

Abstract
Clostridium perfringens TpeL belongs to a family of large clostridial cytotoxins that encompasses Clostridium difficile toxin A (TcdA) and B (TcdB) and Clostridium sordellii lethal toxin (TcsL). We report here the identification of the TpeL-catalyzed modification of small GTPases. A recombinant protein (TpeL1-525) derived from the TpeL N-terminal catalytic domain in the presence of streptolysin O (SLO) induced the rounding of Vero cells and the glycosylation of cellular Rac1. Among several hexoses tested, UDP-N-acetyl-glucosamine (UDP-GlcNAc) and UDP-glucose (UDP-Glc) served as cosubstrates for TpeL1-525-catalyzed modifications. TpeL1-525 catalyzed the incorporation of UDP-Glc into Ha-Ras, Rap1B, and RalA and of UDP-GlcNAc into Rac1, Ha-Ras, Rap1B, and RalA. In Rac1, TpeL and TcdB share the same acceptor amino acid for glycosylation, Thr-35. In Vero cells treated with TpeL1-525 in the presence of SLO, glycosylation leads to a translocation of the majority of Rac1 and Ha-Ras to the membrane. We demonstrate for first time that TpeL uses both UDP-GlcNAc and UDP-Glc as donor cosubstrates and modifies the Rac1 and Ras subfamily by glycosylation to mediate its cytotoxic effects.
Clostridium perfringens type C has been identified as a causative agent of necrotizing enterocolitis associated with diarrhea and dysentery in infant animals (20, 23). In humans, the bacteria cause necrotic enteritis, which is termed “pig-bel” (20). Type C strains produce alpha-toxin, beta-toxin, beta2-toxin, and perfringolysin O. Beta-toxin is lethal, cytotoxic, and related to the pathogenicity of C. perfringens type C (20). Beta2-toxin is also considered important for pathogenicity (22). Furthermore, many type C isolates produce a newly discovered toxin named TpeL, which is a truncated homologue of Clostridium difficile TcdA and TcdB (1). TpeL was identified in the culture supernatant of C. perfringens strain CP4 and is thought to be associated with necrotic enteritis (1).
TpeL is cytotoxic, causing cell rounding (1). The molecular mass calculated from the deduced amino acid sequence was 191 kDa, and a signal peptide region was not found within the open reading frame (1). The deduced amino acid sequence exhibited 30 to 39% homology to Clostridium difficile toxins A (TcdA) and B (TcdB), Clostridium sordellii lethal toxin (TcsL), and Clostridium novyi alpha-toxin (TcnA). All of these toxins are called large clostridial toxins (LCTs) (1). At least four domains, “ABCD,” can be distinguished in LCTs. Respectively, they are putatively involved in N-terminal biological activity (A-domain), C-terminal receptor binding (B-domain), autoproteolytic cleavage during toxin-processing (C-domain), and delivery of the A-domain into the cytosol (D-domain) (2). LCTs bind with their B-domain to the membrane receptor of host cells (2, 24). After endocytosis, the toxin inserts into the endosome membrane, most likely via the D-domain. Cellular inositolhexaphosphate (InsP6) activates the protease C-domain (7, 18). This results in cleavage of the toxin and release of the A-domain into the cytosol. In the cytosol, small GTPases are glycosylated and thereby inactivated. The amino acid sequence of TpeL is shorter than that of other LCTs, and the homologous region was located at the N terminus (1). A DxD motif in LCTs is essential for glycosyltransferase activity, and the amino acid W102 in TcsL is needed for the enzymatic activity (5, 6). The DXD motif and W102 of TcsL (1) are conserved in TpeL. However, the C-terminal carbohydrate-binding sites of LCTs are not (2, 24), and it is not known how TpeL binds to its receptor. LCTs monoglycosylate the small GTPases TcdA, TcdB, and TcnA glycosylate Rho, Rac, and Cdc42, whereas TcsL modifies Rac, Ras, Ral, and Rap (24). TcdA, TcdB, and TcsL use UDP-Glc as a donor for glycosylation of small GTPase (2, 24). In contrast, TcnA N-acetylglucosaminylates small GTPases by using UDP-GlcNAc, indi- cating that most clostridial toxins utilize UDP-Glc as a cosubstrate with the exception of the TcnA, which uses UDP-GlcNAc (21). Recently, the crystal structure of the catalytic domain of TcdB was solved (19).
Our understanding of the biological activity of TpeL remains incomplete. Native TpeL is labile and is difficult to purify from the culture supernatant of C. perfringens type C. Furthermore, since the recombinant full-length TpeL was poorly expressed in Escherichia coli, full-length TpeL is not available. To clarify the catalytic activity of TpeL, we prepared a recombinant glycosyltransferase domain, TpeL1-525 (covering amino acids 1 to 525). To transport enzyme domain into cells, we used the streptolysin O (SLO) delivery systems (25). Here, we demonstrate that among LCTs, TpeL can utilize both UDP-Glc and UDP-GlcNAc as cosubstrates and mainly glycosylates the Ras subfamily.
MATERIALS AND METHODS
Materials.
UDP-GalNAc, UDP-Man, UDP-GlcNAc, UDP-Glc, UDP-Gal, and SLO were obtained from Sigma (St. Louis, MO). UDP-[14C]Glc and UDP-[14C]GlcNAc were obtained from Perkin-Elmer Life Sciences (Boston, MA). The anti-Rac1 (Mab23A8), Rac1 (Mab102), RhoA, Cdc-42, and Ras antibodies were from Millipore (Billerica, MA), BD biosciences (Franklin Lakes, NJ), Santa Cruz (Santa Cruz, CA), BD biosciences, and Cell signaling (Danvers, MA), respectively. Horseradish peroxidase-labeled anti-rabbit immunoglobulin G (IgG), horseradish peroxidase-labeled anti-mouse IgG, and an enhanced chemiluminescence kit were obtained from GE Healthcare (Tokyo, Japan). Dulbecco modified Eagle medium (DMEM) was purchased from Gibco-BRL (New York, NY). C. difficile TcdB was purchased from List Biol. Lab. (Campbell, CA). Plasmids encoding GST-RhoA, GST-Rac1, GST-Cdc42, and GST-Ha-Ras were provided by Y. Horiguchi (Osaka University) and plasmids encoding GST-RalA and GST-Rap1B, by M. Matsuda (Kyoto University). A monoclonal anti-TpeL antibody was prepared as previously described (1). All other chemicals were of the highest grade available from commercial sources.
Construction of TpeL1-525.
To prepare the catalytic domain of TpeL, we constructed TpeL1-525, from which 1,125 amino acids have been from the C terminus of TpeL (residues 526 to 1,651), by a PCR using the DNA of the C. perfringens type C strain MC18 (1), a forward primer (5′-AGGGGATCCATGGGGTTAATGTCAAAAGAA-3′) encoding a BamHI site (indicated in boldface), and a reverse primer (5′-TCCCTCGAGTTATTTAACTTGATTAAATGTCCA-3′) encoding a downstream XhoI restriction site (indicated in boldface). PCR products were digested with BamHI and XhoI and ligated into BamHI- and XhoI-digested pGEX-4T-1 (GE Healthcare) so that the correct reading frame was maintained with the thrombin cleavage site under the glutathione S-transferase (GST) gene (pGEX-TpeL1-525). The construct was checked by sequencing with an ABI Prism dye terminator cycle sequencing ready reaction kit and ABI 310 cycle sequencer (Life Technologies, Carlsbad, CA).
Expression and purification of TpeL1-525.
The recombinant TpeL1-525 was expressed as a protein fused with GST in E. coli BL21 as described previously (15). After growth (at 30°C) and induction (with IPTG [isopropyl-β-d-thiogalactopyranoside] to 0.5 mM) of a large culture, the cells were centrifuged and disrupted by a sonicator on ice in a short burst. Centrifugation of the lysate and passage of the soluble fraction over a glutathione-Sepharose (GE Healthcare) column yielded the GST-TpeL1-525 fusion protein at ∼2 mg/liter. The purified GST-TpeL1-525 was cleaved with thrombin, passed through the glutathione-Sepharose column, and then subjected to anion-exchange chromatography (Mono-Q HR10/10; GE Healthcare). The protein was eluted with a 0 to 1.0 M NaCl gradient. Fractions containing TpeL1-525 were collected. The purified proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1). The protein assay was performed by the method of Bradford (4).
FIG. 1.
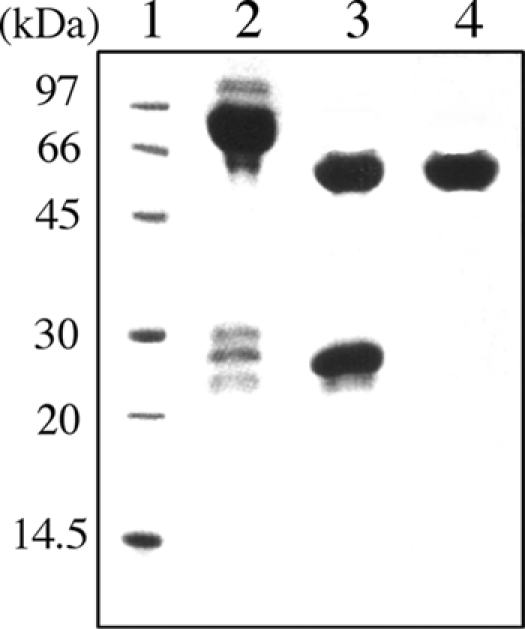
SDS-PAGE analysis of purified TpeL1-525. Lanes: 1, molecular mass standards; 2, GST-TpeL1-525 (7.5 μg); 3, thrombin-digested GST-TpeL1-525 (7.5 μg); 4, purified TpeL1-525 (7.5 μg).
Assay of cytotoxicity.
Vero cells were obtained from the Riken Cell Bank (Tsukuba, Japan). They were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 U of penicillin/ml, 100 μg of streptomycin/ml, and 2 mM glutamine (FCS-DMEM). All incubation steps were performed at 37°C in a 5% CO2 atmosphere. The test for cytotoxicity was done with Vero cells. Cells in 48-well plates were incubated with 0.2 ml of Hanks buffered salt solution (HBSS) without Ca2+ containing 30 mM HEPES (pH 7.2), for 15 min at 37°C. SLO (100 ng/ml) was then added together with various concentrations of TpeL1-525 as indicated for 15 min at 37°C and 5% CO2. To reseal, 0.5 ml of ice-cold HBSS containing 30 mM HEPES and 2 mM Ca2+ (pH 7.2), was added. After incubation for 1 h at 37°C, HBSS was replaced with full growth medium (25). The cells were observed for morphological alterations 4 h after inoculation by phase-contrast microscopy. For cytotoxicity assays, the cells (5 × 104) were inoculated in 96-well plates. Cell viability was determined by using a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt conversion assay (Promega, Madison, WI). The absorbance, which is proportional to the number of viable cells, was read at 490 nm by using an enzyme-linked immunosorbent assay (ELISA) plate reader, using the reference wavelength of 630 nm. The cell viability of a toxin group was calculated as the percentage compared to an untreated control, which was calculated by comparing the mean absorbance value of a toxin group to that of an untreated control for three determinations (16).
Toxin treatment and lysis of cells.
Cell cultures of 6-cm dishes were treated with SLO only or SLO plus TpeL1-525 at 37°C for 15 min. After 120 min of resealing, the cells were rinsed with 5 ml of ice-cold phosphate-buffered saline and scraped off in 300 μl of lysis buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 2.5 mM MgCl2, 40 μg of aprotinin/ml, 0.1 mM phenylmethylsulfonyl fluoride, 20 μg of leupeptin/ml, 80 μg of benzamidine/ml) per dish. The cells were disrupted mechanically by sonication (five times on ice) and then centrifuged for 10 min at 1,000 × g to remove the nuclear fraction and intact cells. The supernatant (1 mg/ml) was used as the cell lysate (8).
Fractionation of cell lysates.
Lysates were centrifuged at 100,000 × g for 1 h to prepare cytosolic and total particulate fractions. The high-speed pellet, which consists of membrane fractions, was washed with lysis buffer and resuspended in the original volume of lysis buffer (8).
Immunoblotting.
Proteins were separated on 12.5% polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Millipore) for 2 h at 250 mA. This was followed by blocking with Tris-buffered saline containing 5% (wt/vol) bovine serum albumin and 1% Tween 20 (blocking buffer) for 1 h. Blots were incubated for 2 h with the appropriate primary antibody in the blocking buffer, then for 1 h with a horseradish peroxidase-conjugated secondary antibody, and finally with the reagents from an enhanced chemiluminescence analysis kit. The intensity of the bands was quantified with a densitometer (Atto Co., Ltd., Tokyo, Japan).
Preparation of recombinant GTP-binding proteins.
RhoA, Cdc42, Rac1, Ha-Ras, RalA, and Rap1B were prepared from their fusion proteins (e.g., RhoA-GST). GST fusion proteins from the E. coli expression vector pGEX-2T were isolated by affinity purification with GST-Sepharose (GE Healthcare). GTP-binding proteins were then cleaved from the GST fusion proteins by thrombin treatment (100 μg/ml for 60 min at 22°C). The thrombin was removed by binding to benzamide-Sepharose, and the GTP-binding proteins were concentrated with Centricon (Millipore).
Glycosylation reaction.
Recombinant GTP-binding proteins (50 μg/ml) or cell lysates were incubated with TpeL1-525 (20 μg/ml) or TcdB (10 μg/ml) in a buffer containing 30 μM UDP-[14C]GlcNAc or 30 μM UDP-[14C]Glc, 2 mM MgCl2, 150 mM KCl, and 50 mM HEPES (pH 7.4) for the periods indicated at 37°C (21). Labeled proteins were subjected to SDS-PAGE and autoradiographed with a FLA-2000 system (Fuji Photo Film Co., Ltd., Tokyo, Japan).
RESULTS
Cytotoxic effect of TpeL1-525.
TpeL was toxic to Vero cells (1). We tested the cytotoxicity of the recombinant TpeL1-525, a glycosyltransferase domain of TpeL, to Vero cells. TpeL1-525 alone was without any cytotoxic effects (Fig. 2C). This finding indicates that TpeL induces cytotoxic effects through the binding of its C-terminal region to cell surface receptors. To confirm this, TpeL1-525 was transported into Vero cells using the pore-forming toxin SLO as a delivery system (25). As shown in Fig. 2B, in the presence of SLO, TpeL1-525 caused the rounding of cells like the native toxin as reported previously (1). TpeL1-525 at 1 to 10 μg/ml in the presence of SLO induced cell rounding in a dose-dependent manner (Fig. 2C). On the other hands, The cells eventually detached from the well. Furthermore, when TpeL1-525 at a concentration of 1 to 10 μg/ml was delivered to the cells by SLO, cell viability decreased in a dose-dependent manner (data not shown). The cytotoxicity induced by TpeL1-525 was completely neutralized by a monoclonal anti-TpeL antibody and heat-inactivated TpeL1-525 did not induce cell rounding.
FIG. 2.

Morphological changes of Vero cells upon treatment with SLO plus TpeL1-525. Vero cells were incubated with SLO (100 ng/ml) alone (A) or a combination of TpeL1-525 (20 μg/ml) with SLO (100 ng/ml) (B) at 37°C for 15 min. Pictures were taken after 120 min of resealing. (C) Vero cells were incubated with various amounts of TpeL alone (○) or a combination of various amounts of TpeL1-525 with SLO (100 ng/ml) (•) at 37°C for 15 min. After 120 min of resealing, pictures of cells were taken and the percentage of rounded cells was determined. Values of three experiments were given a mean ± the standard deviation (SD).
Glycosylation of Rac1.
Rac1 is the only substrate GTPase inactivated by all LCTs. We investigated whether TpeL1-525 in the presence of SLO glycosylates Rac1 in Vero cells. Genth et al. (9) reported that the recognition of Rac1 by anti-Rac1 (Mab102) is impaired by the glycosylation of Thr-35 in Rac1. The intracellular level of unmodified Rac1 in Vero cells treated with TpeL1-525 in the presence of SLO was determined using anti-Rac1 (Mab102) (Fig. 3). TpeL1-525 caused a decrease in the cellular level of unmodified Rac1 (Fig. 3A). In addition, a quantitative densitometric analysis indicated that TpeL1-525 in the range of 1 to 10 μg/ml reduced the relative band intensities applying the antibody Rac1 (Mab102) in a dose-dependent manner (Fig. 3B). We applied an alternative anti-Rac1 (Mab23A8) recognizing glycosylated and unglycosylated Rac1 (total Rac1) to analyze cellular Rac1 levels. As shown in Fig. 3A, cellular Rac1 levels did not decrease in the cells treated with TpeL1-525 in the presence of SLO. These observations demonstrated that the decreasing levels of Rac1 in Vero cells treated with TpeL1-525 in the presence of SLO observed with anti-Rac1(Mab102) were due to impaired recognition by anti-Rac1 (Mab102), suggesting that TpeL1-525 glycosylates Rac1 in Vero cells.
FIG. 3.

TpeL1-525-induced glycosylation of Rac1 in Vero cells. (A) Vero cells were incubated with SLO (100 ng/ml) alone or a combination of various amounts of TpeL1-525 with SLO (100 ng/ml) at 37°C for 15 min. After 120 min of resealing, the cells were washed and directly lysed in SDS-sample buffer. The level of Rac1 was analyzed by SDS-PAGE and Western blotting with the anti-Rac1 antibodies Mab102 and Mab23A8. (B) Signal intensities from immunoblots were recorded densitometrically. For quantification, Rac1 signals applying the glycosylation-sensitive antibody Rac1(Mab102) were normalized to the Rac1 signals applying the antibody Rac1 (Mab23A8). The Rac1 level of untreated cells was set to 1. The data shown represent the mean of three independent experiments.
Substrate specificity.
LCTs use UDP-Glc or UDP-GlcNAc as a cosubstrate (2). The specificity of TpeL1-525 was examined by incubating UDP-[14C]Glc or UDP-[14C]GlcNAc and TpeL1-525 with various GTPases. As shown in Fig. 4, Ha-Ras, RalA and Rap1B, the members of the Ras subfamily, were substrates for TpeL1-525-catalyzed glucosylation, whereas other GTPases of the Rho subfamily, namely, RhoA, Cdc42, and Rac1, were not modified in vitro by TpeL1-525 in the presence of UDP-[14C]Glc. As illustrated in Fig. 4, Rac1, Ha-Ras, RalA, and Rap1B were N-acetylglucosaminylated by TpeL1-525, but RhoA and Cdc42 were not. Heat denaturation of either GTPases or TpeL1-525 completely inhibited glycosylation and N-acetylglucosaminylation (data not shown), indicating that the native protein structure is essential for this type of modification. Thus, TpeL1-525 modifies the same recombinant substrates as do TcsL from C. sordellii and TcdB-1470 from C. difficile 1470 (13). To investigate the type of sugar incorporated into Ha-Ras and Rac1, several UDP-hexoses were tested. Because Ha-Ras was incubated with TpeL1-525 and UDP-[14C]Glc in the presence of a 50-fold molar excess of various unlabeled UDP-hexoses, UDP-GlcNAc and UDP-Glc inhibited the TpeL-catalyzed incorporation of [14C]Glc into Ha-Ras by ca. 90 and 95%, respectively (Fig. 5A). [14C]N-acetylglucosamination of Rac1 with TpeL1-525 was inhibited by UDP-GlcNAc (Fig. 5B). Other UDP-hexoses did not inhibit the glycosylation activity of TpeL1-525. Taken together, these results indicated that TpeL is a glycosyltransferase that utilizes UDP-Glc and UDP-GlcNAc as cosubstrates.
FIG. 4.

TpeL1-525-induced glycosylation of small GTPases. Small GTPases (50 μg/ml) were incubated with TpeL1-525 (20 μg/ml) in the presence of UDP-[14C]Glc or UDP-[14C]GlcNAc at 37°C for 120 min. Labeled proteins were analyzed by SDS-PAGE and autoradiography.
FIG. 5.

Glycosyltransferase activity of TpeL1-525. Influence of various nucleotide-hexoses on the TpeL1-525-induced glycosylation of small GTPases. Ha-Ras or Rac1 was incubated with TpeL1-525 (20 μg/ml) and UDP-[14C]Glc (A) or UDP-[14C]GlcNAc (B), respectively, in the presence of various nucleotide-hexoses at 37°C for 120 min. Labeled proteins were analyzed by SDS-PAGE and autoradiography. The amount of glycosylation was calculated as the percentage of the untreated control using densitometric analysis of the autoradiography. One representative experiment from three is shown. (C) Acceptor amino acid. Rac1 was glycosylated with either TpeL1-525 (20 μg/ml) or TcdB (5 μg/ml) in the presence of 30 μM UDP-[14C]GlcNAc (lane 1) or 30 μM UDP-[14C]Glc (lane 2). For sequential glycosylation, Rac1 was glycosylated with TpeL1-525 (lane 3) in the presence of 30 μM unlabeled UDP-GlcNAc for 60 min at 37°C and then with TcdB in the presence of 30 μM UDP-[14C]Glc. In lane 4, glycosylation with TcdB and 30 μM unlabeled UDP-Glc was followed by a second glycosylation with 30 μM UDP-[14C]GlcNAc and TpeL1-525. Labeled proteins were analyzed by SDS-PAGE and autoradiography.
Acceptor amino acid.
To test whether TpeL uses the same acceptor site as TcdB from C. difficile, sequential glycosylation was performed. The modification of Rac1 with TpeL1-525 in the presence of unlabeled UDP-GlcNAc, followed by a second glycosylation in the presence of UDP-[14C]Glc and TcdB resulted in blocked incorporation of [14C]Glc (Fig. 5C). The same was true when the first glycosylation was performed with TcdB and UDP-Glc, followed by a second glycosylation in the presence of UDP-[14C]GlcNAc and TpeL1-525. These results indicated that TpeL shares the same acceptor amino acid in Rac1, namely, Thr-35 (13).
Membrane binding of glycosylated GTPase.
We investigated whether the glycosylation of cellular small GTPases occurred in the cells treated with TpeL1-525 in the presence of SLO. It was reported that GTPases glycosylated by LCTs are accumulated in the cell membranes (2, 8). Therefore, we studied the membrane binding of glycosylated GTPases. After incubation of the cells with SLO alone or SLO plus TpeL1-525, the membranes were prepared from the cells. The membrane levels of Rac1, Ha-Ras, RhoA, and Cdc42 were determined by using anti-Rac1 (Mb23A8), anti-Ha-Ras, anti-RhoA, and anti-Cdc42 antibodies, respectively. As shown in Fig. 6, Rac1 and Ha-Ras in the membranes increased in TpeL1-525-treated cells in the presence of SLO compared to the level in the cells treated with SLO alone. TpeL1-525 caused a 2- to 3-fold increase in membrane levels of Rac1 and Ha-Ras. RhoA and Cdc42 levels were not altered by SLO plus TpeL1-525.
FIG. 6.

Effect of SLO and SLO plus TpeL1-525 on binding of small GTPases to plasma membranes. Vero cells were incubated with SLO (100 ng/ml) alone or in combination with TpeL1-525 (20 μg/ml) at 37°C for 10 min. After 120 min of resealing, the lysate (1 mg/ml) was fractionated into cytosolic and membranous fractions. The membranous fractions were subjected to SDS-PAGE and Western blotting with various anti-small GTPase antibodies. For quantitative analysis, small-GTPase signals were quantified densitometrically. Small GTPase level of untreated cells was set to 1. One representative experiment from three is shown.
DISCUSSION
TpeL is the first LCT shown to utilize UDP-GlcNAc and UDP-Glc to modify small GTP-binding proteins. The substrate specificity of TpeL is similar to that of C. sordellii TcsL. TpeL glycosylates Ha-Ras, RalA, Rap1B, and Rac1.
TpeL1-525 in the presence of SLO induced cell rounding and the detachment of cells from the dish. The cytotoxicity was inhibited by the anti-TpeL antibody, and heat-inactivated TpeL1-525 was not cytotoxic. On the other hand, TpeL1-525 by itself did not have the cytotoxic effects. The results indicated that the N- and C-terminal regions of TpeL plays a role in the cytotoxicity and the C-terminal region is responsible for the binding of cells. The morphological alteration of cultured cells induced by TpeL is similar to that caused by TcdB and TcsL (14, 17). When the cells were treated with TpeL in the presence of SLO, glycosylation of cellular Rac1 was confirmed by Western blotting with the glycosylation-sensitive anti-Rac1 (Mab102) (9). TpeL and TcsL (24) act on Rac1 and the Ras subfamily but not RhoA. Furthermore, the isomeric TcdB from the variant C. difficile serotype F strain 1470 (TcdBF) that glucosylates Rac1 but not RhoA, has a cytopathic effect (13). Halabi-Cabezon et al. (10) reported that the glucosylation of Rac1 rather than RhoA correlates with the cytopathic effect of TcdB. It has been reported that Rac1 plays a critical role in the organization of the actin cytoskeleton (3). These results strongly suggest that glycosylation of Rac1 is critical for the cytopathic effect of TpeL.
TpeL uses UDP-Glc and UDP-GlcNAc as sugar donors. All other LCTs use a single UDP-hexose (2). The crystal structure provides evidence that two amino acids in the vicinity of the catalytic cleft are responsible for the specificity (11). TcdA and TcdB, which both use UDP-Glc, have isoleucine and glutamine in the equivalent positions (Ile-383 and Gln-385 in TcdB), whereas TcnL, which uses UDP-GlcNAc, has serine and alanine residues at the respective positions (11). It has been reported that the bulkier side chains of Ile-383 and Gln-385 in TcdB limit the space of the catalytic cleft for the binding of UDP-GlcNAc and the exchange of these side chains with smaller groups changes the cosubstrate specificity from UDP-Glc to UDP-GlcNAc (11). TpeL has the smaller side chain Ala-383 and the bulkier side chain Gln-385 at the respective positions (1). We speculate that Ala-383 and Gln-385 in TpeL may stabilize the binding of UDP-Glc and UDP-GlcNAc and favor the acceptance of UDP-Glc and UDP-GlcNAc as the cosubstrates.
The sequential glycosylation of Rac1 by TpeL followed by TcdB, and vice versa indicates that both toxins share the same acceptor amino acid in Rac1. The acceptor amino acid of TcdB-glycosylated Rac1 has been determined as Thr-35 (2, 13). TpeL inactivates Rac1 through the glycosylation of Thr-35.
TpeL glycosylated Rac1, as well as the Ras subfamily consisting of Ha-Ras, RalA, and Rap1B, but not RhoA and Cdc42. Important differences in substrate specificity have been detected among the various LCTs. Whereas TcdA, TcdB, and TcnA modify most RhoA, Rac1, and Cdc42 isoforms, TcsL glucosylates Rac1 but not RhoA or Cdc42 (24). On the other hand, TcsL also modifies the Ras subfamily, including Ras, Rap, and Ral isoforms (24). Thus, TpeL modifies similar substrates to TcsL. It was reported that Arg-455, Asp-461, Lys-463, and Glu-472 and residues of helix α17 (e.g., Glu-449) of TcdB are essential for enzyme-RhoA recognition (12). Changing the respective amino acid residues in TcsL to those of TcdB reduced glycosylation of Ras by TcsL (12). Furthermore, the introduction of helix α17 of TcdB into TcsL caused a reduction in the glycosylation of Ras subfamily proteins but permitted the glycosylation of RhoA, indicating that helix α17 is involved in RhoA's recognition by TcdB (12). Glu-449, Lys-463, and Glu-472 in TcdB correspond to Lys, Arg, and Gly residues in TcsL and TpeL. Arg-455 in TcdB corresponds to Lys in TcsL and Gly in TpeL (1). The difference in those amino acid residues may be involved in recognizing small GTPases by TpeL. Additional residues in LCTs are needed for the recognition of small GTPases.
In conclusion, TpeL from C. perfringens has been identified as a glycosyltransferase using UDP-GlcNAc and UDP-Glc as cosubstrates. The substrates of TpeL are confined to Rac1 and Ras subfamily proteins. The modification of Thr-35 on Rac1 induces cytopathic effects.
Acknowledgments
We thank T. Tateishi and M. Satoh for technical assistance.
This study was supported by a grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; MEXT.SENRYAKU, 2009, and MEXT.HAITEKU, 2003-2007; and the Open Research Center Program of MEXT.
Editor: J. B. Bliska
Footnotes
Published ahead of print on 22 November 2010.
REFERENCES
- 1.Amimoto, K., T. Noro, E. Oishi, and M. Shimizu. 2007. A novel toxin homologous to large clostridial cytotoxins found in culture supernatant of Clostridium perfringens type C. Microbiology 153:1198-1206. [DOI] [PubMed] [Google Scholar]
- 2.Belyi, Y., and K. Aktories. 2010. Bacterial toxin and effector glycosyltransferases. Biochim. Biophys. Acta 1800:134-143. [DOI] [PubMed] [Google Scholar]
- 3.Bosco, E. E., J. C. Mulloy, and Y. Zheng. 2009. Rac1 GTPase: a “Rac” of all trades. Cell. Mol. Life Sci. 66:370-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 5.Busch, C., et al. 1998. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J. Biol. Chem. 273:19566-19572. [DOI] [PubMed] [Google Scholar]
- 6.Busch, C., F. Hofmann, R. Gerhard, and K. Aktories. 2000. Involvement of a conserved tryptophan residue in the UDP-glucose binding of large clostridial cytotoxin glycosyltransferases. J. Biol. Chem. 275:13228-13234. [DOI] [PubMed] [Google Scholar]
- 7.Egerer, M., T. Giesemann, C. Herrmann, and K. Aktories. 2009. Autocatalytic processing of Clostridium difficile toxin B. Binding of inositol hexakisphosphate. J. Biol. Chem. 284:3389-3395. [DOI] [PubMed] [Google Scholar]
- 8.Genth, H., K. Aktories, and I. Just. 1999. Monoglucosylation of RhoA at threonine 37 blocks cytosol-membrane cycling. J. Biol. Chem. 274:29050-29056. [DOI] [PubMed] [Google Scholar]
- 9.Genth, H., et al. 2006. Cellular stability of Rho-GTPases glucosylated by Clostridium difficile toxin B. FEBS Lett. 580:3565-3569. [DOI] [PubMed] [Google Scholar]
- 10.Halabi-Cabezon, I., et al. 2008. Prevention of the cytopathic effect induced by Clostridium difficile toxin B by active Rac1. FEBS Lett. 582:3751-3756. [DOI] [PubMed] [Google Scholar]
- 11.Jank, T., D. J. Reinert, T. Giesemann, G. E. Schulz, and K. Aktories. 2005. Change of the donor substrate specificity of Clostridium difficile toxin B by site-directed mutagenesis. J. Biol. Chem. 280:37833-37838. [DOI] [PubMed] [Google Scholar]
- 12.Jank, T., T. Giesemann, and K. Aktories. 2007. Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J. Biol. Chem. 282:35222-35231. [DOI] [PubMed] [Google Scholar]
- 13.Jank, T., T. Giesemann, and K. Aktories. 2007. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology 17:15-22. [DOI] [PubMed] [Google Scholar]
- 14.Kato, H., et al. 1998. Identification of toxin A-negative, toxin B-positive Clostridium difficile by PCR. J. Clin. Microbiol. 36:2178-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagahama, M., K. Nagayasu, K. Kobayashi, and J. Sakurai. 2002. Binding component of Clostridium perfringens iota-toxin induces endocytosis in Vero cells. Infect. Immun. 70:1909-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagahama, M., S. Hayashi, S. Morimitsu, and J. Sakurai. 2003. Biological activities and pore formation of Clostridium perfringens beta toxin in HL 60 cells. J. Biol. Chem. 278:36934-36941. [DOI] [PubMed] [Google Scholar]
- 17.Popoff, M. R. 1987. Purification and characterization of Clostridium sordellii lethal toxin and cross-reactivity with Clostridium difficile cytotoxin. Infect. Immun. 55:35-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reineke, J., et al. 2007. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446:415-419. [DOI] [PubMed] [Google Scholar]
- 19.Reinert, D. J., T. Jank, K. Aktories, and G. E. Schulz. 2005. Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 351:973-981. [DOI] [PubMed] [Google Scholar]
- 20.Sakurai, J., and M. Nagahama. 2006. Clostridium perfringens beta-toxin: characterization and action. Toxin Rev. 25:89-108. [Google Scholar]
- 21.Selzer, J., et al. 1996. Clostridium novyi alpha-toxin-catalyzed incorporation of GlcNAc into Rho subfamily proteins. J. Biol. Chem. 271:25173-25178. [DOI] [PubMed] [Google Scholar]
- 22.Smedley, J. G., III, D. J. Fisher, S. Sayeed, G. Chakrabarti, and B. A. McClane. 2004. The enteric toxins of Clostridium perfringens. Rev. Physiol. Biochem. Pharmacol. 152:183-204. [DOI] [PubMed] [Google Scholar]
- 23.Tweten, R. K. 2001. Clostridium perfringens beta toxin and Clostridium septicum alpha toxin: their mechanisms and possible role in pathogenesis. Vet. Microbiol. 82:1-9. [DOI] [PubMed] [Google Scholar]
- 24.Voth, D. E., and J. D. Ballard. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walev, I., et al. 2001. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin O. Proc. Natl. Acad. Sci. U. S. A. 98:3185-3190. [DOI] [PMC free article] [PubMed] [Google Scholar]