precultured overnight and subcultured for 4 h in tryptic soy broth. Assay mixtures for antibacterial activities contained 1 × 106 CFU of bacteria tested and rmPGLYRP-1 or bovine serum albumin (fraction V; Sigma-Aldrich, Japan) in 1 ml of assay buffer (5 mM Tris-HCl, pH 7.6, 150 mM NaCl, 2.5 mM CaCl2, and 5% glycerol supplemented with 1% LB broth). Where indicated, 1 mM zinc chloride (Wako) or 1 mM EDTA (EDTA, Wako) were added to the assay mix. After 0.5, 1, 3, and 6 h, 100 μl of mixture was recovered, and serially diluted specimens were plated on tryptic soy agar (BD Sciences). The colony number was counted 24 h later. Neutralization by anti-mPGLYRP-1 antibody was accomplished by adding 1 mg of the antibody to the mixture.
In vitro stimulation of spleen cells and macrophages.
Spleen cells and splenic macrophages were plated on 24-well culture plates at a concentration of 1 × 106 cells/ml in DMEM supplemented with 10% (vol/vol) FCS, and 0.4, 2, or 10 μg of rmPGLYRP-1 was added. The culture supernatant fluids were collected 48 h later and used for cytokine assays. In some experiments, the cells were plated on 24-well culture plates (Asahi Glass, Tokyo, Japan) at a concentration of 1 × 106 cells/ml in DMEM supplemented with 10% (vol/vol) FCS with 1 × 107 CFU of L. monocytogenes. After 30 min, the medium was changed to DMEM supplemented with 10% (vol/vol) FCS and 5 μg/ml gentamicin and cultured for 12 h. Culture supernatant fluids were collected and used for cytokine assays.
Construction of PGLYRP-1 with enhanced green fluorescent protein (EGFP) expression plasmid.
pEGFP-C2 vector was purchased from Takara Bio Inc. The gene encoding PGLYRP-1 was amplified using cDNA from spleens of L. monocytogenes-infected mice (described above) by the primers p505 (5′-GAA TTC ATG TTG TTT GCC TGT GCT CTC CTT G-3′) and 507 (5′-GGA TCC TCA CTC TCG GTA GTG TTC CCA GC-3′) with EcoRI and BamHI restriction sites, respectively. This fragment was ligated into pEGFP-C2 by restriction sites, and the plasmid with PGLYRP-1 was designated pEGFP-C2/PGLYRP-1.
Overexpression of PGLYRP-1 in hepatocyte NMuLi cells and observation of L. monocytogenes in the cells.
NMuLi cells were seeded on 24-well culture plates at a concentration of 2 × 105 cells/ml in DMEM supplemented with 10% (vol/vol) FCS for 12 h. pEGFP-C2/PGLYRP-1 and control pEGFP-C2 vectors were transfected with the support of Lipofectamine 2000 (Invitrogen) and incubated for 24 h. Cells were infected with L. monocytogenes at a multiplicity of infection (MOI) of 100. After 30 min of incubation, the extracellular bacteria were eliminated with 5 μg/ml gentamicin and additionally incubated for 6 h. Bacteria were sequentially labeled with rabbit anti-Listeria spp. (ViroStat, Portland, ME) and rhodamine-conjugated goat anti-rabbit immunoglobulin G (MP Bio Japan, Tokyo, Japan) and observed by fluorescent microscopy (Olympus IX71; Olympus, Tokyo, Japan). GFP- or GFP-PGLYRP-1-overexpressing cells and intracellular rhodamine-labeled L. monocytogenes inside the cells were counted for 10 fields and analyzed statistically.
Preparation of organ and cell samples for determination of bacterial numbers and for cytokine assays.
Spleens and livers from infected mice were homogenized in PBS containing 1% (wt/vol) 3-[(cholamidopropyl)-dimethyl-ammonio]-1-propanesulfate (CHAPS; Wako) to prepare 10% (wt/vol) homogenates. NMuli cells seeded on 24-well culture plates at a concentration of 2 × 105 cells/well also were homogenized in 1 ml of PBS containing 1% (wt/vol) CHAPS. The numbers of viable L. monocytogenes cells were established by plating 10-fold serial dilutions of a bacterial solution in PBS on tryptic soy agar plates. Colonies were counted 24 h later.
Cytokine assays.
TNF-α levels were determined by ELISA using a cyto-set Elisa kit (Invitrogen) according to the manufacturer's instrumentation. IFN-γ ELISA was performed as previously described (40).
SDS-PAGE and Western blotting.
SDS-PAGE was performed as previously reported (27). For the confirmation of the purity of rmPGLYRP-1, 2 μg of recombinant protein was applied to SDS-PAGE. Endogenous PGLYRP-1 in sera of mice were detected at various time points after L. monocytogenes infection. Exogenous rmPGLYRP-1 was detected in sera, spleens, and livers of mice at various time points after the injection of 100 μg of rmPGLYRP-1. Protein concentration was determined using a Bio-Rad protein assay (Bio-Rad Japan) according to the manufacturer's instructions and normalized to 20 μg/well. Separated proteins were transferred to Immobillon-P membranes (Millipore Japan, Tokyo, Japan) by electrophoresis. The polyclonal antibody prepared in this study was used to detect mPGLYRP-1. Horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (MP Bio, Japan) was used as a second antibody, and color reaction was performed with 3,3′-diaminobenzidine, tetrahydrochloride (Wako) as a substrate.
Statistical analysis.
Data were expressed as means ± standard deviations (SD), and the Mann-Whitney U test was used to determine the significance of the differences.
RESULTS
PGLYRP-1 was induced by L. monocytogenes infection in mice.
Mouse PGLYRP-1 is known to be highly produced in neutrophils (32). To estimate whether tag7 gene expression and the production of PGLYRP-1 are regulated by bacterial infections, we investigated the expression of PGLYRP-1 in spleens and livers of mice infected with L. monocytogenes. After infection, mice were sacrificed and RNA was obtained from the spleens and livers for RT-PCR. The expression of PGLYRP-1 mRNA was upregulated 30 min to 6 h after infection (Fig. 1A). Quantitative real-time RT-PCR also was carried out, and a consistent expression pattern was observed (see Fig. S2 in the supplemental material). Moreover, to investigate that PGLYRP-1 is secretory, sera were taken from mice infected with L. monocytogenes and PGLYRP-1 was detected by anti-mPGLYRP-1 antibody. PGLYRP-1 protein was detected 3 h after infection in the sera and continued to 12 h (Fig. 1B). These results indicated that PGLYRP-1 was induced in the early stages of L. monocytogenes infection in mice.
FIG. 1.

Expression of PGLYRP-1-coding tag7 gene and PGLYRP-1 protein after L. monocytogenes infection. (A) Mice were infected with 5 × 105 CFU of L. monocytogenes, the spleens and livers were obtained 0, 0.5, 1, 2, 4, 6, and 12 h after infection, and RNA was extracted from the organs. The expression of tag7 and gapdh was estimated by RT-PCR. Representative data are shown for three independent experiments. (B) Mice were infected with 5 × 105 CFU of L. monocytogenes, and the sera were obtained 0, 3, 6, and 12 h after infection. The protein concentration was adjusted to 20 μg/well and applied to Western blotting. Representative data are shown for three independent experiments.
Anti-PGLYRP-1 antibody increased susceptibility to L. monocytogenes infection and reduced TNF-α and IFN-γ production.
We assessed whether endogenous PGLYRP-1 is involved in host resistance against L. monocytogenes infection. Recombinant protein was prepared and purified as a dimer (see Fig. S1), consistently with the previous report (34). Polyclonal antibodies against mPGLYRP-1 were prepared by the immunization of rabbits with this protein, and the specificity was confirmed by Western blotting, although minor nonspecific bands were detected (Fig. 1B; also see Fig. S3 in the supplemental material). Mice were injected with anti-PGLYRP-1 antibody or NRG 24 h before infection with a sublethal dose of L. monocytogenes. The viable bacterial numbers in the spleens and livers were measured on days 1, 2, 3, and 5 after infection. The numbers in both organs increased in anti-PGLYRP-1 antibody-injected mice on days 2 and 3 after infection compared to that in NRG-injected animals (Fig. 2 A and B). However, the effect of anti-PGLYRP1 antibody was not observed on day 5 of infection. The levels of IFN-γ and TNF-α in the sera and spleens were determined next, because TNF-α and IFN-γ are critical in host resistance against L. monocytogenes infection (2, 11, 21, 22, 41, 42, 46). The titers of both cytokines in the organs tested were significantly decreased in the anti-PGLYRP-1 antibody-treated group compared to that of the NRG-treated group 24 h after infection (Fig. 2C and D). These results indicated that endogenous PGLYRP-1 is involved in host resistance against L. monocytogenes infection and the production of IFN-γ and TNF-α.
FIG. 2.

Inhibition of elimination of L. monocytogenes and endogenous cytokine production by administration of anti-mPGLYRP-1 antibody in mice. C57BL/6 mice were injected with 1 mg of anti-PGLYRP-1 antibody or normal rabbit globulin (NRG) 24 h before infection with 5 × 105 CFU of L. monocytogenes, and viable bacterial numbers in the spleens (A) and livers (B) from mice were determined 1, 2, 3, and 5 days after infection. Similarly, titers of IFN-γ (C) and TNF-α (D) in the sera and spleens 24 h after infection were determined in an anti-PGLYRP-1 antibody-treated group and NRG-treated group. The threshold of detection is indicated by <12.5. Each group consists of six mice. Bars show SD. An asterisk indicates a significant difference from the NRG-treated group at P < 0.05.
rmPGLYRP-1 enhanced the elimination of L. monocytogenes from the organs of mice.
We investigated the effect of the administration of rmPGLYRP-1 on antilisterial resistance. We first assessed whether rmPGLYRP-1 is able to induce cytokines in vivo. Naive mice were administered rmPGLYRP-1, and the levels of IFN-γ and TNF-α in the sera and spleens were measured 24 h later. The concentration of IFN-γ and TNF-α in sera increased in rmPGLYRP-1-treated mice compared to that of the PBS-treated group (Fig. 3 A and B). Mice then were injected with rmPGLYRP-1 followed by L. monocytogenes infection 6 h later, and bacterial numbers were determined in the spleens and livers on days 1, 2, 3, and 5 of infection. The numbers of L. monocytogenes cells in the organs significantly decreased when rmPGLYRP-1 had been administered on days 1, 2, 3, and 5 after infection (Fig. 3C and D). We next carried out the detection of exogenous PGLYRP-1 in the bloodstream and organs of mice. The thick band of PGLYRP-1 was detected at 12 and 24 h in sera and rapidly disappeared (see Fig. S3 in the supplemental material). In contrast, PGLYRP-1 still could be detected in livers and spleens thereafter (see Fig. S3). The titers of TNF-α and IFN-γ in the sera and spleens also were measured 24 h after infection. IFN-γ and TNF-α production in L. monocytogenes-infected mice increased significantly at 24 h in the rmPGLYRP-1-injected groups compared to that of the PBS-injected group (Fig. 3E and F).
FIG. 3.

Enhancement of host resistance against L. monocytogenes infection and cytokine production by administration of rmPGLYRP-1. C57BL/6 mice were injected with 100 μg of rmPGLYRP-1, heat-inactivated rmPGLYRP-1, or PBS, and titers of IFN-γ (A) and TNF-α (B) in the sera were determined 24 h after administration. Similarly, C57BL/6 mice were injected with 100 μg of rmPGLYRP-1, heat-inactivated rmPGLYRP-1, or PBS 6 h before infection with 5 × 106 CFU of L. monocytogenes, and viable bacterial numbers in the spleens (C) and livers (D) from mice were determined 1, 2, 3, and 5 days after infection. Titers of IFN-γ (E) and TNF-α (F) in the sera and spleens were determined for the rmPGLYRP-1-treated group, heat-inactivated rmPGLYRP-1-treated group, and PBS-treated group 24 h after infection. The threshold of detection is indicated by <12.5. Each group consists of six mice. Bars show SD. An asterisk indicates a significant difference from the PBS-treated group and heat-inactivated rmPGLYRP-1-treated group at P < 0.05.
rmPGLYRP-1 showed antibacterial activities.
Mammalian PGLYRP-1 is known to show bactericidal activities against Gram-positive bacteria (31, 32). In this study, we investigated the antibacterial activities of rmPGLYRP-1 against some species of Gram-positive and Gram-negative bacteria. Two hundred μg of rmPGLYRP-1 was added to 1 ml of the bacterial suspension, and viable bacterial numbers were determined during 6 h of incubation. rmPGLYRP-1 showed no antibacterial activity against the Gram-negative bacteria examined (data not shown). In contrast, rmPGLYRP-1 revealed antibacterial activities against Gram-positive S. aureus, S. epidermidis, and L. monocytogenes (Fig. 4 A, B, and C). We investigated whether anti-mPGLYRP-1 antibody could neutralize the antibacterial activities of rmPGLYRP-1. No antibacterial activity was observed against L. monocytogenes when rmPGLYRP-1 had been treated with anti-PGLYRP-1 antibody (Fig. 4C). The antibacterial activity was not significantly enhanced by the addition of zinc chloride (Fig. 4D and E). However, the addition of EDTA inhibited the antibacterial activity of PGLYRP-1 (Fig. 4D and E).
FIG. 4.

Antibacterial activities of rmPGLYRP-1 against Gram-positive and Gram-negative bacteria. (A to C) An assay mixture for antibacterial activity contained 1 × 106 CFU of S. aureus (A), S. epidermidis (B), and L. monocytogenes (C) rmPGLYRP-1 or bovine serum albumin (BSA) as a control in 1 ml of the assay buffer with or without anti-mPGLYRP-1 antibody (1 mg/ml). After 0.5, 1, 3, and 6 h of incubation, 100 μl of the mixture was taken and plated on tryptic soy agar plates after serial dilution. Each result was obtained from six independent assays. An asterisk indicates that a value is significantly different from the value obtained from BSA controls at P < 0.01. (D, E) An assay mixture for antibacterial activity contained 1 × 106 CFU of S. aureus (D) and L. monocytogenes (E) rmPGLYRP-1 or BSA as a control in 1 ml of the assay buffer with or without 1 mM zinc chloride or 1 mM EDTA. Each result was obtained from six independent assays. An asterisk indicates that a value is significantly different from the value obtained from BSA controls at P < 0.01.
rmPGLYRP-1 induced cytokine production in spleen cells.
We next investigated the involvement of PGLYRP-1 in IFN-γ and TNF-α production in vitro. Naïve spleen cells and splenic macrophages were stimulated with 0.4, 2, or 10 μg of rmPGLYRP-1 for 48 h. Cytokine titers in the culture supernatant fluids were measured. Spleen cells produced IFN-γ and TNF-α by stimulation with rmPGLYRP-1 in a dose-dependent manner (Fig. 5). To investigate whether PGLYRP-1 could activate spleen cells in vivo, mice then were injected with 100 μg of rmPGLYRP-1, spleen cells and splenic macrophages were obtained 6 h later, and these cells were stimulated with 1 × 106 CFU of viable L. monocytogenes in vitro. After 12 h of incubation in the presence of gentamicin, TNF-α and IFN-γ levels in culture supernatant fluids were measured. TNF-α and IFN-γ production was significantly enhanced in rmPGLYRP-1-injected mice compared to that of the PBS-treated group (Fig. 6). These results indicated that PGLYRP-1 could induce TNF-α and IFN-γ production in vitro and in vivo.
FIG. 5.
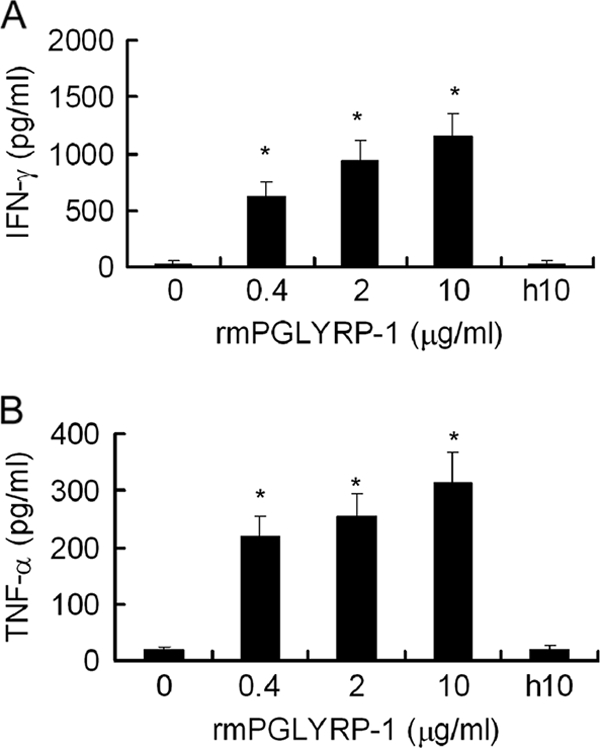
Cytokine production by stimulation with rmPGLYRP-1 in vitro. (A) Naïve mouse spleen cells were stimulated with 0.4, 2, or 10 μg of rmPGLYRP-1 or 10 μg of heat-inactivated rmPGLYRP-1 for 48 h, and IFN-γ in culture supernatant fluids was assayed. (B) Splenic macrophages were stimulated with 0.4, 2, or 10 μg of rmPGLYRP-1 or 10 μg of heat-inactivated rmPGLYRP-1 for 48 h, and TNF-α in culture supernatant fluids was assayed. h10 means 10 μg of heat-inactivated rmPGLYRP-1. Each group consists of six wells of the plates. Bars show SD. An asterisk indicates that the value is significantly different from that of the unstimulated culture and heat-inactivated rmPGLYRP-1 at P < 0.01.
FIG. 6.

Enhancement of cytokine production in spleen cells and macrophages by systemic administration of rmPGLYRP-1. (A) Spleen cells were obtained from C57BL/6 mice 6 h after the injection of 100 μg of rmPGLYRP-1 or PBS in vivo and then were stimulated with PBS or 1 × 107 CFU of viable L. monocytogenes cells for 12 h in vitro. IFN-γ titers in culture supernatant fluids were assayed. (B) Splenic macrophages from C57BL/6 mice pretreated with rmPGLYRP-1 or PBS were stimulated with PBS or viable L. monocytogenes cells for 12 h in vitro. TNF-α titers in culture supernatant fluids were assayed. The threshold of detection is indicated by <12.5. Each group consists of six wells of the plates. Bars show SD. An asterisk indicates that the value is significantly different from that obtained from PBS-pretreated groups at P < 0.01.
NMuLi hepatocyte cells overexpressing PGLYRP-1 showed increased resistance to L. monocytogenes infection.
We showed that mPGLYRP-1 induces TNF-α and IFN-γ production in vitro (Fig. 5 and 6). To investigate further whether PGLYRP-1 enhances the elimination of L. monocytogenes, we constructed NMuLi hepatocyte cells overexpressing PGLYRP-1 with GFP as a marker of successful transfection. Transfected hepatocytes were infected with L. monocytogenes. Rhodamine-labeled L. monocytogenes cells were observed in green-labeled cells under microscopy 6 h after infection, and the intracellular bacterial number in GFP- or GFP-PGLYRP-1-overexpressing cells was counted. The number of L. monocytogenes cells inside PGLYRP-1-overexpressing cells decreased compared to that of GFP-overexpressing control cells (Fig. 7).
FIG. 7.
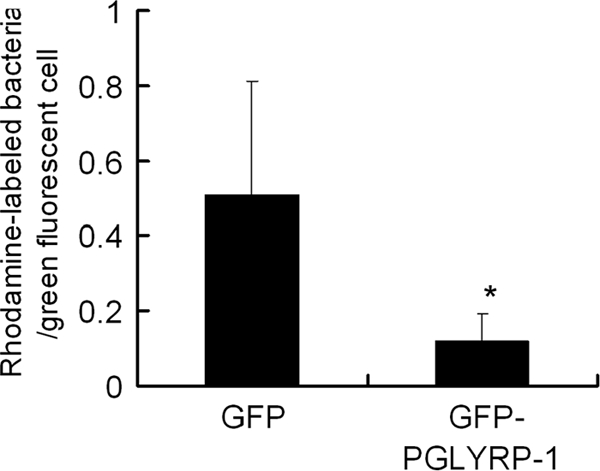
Increased elimination of L. monocytogenes in PGLYRP-1-overexpressing hepatocytes. NMuLi hepatocyte cells seeded at a concentration of 2 × 105 cells/well were transfected with pEGFP-C2/PGLYRP-1 or pEGFP-C2 control vector. Both transfected cells were infected with L. monocytogenes for 30 min, and cell cultivation was continued with the elimination of extracellular bacteria by gentamicin. After 6 h of cultivation, L. monocytogenes cells were labeled with rhodamine, and rhodamine-labeled bacterial numbers in GFP-labeled cells were counted and the ratio of the bacterial number to the cell number was calculated. An asterisk indicates a significant difference from the pEGFP-C2-transfected group at P < 0.05.
rmPGLYRP-1 failed to enhance host resistance against L. monocytogenes infection in TNF-α−/− mice.
To confirm the role of enhanced IFN-γ and TNF-α production induced by PGLYRP-1, we investigated the effect of rmPGLYRP-1 administration on host resistance against L. monocytogenes infection in IFN-γ−/− and TNF-α−/− mice. These mice were injected with 100 μg of rmPGLYRP-1 prior to infection with L. monocytogenes, and the viable bacterial number was determined on day 3 after infection. The bacterial number in the organs of IFN-γ−/− mice decreased significantly in the rmPGLYRP-1-injected group to a level comparable to that of wild-type mice (Fig. 8A). Conversely, the bacterial numbers were not significantly different between the rmPGLYRP-1-injected group and PBS control group in TNF-α−/− mice (Fig. 8B). These results indicated that TNF-α, but not IFN-γ, is involved in the enhancement of antilisterial resistance by PGLYRP-1.
FIG. 8.
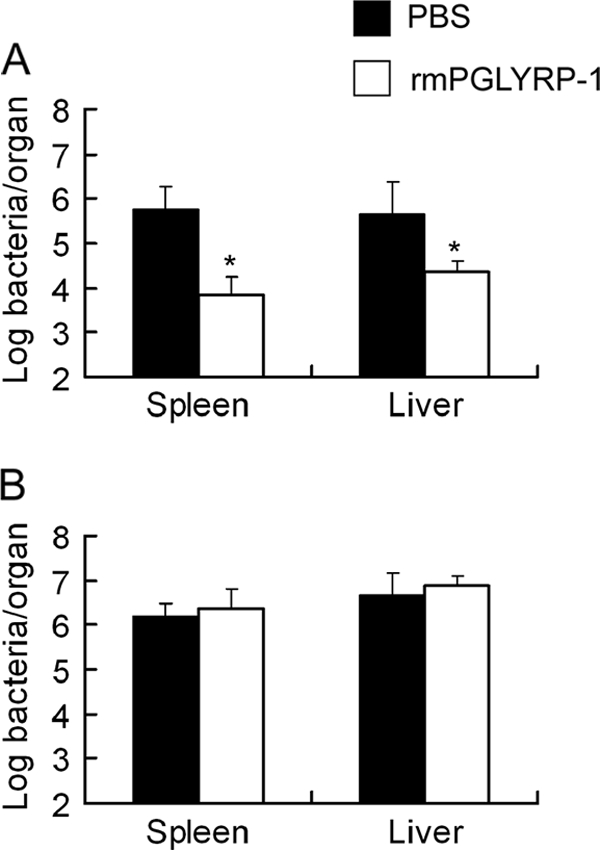
Effect of rmPGLYRP-1 on host resistance against L. monocytogenes infection in IFN-γ−/− mice and TNF-α−/− mice. IFN-γ−/− mice and TNF-α−/− mice were injected with 100 μg of rmPGLYRP-1 or PBS 6 h before infection with 1 × 105 CFU of L. monocytogenes. The number of viable L. monocytogenes cells in organs from IFN-γ−/− mice (A) and TNF-α−/− mice (B) was determined on day 3 after infection. Each group consists of six mice. An asterisk indicates the significant difference from the PBS-treated group at P < 0.01.
Possibility of endotoxin contamination in rmPGLYRP-1 was excluded.
To exclude the possibility of endotoxin contamination of the rmPGLYRP-1 preparation, LAL tests were carried out. The endotoxin level was below 3 pg/100 μg rmPGLYRP-1. We investigated whether the low dose of LPS could modulate antilisterial resistance. Mice were injected with 3 pg of LPS 6 h before L. monocytogenes infection. This amount of LPS failed to affect bacterial proliferation in the organs on day 2 of infection (data not shown). LPS is known to be heat stable. Therefore, we assessed whether heat-inactivated rmPGLYRP-1 could modulate cytokine production and antilisterial resistance. Naive mice were injected with heat-inactivated rmPGLYRP-1, and serum IFN-γ and TNF-α levels were determined. The titers of both cytokines were under the detectable level at 24 h (Fig. 3A and B). IFN-γ and TNF-α in the sera also were determined 4 h after injection with heat-inactivated rmPGLYRP-1, and both cytokines were under the detectable level (data not shown). Moreover, mice were injected with heat-inactivated rmPGLYRP-1 6 h before L. monocytogenes infection, and the bacterial number in the spleens and livers was determined on days 1, 2, 3, and 5 after infection. The viable bacterial numbers were similar between heat-inactivated rmPGLYRP-1-treated mice and PBS-treated animals (Fig. 3C and D). The induction of IFN-γ and TNF-α production by heat-inactivated rmPGLYRP treatment was estimated in L. monocytogenes-infected mice. The titers of both cytokines in heat-inactivated rmPGLYRP-1-treated mice were at the same level as that for the PBS-treated group 24 h after infection (Fig. 3E and F). We also assessed the involvement of endotoxin contamination in stimulation with rmPGLYRP-1 in vitro. The production of IFN-γ and TNF-α did not occur when heat-inactivated rmPGLYRP-1 was added (Fig. 5). The enhanced elimination of L. monocytogenes in pEGFP-C2/PGLYRP-1-transfected hepatocytes also excluded the possibility of LPS, because PGLYRP-1 expressed in NMuLi hepatocyte cells caused reduced numbers of L. monocytogenes (Fig. 7). These results suggested that the activation of antilisterial resistance and cytokine induction by rmPRLYRP-1 is not affected under the detectable dose of endotoxin in the recombinant preparation.
DISCUSSION
Enzymatic characteristics of mammalian PGRPs, including structure biological studies, have been investigated (5, 16-19, 26, 49). However, the actual physiological functions of PGRPs remain unclear (8). In this report, we studied the functions of mouse PGLYRP-1 from the point of innate immunity during L. monocytogenes infection.
The expression of the tag7 gene was upregulated in spleens and livers soon after L. monocytogenes infection (Fig. 1A), and PGLYRP-1 was released into the bloodstream beginning 3 h after infection (Fig. 1B). Endogenous PGLYRP-1 reduced bacterial numbers until day 3 of infection (Fig. 2A and B), and the administration of rmPGLYRP-1 before infection enhanced the elimination of L. monocytogenes from the organs until day 5 of infection (Fig. 3C and D). However, the effect of anti-PGLYRP-1 antibody or rmPGLYRP-1 was not observed on day 5 of infection (Fig. 2A and B). CD8+ T cells reportedly are critical in adaptive host defense against L. monocytogenes infection (38). Previous studies of adaptive immune responses to L. monocytogenes infection demonstrated that H2-M3-restricted CD8+ T cells reach high frequencies in the spleen on days 5 to 6, and the number of major histocompatibility complex class I-restricted CD8+ T cells peaks on days 7 to 9 of sublethal infection (38). In this study, the effects of anti-PGLYRP-1 antibody or rmPGLYRP-1 were restricted in the early stage of infection. Neutrophils as well as macrophages play a critical role in host resistance against early-stage L. monocytogenes infection (45). Previous studies reported that the expression of mammalian PGLYRP-1 is restricted to neutrophils (32, 53, 54). Although the accurate cellular source of PGLYRP-1 is not identified in this study, it is possible that macrophages are able to produce PGLYRP-1 as well as neutrophils (unpublished observation). Moreover, we showed that PGLYRP-1 without a secretion signal sequence autonomously activated NMuLi hepatocyte cells (Fig. 7). These results suggest that PGLYRP-1 plays a role in innate host defense against L. monocytogenes infection in the early stage.
PGLYRP-1 is a secretory protein (8, 9, 33), and mammalian PGLYRP-1 has antibacterial activities against Gram-positive bacteria (31-33), suggesting the contribution to protection from bacterial infections by its antibacterial activity (32). In this study, rmPGLYRP-1 showed antibacterial activities against Gram-positive bacteria such as S. aureus, S. epidermidis, and L. monocytogenes but not against Gram-negative bacteria (Fig. 4). These results were almost comparable to those of the previous report (34), although the antibacterial activity of rmPGLYRP-1 against S. aureus was lower than that of human PGLYRP-1 (34) and L. monocytogenes is sensitive to PGLYRP-1 (34). This contradiction may be due to the difference of species or preparation of proteins. The fact that N-glycosylation is required for the bactericidal activity of human PGLYRP-1 (34) might explain the weak activity of rmPGLYRP-1, because the rmPGLYRP-1 used herein was prepared by an E. coli overexpression system and was not N-glycosylated.
In D. melanogaster, PGRP-SA, the homologue of PGLYRP-1, recognizes PGNs through the Toll signaling pathway and induces antibacterial peptide production (6, 39), thus PGLYRP-1 is involved in insect innate immunity. We investigated the possibility that mPGLYRP-1 is involved in cytokine-mediated innate immunity against L. monocytogenes infection. TNF-α and IFN-γ are known to be crucial cytokines in innate immunity against L. monocytogenes infection and following macrophage activation in mice (2, 11, 21, 22, 40-42, 46). TNF-α and IFN-γ production in the spleens and sera was suppressed when endogenous PGLYRP-1 had been neutralized by anti-PGLYRP-1 antibody (Fig. 2C and D), suggesting that PGLYRP-1 is involved in the induction of the production of both cytokines. The inhibition of IFN-γ production was incomplete by anti-PGLYRP-1 antibody treatment (Fig. 2C). This effect might lead to the result that the increase of bacterial numbers in spleens and livers by anti-PGLYRP-1 antibodies was marginal (Fig. 2A and B). Therefore, to ensure that PGLYRP-1 enhanced IFN-γ and TNF-α production, IFN-γ and TNF-α responses were examined after the administration of rmPGLYRP-1. Indeed, TNF-α and IFN-γ production was induced by the administration of rmPGLYRP-1 (Fig. 3A and B), and the production of both cytokines was enhanced by L. monocytogenes infection (Fig. 3E and F). The induction of TNF-α and IFN-γ production by rmPGLYRP-1 also was shown by experiments with spleen cell cultures (Fig. 5 and 6). These results support that PGLYRP-1 is involved in protection from L. monocytogenes infection by inducing TNF-α and IFN-γ production.
We next addressed whether both TNF-α and IFN-γ are critically involved in the enhancement of host resistance against L. monocytogenes infection by PGLYRP-1. The bacterial numbers in spleens and livers 3 days after infection were significantly reduced by the administration of PGLYRP-1 before infection in IFN-γ−/− mice but not in TNF-α−/− mice (Fig. 8), suggesting that TNF-α, but not IFN-γ, induced by PGLYRP-1 mainly contributes to the protection against L. monocytogenes infection. This result is consistent with our previous study that TNF-α is more important in the protection of L. monocytogenes than IFN-γ (40).
It is unclear whether the rmPGLYRP-1 preparation used in this study was correctly refolded and has the same specific activity as native proteins. The heat-treated rmPGLYRP-1 lost the inducing ability of TNF-α and IFN-γ production (Fig. 3E and F and 5C and D), suggesting that rmPGLYRP-1 is active in the nondenatured condition, although the antibacterial activities of rmPGLYRP-1 were lower than those of human PGLYRP-1 because of the possibility of the incomplete refolding and the absence of N-glycosylation (34). We also examined the effect of zinc ions on the antibacterial activities. Zinc is important for antibacterial activities of human PGRPs (34). However, the addition of zinc ions did not cause increased antibacterial activities (Fig. 4D and E), suggesting that rmPGLYRP-1 already contains metal cationic ions such as a zinc ion (34). This suggestion was strongly supported by the addition of EDTA, a metal cation chelator, which suppressed antibacterial activities (Fig. 4D and E). We also showed that rmPGLYRP-1 as prepared herein was a dimer (see Fig. S1 and S3 in the supplemental material). This result supports the idea that the recombinant proteins were recovered in the native form.
The administration of 100 μg of rmPGLYRP-1 may not reflect physiological conditions (Fig. 1B; also see Fig. S3 in the supplemental material). Indeed, the administration of 10 μg of rmPGLYRP-1 failed to affect the bacterial number and cytokine responses in L. monocytogenes infection in mice (data not shown). The administration of antibodies against PGLYRP-1 incompletely inhibited cytokine responses (Fig. 2C and D). These results suggest that an additional mechanism that is involved in protection by PGLYRP-1 from L. monocytogenes infection should be considered (3, 10, 12).
We also noticed endotoxin contamination, because we prepared rmPGLYRP-1 by using an E. coli overexpression system. LPS induces inflammatory cytokine production via the Toll-like receptor 4-NF-κB signaling pathway (3). In addition to LAL tests, which proved no contamination of LPS in rmPGLYRP-1, we showed that heat-inactivated rmPGLYRP-1 lost the ability to induce cytokine production and to modulate antilisterial resistance (Fig. 3 and 5). The enhanced elimination of L. monocytogenes from pEGFP-C2/PGLYRP-1-transfected hepatocytes supports the evidence, because the contamination of LPS can be eliminated in this system (Fig. 7).
Recent studies demonstrated that the expression of PGLYRP-1 was upregulated by transcription factor NF-κB in brain ischemia (28) and that PGLYRP-1 may regulate the inflammatory effect of PGLYRP-2 (46). The next subject of investigation should be to elucidate the precise role of PGLYRP-1 in L. monocytogenes infection. In summary, for the first time we demonstrated that the expression of PGLYRP-1 is induced by L. monocytogenes infection and that PGLYRP-1 plays a role in the protection from L. monocytogenes infection through the induction of TNF-α and IFN-γ, especially TNF-α.
Supplementary Material
Acknowledgments
We thank Yu Sawaya for technical assistance with the experiments.
This work was supported by a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (18659122 to A.N.), Grants-in-Aid for Challenging Exploratory Research from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (21659107 to A.N.), and a grant for Hirosaki University institutional research.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 6 December 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Bischoff, V., et al. 2004. Function of the drosophila pattern-recognition receptor PGRP-SD in the detection of Gram-positive bacteria. Nat. Immunol. 5:1175-1180. [DOI] [PubMed] [Google Scholar]
- 2.Buchmeier, N. A., and R. D. Schreiber. 1985. Requirement of endogenous interferon-γ production for resolution of Listeria monocytogenes infection. Proc. Natl. Acad. Sci. U. S. A. 82:7404-7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carpenter, S., and L. A. O'Neill. 2007. How important are Toll-like receptors for antimicrobial responses? Cell. Microbiol. 9:1891-1901. [DOI] [PubMed] [Google Scholar]
- 4.Chang, C. I., et al. 2005. Structure of the ectodomain of Drosophila peptidoglycan-recognition protein LCa suggests a molecular mechanism for pattern recognition. Proc. Natl. Acad. Sci. U. S. A. 102:10279-10284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho, S., et al. 2007. Structural insights into the bactericidal mechanism of human peptidoglycan recognition proteins. Proc. Natl. Acad. Sci. U. S. A. 104:8761-8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choe, K. M., T. Werner, S. Stöven, D. Hultmark, and K. V. Anderson. 2002. Requirement for a peptidoglycan recognition protein (PGRP) in Relish activation and antibacterial immune responses in Drosophila. Science 296:359-362. [DOI] [PubMed] [Google Scholar]
- 7.Dziarski, R., K. A. Platt, E. Gelius, H. Steiner, and D. Gupta. 2003. Defect in neutrophil killing and increased susceptibility to infection with nonpathogenic gram-positive bacteria in peptidoglycan recognition protein-S (PGRP-S)-deficient mice. Blood 102:689-697. [DOI] [PubMed] [Google Scholar]
- 8.Dziarski, R. 2004. Peptidoglycan recognition proteins (PGRPs). Mol. Immunol. 40:877-886. [DOI] [PubMed] [Google Scholar]
- 9.Dziarski, R., and D. Gupta. 2006. Mammalian PGRPs: novel antibacterial proteins. Cell Microbiol. 8:1059-1069. [DOI] [PubMed] [Google Scholar]
- 10.Fearon, D. T., and R. M. Locksley. 1996. The instructive role of innate immunity in the acquired immune response. Science 272:50-53. [DOI] [PubMed] [Google Scholar]
- 11.Flannagan, R. S., G. Cosío, and S. Grinstein. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7:355-366. [DOI] [PubMed] [Google Scholar]
- 12.Franchi, L., et al. 2008. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell Microbiol. 10:1-8. [DOI] [PubMed] [Google Scholar]
- 13.Gelius, E., C. Persson, J. Karlsson, and H. Steiner. 2003. A mammalian peptidoglycan recognition protein with N-acetylmuramoyl-L-alanine amidase activity. Biochem. Biophys. Res. Commun. 306:988-994. [DOI] [PubMed] [Google Scholar]
- 14.Gottar, M., et al. 2002. The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature 416:640-644. [DOI] [PubMed] [Google Scholar]
- 15.Gregory, S. H., and E. J. Wing. 2002. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J. Leukoc. Biol. 72:239-248. [PubMed] [Google Scholar]
- 16.Guan, R., E. L. Malchiodi, Q. Wang, P. Schuck, and R. A. Mariuzza. 2004. Crystal structure of the C-terminal peptidoglycan-binding domain of human peptidoglycan recognition protein Iα. J. Biol. Chem. 279:31873-31882. [DOI] [PubMed] [Google Scholar]
- 17.Guan, R., et al. 2004. Structural basis for peptidoglycan binding by peptidoglycan recognition proteins. Proc. Natl. Acad. Sci. U. S. A. 101:17168-17173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan, R., Q. Wang, E. J. Sundberg, and R. A. Mariuzza. 2005. Crystal structure of human peptidoglycan recognition protein S (PGRP-S) at 1.70 A resolution. J. Mol. Biol. 347:683-691. [DOI] [PubMed] [Google Scholar]
- 19.Guan, R., et al. 2006. Crystal structure of human peptidoglycan recognition protein Iα bound to a muramyl pentapeptide from Gram-positive bacteria. Protein Sci. 15:1199-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan, R., and R. A. Mariuzza. 2007. Peptidoglycan recognition proteins of the innate immune system. Trends Microbiol. 15:127-134. [DOI] [PubMed] [Google Scholar]
- 21.Havell, E. A. 1989. Evidence that tumor necrosis factor has an important role in antibacterial resistance. J. Immunol. 143:2894-2899. [PubMed] [Google Scholar]
- 22.Huang, S., et al. 1993. Immune response in mice that lack the interferon-γ receptor. Science 259:1742-1745. [DOI] [PubMed] [Google Scholar]
- 23.Kang, D., G. Liu, A. Lundström, E. Gelius, and H. Steiner. 1998. A peptidoglycan recognition protein in innate immunity conserved from insects to humans. Proc. Natl. Acad. Sci. U. S. A. 95:10078-10082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim, M. S., M. Byun, and B. H. Oh. 2003. Crystal structure of peptidoglycan recognition protein LB from Drosophila melanogaster. Nat. Immunol. 4:787-793. [DOI] [PubMed] [Google Scholar]
- 25.Kiselev, S. L., et al. 1998. Molecular cloning and characterization of the mouse tag7 gene encoding a novel cytokine. J. Biol. Chem. 273:18633-18639. [DOI] [PubMed] [Google Scholar]
- 26.Kumar, S., et al. 2005. Selective recognition of synthetic lysine and meso-diaminopimelic acid-type peptidoglycan fragments by human peptidoglycan recognition proteins Iα and S. J. Biol. Chem. 280:37005-37012. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 28.Lang, M. F., et al. 2008. Peptidoglycan recognition protein-S (PGRP-S) is upregulated by NF-κB. Neurosci. Lett. 2430:138-141. [DOI] [PubMed] [Google Scholar]
- 29.Leone, P., et al. 2008. Crystal structure of Drosophila PGRP-SD suggests binding to DAP-type but not lysine-type peptidoglycan. Mol. Immunol. 45:2521-2530. [DOI] [PubMed] [Google Scholar]
- 30.Leulier, F., et al. 2003. The Drosophila immune system detects bacteria through specific peptidoglycan recognition. Nat. Immunol. 4:478-484. [DOI] [PubMed] [Google Scholar]
- 31.Li, X., S. Wang, H. Wang, and D. Gupta. 2006. Differential expression of peptidoglycan recognition protein 2 in the skin and liver requires different transcription factors. J. Biol. Chem. 281:20738-20748. [DOI] [PubMed] [Google Scholar]
- 32.Liu, C., E. Gelius, G. Liu, H. Steiner, and R. Dziarski. 2000. Mammalian peptidoglycan recognition protein binds peptidoglycan with high affinity, is expressed in neutrophils, and inhibits bacterial growth. J. Biol. Chem. 275:24490-24499. [DOI] [PubMed] [Google Scholar]
- 33.Liu, C., Z. Xu, D. Gupta, and R. Dziarski. 2001. Peptidoglycan recognition proteins: a novel family of four human innate immunity pattern recognition molecules. J. Biol. Chem. 276:34686-34694. [DOI] [PubMed] [Google Scholar]
- 34.Lu, X., et al. 2006. Peptidoglycan recognition proteins are a new class of human bactericidal proteins. J. Biol. Chem. 281:5895-5907. [DOI] [PubMed] [Google Scholar]
- 35.Mathur, P., et al. 2004. Murine peptidoglycan recognition proteins PglyrpIα and PglyrpIβ are encoded in the epidermal differentiation complex and are expressed in epidermal and hematopoietic tissues. Genomics 83:1151-1163. [DOI] [PubMed] [Google Scholar]
- 36.Mellroth, P., J. Karlsson, and H. Steiner. 2003. A scavenger function for a Drosophila peptidoglycan recognition protein. J. Biol. Chem. 278:7059-7064. [DOI] [PubMed] [Google Scholar]
- 37.Mellroth, P., and H. Steiner. 2006. PGRP-SB1: an N-acetylmuramoyl L-alanine amidase with antibacterial activity. Biochem. Biophys. Res. Commun. 350:994-999. [DOI] [PubMed] [Google Scholar]
- 38.Messingham, K. A. A., and J. T. Harty. 2007. Adaptive immunity to Listeria monocytogenes, p. 225-249. In H. Goldfine and H. Shen (ed.), Listeria monocytogenes: pathogenesis and host response. Springer, New York, NY.
- 39.Michel, T., J. M. Reichhart, J. A. Hoffmann, and J. Royet. 2001. Drosophila Toll is activated by Gram-positive bacteria through a circulating peptidoglycan recognition protein. Nature 414:756-759. [DOI] [PubMed] [Google Scholar]
- 40.Mizuki, M., A. Nakane, K. Sekikawa, Y. I. Tagawa, and Y. Iwakura. 2002. Comparison of host resistance to primary and secondary Listeria monocytogenes infections in mice by intranasal and intravenous routes. Infect. Immun. 70:4805-4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakane, A., T. Minagawa, and K. Kato. 1988. Endogenous tumor necrosis factor (cachectin) is essential to host resistance against Listeria monocytogenes infection. Infect. Immun. 56:2563-2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfeffer, K., et al. 1993. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73:457-467. [DOI] [PubMed] [Google Scholar]
- 43.Rehman, A., P. Taishi, J. Fang, J. A. Majde, and J. M. Krueger. 2001. The cloning of a rat peptidoglycan recognition protein (PGRP) and its induction in brain by sleep deprivation. Cytokine 13:8-17. [DOI] [PubMed] [Google Scholar]
- 44.Reiser, J. B., L. Teyton, and I. A. Wilson. 2004. Crystal structure of the Drosophila peptidoglycan recognition protein (PGRP)-SA at 1.56 Å resolution. J. Mol. Biol. 340:909-917. [DOI] [PubMed] [Google Scholar]
- 45.Rogers, H. W., and E. R. Unanue. 1993. Neutrophils are involved in acute, nonspecific resistance to Listeria monocytogenes in mice. Infect. Immun. 61:5090-5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rothe, J., et al. 1993. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 364:798-802. [DOI] [PubMed] [Google Scholar]
- 47.Royet, J., and R. Dziarski. 2007. Peptidoglycan recognition proteins: pleiotropic sensors and effectors of antimicrobial defences. Nat. Rev. Microbiol. 5:264-277. [DOI] [PubMed] [Google Scholar]
- 48.Saha, S., et al. 2009. PGLYRP-2 and Nod2 are both required for peptidoglycan-induced arthritis and local inflammation. Cell Host Microbe 5:137-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma, P., et al. 2008. Crystal structure of the peptidoglycan recognition protein at 1.8 Å resolution reveals dual strategy to combat infection through two independent functional homodimers. J. Mol. Biol. 378:921-930. [DOI] [PubMed] [Google Scholar]
- 50.Tagawa, Y., K. Sekikawa, and Y. Iwakura. 1997. Suppression of concanavalin A-induced hepatitis in IFN-γ−/− mice, but not in TNF-α−/− mice: role for IFN-γ in activating apoptosis of hepatocytes. J. Immunol. 159:1418-1428. [PubMed] [Google Scholar]
- 51.Takehana, A., et al. 2004. Peptidoglycan recognition protein (PGRP)-LE and PGRP-LC act synergistically in Drosophila immunity. EMBO J. 23:4690-4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taniguchi, T., M. Takata, A. Ikeda, E. Momotani, and K. Sekikawa. 1997. Failure of germinal center formation and impairment of response to endotoxin in tumor necrosis factor alpha-deficient mice. Lab. Investig. 77:647-658. [PubMed] [Google Scholar]
- 53.Tydell, C. C., N. Yount, D. Tran, J. Yuan, and M. E. Selsted. 2002. Isolation, characterization, and antimicrobial properties of bovine oligosaccharide-binding protein. A microbicidal granule protein of eosinophils and neutrophils. J. Biol. Chem. 277:19658-19664. [DOI] [PubMed] [Google Scholar]
- 54.Tydell, C. C., J. Yuan, P. Tran, and M. E. Selsted. 2006. Bovine peptidoglycan recognition protein-S: antimicrobial activity, localization, secretion, and binding properties. J. Immunol. 176:1154-1162. [DOI] [PubMed] [Google Scholar]
- 55.Wang, Z. M., et al. 2003. Human peptidoglycan recognition protein-L is an N-acetylmuramoyl-L-alanine amidase. J. Biol. Chem. 278:49044-49052. [DOI] [PubMed] [Google Scholar]
- 56.Werner, T., et al. 2000. A family of peptidoglycan recognition proteins in the fruit fly Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 97:13772-13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Werner, T., K. Borge-Renberg, P. Mellroth, H. Steiner, and D. Hultmark. 2003. Functional diversity of the Drosophila PGRP-LC gene cluster in the response to lipopolysaccharide and peptidoglycan. J. Biol. Chem. 278:26319-26322. [DOI] [PubMed] [Google Scholar]
- 58.Xu, M., Z. Wang, and R. M. Locksley. 2004. Innate immune responses in peptidoglycan recognition protein L-deficient mice. Mol. Cell. Biol. 24:7949-7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yano, T., et al. 2008. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat. Immunol. 9:908-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin, J., and T. A. Ferguson. 2009. Identification of an IFN-γ-producing neutrophil early in the response to Listeria monocytogenes. J. Immunol. 182:7069-7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshida, H., K. Kinoshita, and M. Ashida. 1996. Purification of a peptidoglycan recognition protein from hemolymph of the silkworm, Bombyx mori. J. Biol. Chem. 271:13854-13860. [DOI] [PubMed] [Google Scholar]
- 62.Zhang, Y., et al. 2005. Identification of serum N-acetylmuramoyl-l-alanine amidase as liver peptidoglycan recognition protein 2. Biochim. Biophys. Acta 1752:34-46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.