

Abstract
Activation of complement represents one means of natural resistance to infection from a wide variety of potential pathogens. Recently, properdin, a positive regulator of the alternative pathway of complement, has been shown to bind to surfaces and promote complement activation. Here we studied whether properdin-mediated complement activation occurs on the surface of Chlamydia pneumoniae, an obligate intracellular Gram-negative bacterium that causes 10 to 20% of community-acquired pneumonia. We have determined for the first time that the physiological P2, P3, and P4 forms of human properdin bind to the surface of Chlamydia pneumoniae directly. The binding of these physiological forms accelerates complement activation on the Chlamydia pneumoniae surface, as measured by C3b and C9 deposition. Finally, properdin-depleted serum could not control Chlamydia pneumoniae infection of HEp-2 cells compared with normal human serum. However, after addition of native properdin, the properdin-depleted serum recovered the ability to control the infection. Altogether, our data suggest that properdin is a pattern recognition molecule that plays a role in resistance to Chlamydia infection.
Chlamydiae are obligate intracellular Gram-negative bacteria that develop in a host cell within a membrane-bound compartment termed an inclusion. In humans, Chlamydia pneumoniae causes diseases of the respiratory tract, e.g., bronchitis, sinusitis, or pneumonia, with potentially severe sequelae that include atherosclerosis and chronic obstructive pulmonary disease (2, 8, 20).
Chlamydia's unique developmental cycle starts when the infectious form of the bacterium, the elementary body (EB), targets the mucosal respiratory epithelium and remains within a nonacidified vacuole known as an inclusion (13). Soon after the infection, the EB differentiates into a noninfectious, but metabolically active, reticulate body (RB), which proliferates by binary fission within the expanding inclusion. The generated progeny differentiates back into EBs that are released upon host cell lysis to infect other cells. During the infection process, Chlamydia does not necessarily remain confined to these primary sites; it also has the propensity to disseminate to extramucosal tissues. For instance, it has been suggested that C. pneumoniae multiplies and survives within macrophages (16, 35, 40) and polymorphonuclear neutrophils (PMN) (42, 50) in order to propagate to the rest of the body and reach endothelial cells. During this journey, from circulating cells to endothelial cells, C. pneumoniae encounters the innate immune system, in which the complement system may play a fundamental role in controlling Chlamydia infection.
The complement system is a central component of the innate immune response and is involved in many functions, including recognition, opsonization, phagocytosis, and destruction of foreign cells, as well as generation of chemotactic fragments (C3a and C5a) and activation of adaptive immunity (30, 32, 51) Three pathways of complement activation are known: the classical, lectin, and alternative pathways. Although each uses its own unique mechanism for target versus host discrimination, all pathways result in covalent attachment of C3b to the target and can potentially assemble pores in the bilipid layer of the cell being attacked. The alternative pathway initiates when spontaneously hydrolyzed C3 binds to activating surfaces (i.e., certain bacteria and viruses). Therefore, this pathway does not require a specific antibody response for activation and may play an important role in controlling primary infections with pathogens. Although it is has been described that Chlamydia trachomatis activates the alternative pathway (21, 31), little is known about the effect of this pathway on C. pneumoniae. Moreover, the mechanisms involved in alternative pathway-mediated complement activation or its consequences on infection in C. pneumoniae remain unknown.
Properdin is the only positive regulator of the alternative pathway. It is a plasma protein synthesized by monocytes, hepatocytes, mast cells, T cells (45, 46, 49), and shear-stressed endothelial cells (6) and is also a component of the secondary granules in neutrophils (PMN) (53). Patients with properdin deficiency have a higher risk of meningococcal disease than the general population (14), and an association has also recently been found with recurrent otitis media and pneumonia (44). Properdin facilitates alternative pathway complement activation and amplification by extending the half-life of the C3b,Bb convertase (11). The stabilized C3bBb convertase then rapidly cleaves more C3 to C3b, which acts as an opsonin and can reinitiate the pathway in an amplification loop that proceeds on the pathogenic cell.
Properdin is composed of cyclic dimers (P2), trimers (P3), and tetramers (P4) of a 53-kDa monomeric subunit (37, 47). Biochemical studies of purified properdin indicate that this protein can form nonphysiological higher-level polymers during events such as long-term storage and freeze-thawing (10, 37). This form, also known as “activated properdin or Pn,” has the abnormal ability to activate complement in solution (37). Recent studies using purified properdin have reported that properdin can act as a pattern recognition molecule and bind directly to surfaces such as dying cells and Neisseria gonorrhoeae in the absence of C3b, serving as a platform for de novo C3bBb assembly (15, 25, 26, 48, 56). Although the data from these studies are consistent with the complement initiation function proposed over 50 years ago (38), we have recently shown that physiological forms of properdin in the absence of artifactual aggregates do bind to late apoptotic and necrotic cells (12, 56) but do not bind to Neisseria spp. (1). Therefore, properdin is likely very selective in its recognition of surfaces.
Considering the importance of the recent findings that implicate properdin as a complement initiator on surfaces, we sought to determine whether the physiological forms of properdin (P2, P3, and P4) have the ability to bind to and promote complement activation on the C. pneumoniae surface. Herein, we provide evidence that properdin plays an active role in alternative pathway activation on the C. pneumoniae surface and in controlling infection, suggesting a role for properdin as a specific pattern recognition molecule.
MATERIALS AND METHODS
Chlamydia and cell culture.
Elementary bodies (EBs) of C. pneumoniae isolate Kajaani 6 were propagated within HL cells (29) in Chlamydia medium (complete Iscove's modified Dulbecco's medium containing 10% heat-inactivated fetal bovine serum, 20 mM HEPES, 2 mM l-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 20 μg/ml gentamicin, 0.5 mg/ml glucose, 0.26 mg/ml sodium bicarbonate, and 1.5 μg/ml cycloheximide) as described previously (9, 54). EBs were aliquoted in a sucrose-phosphate-glutamate buffer and stored at −80°C. The infectivity, as measured by the number of inclusion-forming units (IFU) of purified organisms, was titrated in cycloheximide-treated (1 μg/ml) HL cell monolayers. HEp-2 cells (ATCC CCL 23; American Type Culture Collection, Manassas, VA) were grown in complete RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT) and 20 mM HEPES, 2 mM l-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, and 20 μg/ml gentamicin (all from Invitrogen, Carlsbad, CA) at 37°C in 5% CO2. When infected with C. pneumoniae, HEp-2 cell monolayers were cultured in Chlamydia medium.
Antibodies.
Anti-human C3-phycoerythrin (PE) (clone 6C9; Cedarline), anti-human C3-fluorescein isothiocyanate (FITC; Cappel), FITC-labeled anti-Chlamydia EBs (Fitzgerald Industries International, Concord, MA), mouse IgG2a anti-C. pneumoniae (clone 57/062; ABD Serotec), mouse anti-major outer membrane protein (anti-MOMP) (clone RR402) (39), mouse IgG1 anti-human properdin 1 (Quidel), mouse IgG1 and IgG2a isotype controls (eBioscience), and anti-mouse IgG-FITC were purchased from the indicated sources. The anti-properdin and isotype control antibodies were added to the samples at a concentration of 10 μg/ml.
Serum and complement reagents.
Complement component C9 was purified as previously described (5) and was labeled with Alexa Fluor 488 (Molecular Probes) by following the manufacturer's instructions. Properdin was purified from normal human serum (NHS) as described previously (12). Properdin-depleted serum was purchased from CompTech. The alternative pathway was selectively activated by adding EGTA and Mg2+ to serum, both to a final concentration of 5 mM. The addition of a final concentration of 10 mM EDTA was used to inhibit both classical and alternative pathways.
Separation of the polymeric forms of properdin.
Properdin was thawed and subfractionated as described previously (12, 37). Briefly, the polymeric forms of properdin (P2, P3, P4, and Pn) were separated by cation-exchange chromatography on a Mono S column (0.5 by 5 cm). The sample was diluted 2-fold with buffer A (50 mM sodium phosphate, pH 6) and loaded onto a 1-ml Mono S column (GE Healthcare). The column was washed with 20% buffer B (50 mM sodium phosphate. 0.5 M NaCl, pH 6), and the properdin was eluted with a 20-ml gradient from 20 to 45% buffer B. Individual properdin peaks were collected and dialyzed into phosphate-buffered saline (PBS). Subsequently, or alternatively, the properdin was purified by gel filtration on a Phenomenex BioSep-Sec-S4000 column (600 by 7.8 mm) with a guard column (75 by 7.8 mm) at a flow rate of 0.5 ml/min at 21°C in PBS (10 mM sodium phosphate, 140 mM NaCl, pH 7.4).
Measurement of properdin binding to C. pneumoniae.
Properdin binding to EBs was assessed by incubating 107 infection-forming units (IFU) of C. pneumoniae with properdin (15 μg/ml) in Hanks balanced salt solution (HBSS) plus 0.1% bovine serum albumin (BSA) in a final reaction volume of 100 μl for 30 min at 4°C. C. pneumoniae bacteria were washed two times as indicated previously and incubated with either anti-properdin or isotype control antibodies for 45 min at 4°C. C. pneumoniae bacteria were washed and stained with FITC-conjugated goat anti-mouse IgG and analyzed by flow cytometry. A minimum of 5,000 events was acquired, and the data were analyzed using FlowJo software.
Measurement of C3b deposition by flow cytometry.
To determine the time course of C3b deposition on C. pneumoniae, EBs were incubated in the indicated percentage of NHS for 60 min at 37°C. EBs were incubated with serum in the presence of 5 mM Mg-EGTA to selectively measure alternative pathway activation. Negative controls were incubated with NHS in the presence of 10 mM final EDTA or heat-inactivated NHS (HI-NHS) to inhibit the complement system. To stop further complement activation at the end of the incubation, the samples were placed on ice and HBSS-0.1% BSA-EDTA was added. Samples were washed two times with cold HBSS-0.1% BSA-EDTA and centrifuged at 12,000 × g for 10 min at 4°C to pellet the EBs. Deposition of C3b was detected by incubating C. pneumoniae with either PE- or FITC-labeled anti-C3b (1:100) for 40 min at 4°C in HBSS-0.1% BSA-EDTA. After being washed, EBs were analyzed by flow cytometry. When the PE-labeled anti-C3b antibody was used, deposition of C3b was measured on a gated positive C. pneumoniae isolate stained with the FITC-labeled anti-EB antibody. A minimum of 5,000 gated events was acquired. To determine whether properdin bound to C. pneumoniae has the ability to promote complement activation, EBs were preincubated with or without purified properdin form P3 (15 μg/ml) for 30 min at 4°C in HBSS-0.1% BSA. Then, EBs were washed 3 times with HBSS-0.1% BSA and incubated at 37°C for 0, 5, 10, 15, 30, and 60 min with 15% properdin-depleted serum or 5% NHS in the presence of 5 mM Mg-EGTA or 10 mM EDTA. C3b deposition was determined as described above, using PE-labeled anti-C3b antibody.
C9 deposition on Chlamydia.
Purified properdin was added to C. pneumoniae to a final concentration of 15 μg/ml as described above. C. pneumoniae bacteria were washed and incubated for 0, 15, and 60 min with 15% properdin-depleted serum containing Alexa Fluor 488-labeled C9 (9 μg/ml) and incubated for 0 to 60 min. C. pneumoniae bacteria were then washed twice with HBSS-0.1% BSA-EDTA and immediately analyzed by flow cytometry. Serum in the presence of a 10 mM EDTA final concentration was included to prevent complement activation in negative controls.
Infection assay.
EBs were preincubated with 30% NHS or 30% properdin-depleted serum with or without P3 (15 μg/ml) in RPMI 1640 for 1 h at 37°C in a total volume of 50 μl. EBs were diluted 1:10 in cold RPMI 1640, placed on ice to further inhibit complement activation, and kept on ice no more than 5 min before infection of HEp-2 cells. HEp-2 monolayers, prepared 1 day earlier in 48-well plates (0.55 × 105/well) in complete RPMI medium, were washed with plain RPMI 1640 to remove traces of fetal bovine serum. Next, 10 μl of EBs was added to the wells containing 500 μl of RPMI 1640. Plates were centrifuged at 300 × g at 35°C for 1 h. The plates were then placed for 1 h at 37°C in 5% CO2. The supernatant was discarded, and Chlamydia medium was added. Infected HEp-2 cells were incubated for 48 h, washed twice with PBS, and fixed with 100% cold methanol for 10 min at room temperature. Cells were then incubated with PBS-1% BSA for 1 h to reduce background antibody staining. For antibody labeling, fixed cells were incubated for 1 h with anti-MOMP (1:200) antibody in PBS-0.2% BSA and F(ab′)2 goat anti-mouse IgG conjugated to Alexa Fluor 488 (1:200) sequentially, for 60 min each, at room temperature. The numbers of C. pneumoniae inclusions in each well were counted in 30 fields with an inverted microscope using ×600 magnification. Triplicate wells were tested for each condition. Images for Fig. 6C were acquired using an Olympus FSX100 microscope and analyzed using Adobe Photoshop CS, version 8.0.
Statistical analysis.
Student's t tests (two-tailed) were used to assess the statistical significance of the difference between C. pneumoniae infection in the presence of HI-NHS and that in the presence of properdin-depleted serum or the difference between C. pneumoniae infection in the presence of HI-NHS and that in the presence of properdin-depleted serum plus properdin. P values of <0.05 were considered statistically significant.
RESULTS
Complement activation on the C. pneumoniae surface.
Complement activation on the C. pneumoniae surface, as measured by deposition of C3b, was examined. The cells were incubated with normal human serum (0 to 40%). Then, C. pneumoniae bacteria were incubated with anti-C3b PE-conjugated and anti-C. pneumoniae FITC-conjugated antibodies, followed by flow cytometry analysis. Detectable C3b deposition by NHS was seen on C. pneumoniae at low concentrations of NHS (0.6%), and C3b deposition began to plateau at 10% NHS (Fig. 1). When C. pneumoniae bacteria were incubated with serum in the presence of Mg-EGTA to selectively measure alternative pathway activation, similar maximum levels of C3 deposition were achieved, although detectable C3 deposition was observed at 5% NHS and above. The behavior of activation of the alternative pathway was as expected, since this pathway is more dependent on serum concentration than the classical pathway. No C3b deposition was detected in the presence of 10 mM EDTA or when HI-NHS was used.
FIG. 1.

Activation of the human complement system by Chlamydia pneumoniae. C. pneumoniae bacteria (25 × 106 IFU) were incubated with the indicated concentrations of normal human serum (NHS) in the presence of Ca2+ and Mg2+ to evaluate all complement pathways, with Mg-EGTA to evaluate the alternative pathway, or with EDTA or heat-inactivated (HI) serum (56°C at 30 min) to inhibit all complement system pathways. C. pneumoniae bacteria were incubated for 1 h at 37°C, and complement activation was stopped by adding cold HBSS-EDTA. C. pneumoniae bacteria were then washed, and C3b deposition was assessed by flow cytometry using a PE-labeled anti-C3b antibody. EB-positive cells were gated using an FITC-labeled anti-C. pneumoniae antibody. The mean fluorescence intensity of C3b deposition versus the percentage of serum was plotted. No C3b deposition was detected in the presence of EDTA or when HI-NHS was used.
C. pneumoniae directly binds properdin.
Properdin is a positive regulatory factor that facilitates activation and amplification of the alternative pathway complement by extending the half-life of the C3bBb convertase (11). Recent studies have reported that properdin can bind directly to early or late apoptotic or necrotic cells and to N. gonorrhoeae (26, 48, 56), providing a platform for de novo convertase assembly and complement activation. Considering the importance of the recent findings that implicate properdin as a pattern recognition molecule of the alternative pathway and a complement initiator, we examined the ability of properdin to bind C. pneumoniae. In order to carry out this objective, the native physiological properdin forms (P2, P3, and P4) were separated from the nonphysiological higher-molecular-weight forms (Pn) by ion-exchange and gel filtration chromatographies as previously described (10, 12, 37). These steps are crucial, since commercially available properdin and any properdin stored frozen contains Pn, which is an anomalous form resulting from storage and freeze-thawing (10, 37). We have recently shown that these Pn forms bind nonspecifically to certain surfaces such as live cells (12) and Neisseria spp. (1). As shown in Fig. 2, native properdin forms P2, P3, and P4 bind C. pneumoniae, suggesting a possible role for properdin as a pattern recognition molecule for this organism.
FIG. 2.

Native P2, P3, and P4, forms of properdin bind to Chlamydia pneumoniae. C. pneumoniae bacteria (50 × 106 IFU) were incubated with 3 μg of each form of properdin (purified by ion-exchange chromatography, followed by size exclusion chromatography) in 100 μl HBSS plus 0.1% BSA for 30 min at 4°C. C. pneumoniae bacteria were then washed and analyzed by FACS analysis using an anti-properdin monoclonal antibody or an IgG1 monoclonal antibody isotype control (shaded), followed by an FITC-conjugated anti-mouse IgG antibody. (Inset) The purity of the C. pneumoniae elementary body preparation was assayed by FACS analysis using a C. pneumoniae-specific monoclonal antibody or an IgG2a monoclonal antibody isotype control, followed by an FITC-conjugated anti-mouse IgG antibody. The number of particles versus properdin binding (fluorescence intensity) is shown. Cpn, C. pneumoniae.
Properdin bound to C. pneumoniae accelerates complement activation on the surface of the bacterium.
In order to investigate whether properdin bound to C. pneumoniae has the ability to promote complement activation, EBs were preincubated with purified properdin form P3, washed extensively, and subsequently incubated with properdin-depleted serum. Properdin-depleted serum was used to avoid competition with native properdin in order to determine whether alternative pathway activation, measured as C3b deposition, was promoted by properdin bound directly to C. pneumoniae. As shown in Fig. 3, more rapid C3b deposition occurred with C. pneumoniae preincubated with P3 than with C. pneumoniae alone. C3b deposition was seen as early as 5 min with properdin, while none was seen on C. pneumoniae without properdin. At 15 min, ∼700-fold-greater C3b deposition was seen on C. pneumoniae preincubated with properdin than on C. pneumoniae alone. Maximum C3b deposition was reached after 30 min of incubation on C. pneumoniae preincubated with properdin, while C3b deposition on C. pneumoniae without properdin opsonization was much slower (Fig. 3). We examined the consequences of having properdin bound to Chlamydia followed by exposure to normal human serum (Fig. 4), as this may mimic what is happening when Chlamydia encounters sera of normal individuals during infection. C. pneumoniae bacteria were preincubated with or without P3 and then exposed to a low percentage of normal human serum (5%) to detect C3b deposition. Compared with serum alone, properdin increased C3b deposition 800-fold after 15 min of incubation with serum. This difference was reduced to 1.5-fold at 60 min, at which time C3b deposition on C. pneumoniae alone had almost reached the level of properdin-stimulated activation.
FIG. 3.
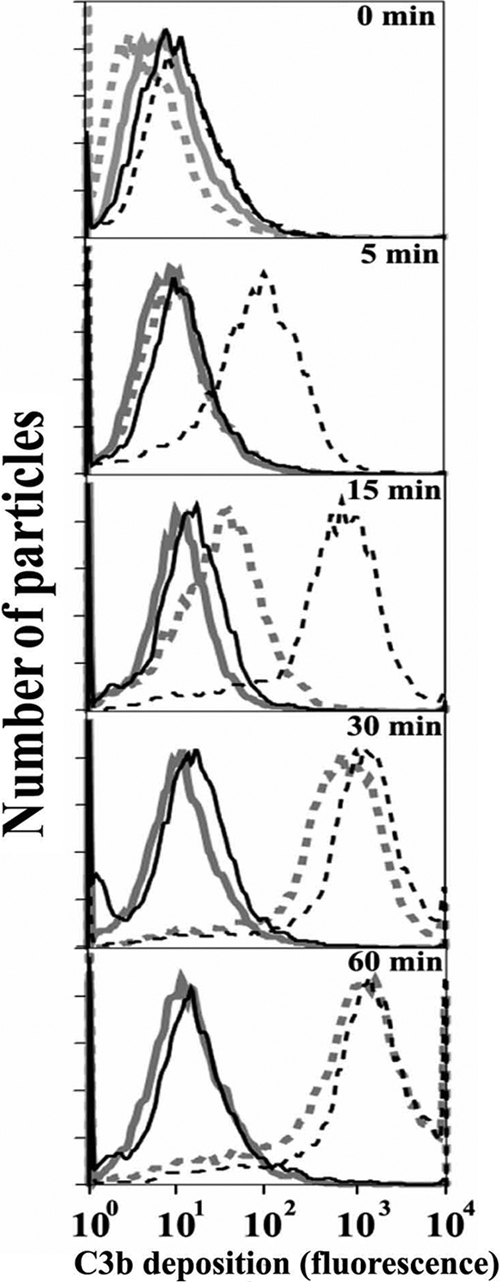
A physiological form of properdin (P3), when bound to C. pneumoniae, induces complement activation using properdin-depleted serum. C. pneumoniae bacteria (50 × 106 IFU) were incubated with (black lines) or without (gray lines) 3 μg of P3 in 100 μl HBSS-0.1% BSA for 30 min at 4°C. C. pneumoniae bacteria were then washed, and 15% properdin-depleted serum was added in the presence of HBSS-0.1% BSA-Mg-EGTA (dashed lines) or HBSS-0.1% BSA-EDTA (solid lines) and incubated at 37°C for the indicated times. The particles were then washed with cold HBSS-EDTA, and C3b deposition was assessed by FACS analysis using an FITC-labeled anti-C3b antibody. The number of particles versus C3b deposition (fluorescence intensity) is shown.
FIG. 4.

The physiological form of properdin (P3), when bound to C. pneumoniae, induces complement activation using normal human serum. The P3 form of properdin was isolated from purified properdin by ion-exchange chromatography, followed by size exclusion chromatography, as described in Materials and Methods. (A and B) C. pneumoniae bacteria (50 × 106 IFU) were incubated with (Cpn + P3) or without (Cpn) 3 μg P3 in 100 μl HBSS-0.1% BSA for 30 min at 4°C. C. pneumoniae bacteria were then washed, and 5% normal human serum was added in the presence of HBSS-0.1% BSA-Mg-EGTA or HBSS-0.1% BSA-EDTA. These mixtures were incubated for the indicated times at 37°C. The particles were then washed with cold HBSS plus EDTA, and C3b deposition was assessed by flow cytometry using an FITC-labeled anti-C3 antibody. Red, 0 min; green, 5 min; dark blue, 10 min; brown, 15 min; purple, 30 min; light blue, 60 min. (C) C3b deposition was graphed as mean fluorescence intensity, obtained from panels A and B, versus time.
We also examined whether properdin bound to C. pneumoniae accelerates the deposition of C9, which forms part of the membrane attack complex (MAC), on C. pneumoniae. Alexa Fluor-labeled C9 was added to properdin-depleted serum to track MAC formation on C. pneumoniae. As shown in Fig. 5, significant C9 deposition was seen at 15 min on C. pneumoniae preincubated with properdin, while none was detectable on C. pneumoniae alone. Maximum C9 deposition was achieved at 60 min on C. pneumoniae preincubated with P3, and a lower level of C9 deposition was seen on C. pneumoniae alone. This suggests that faster properdin-mediated C3b deposition is accompanied by faster MAC formation.
FIG. 5.
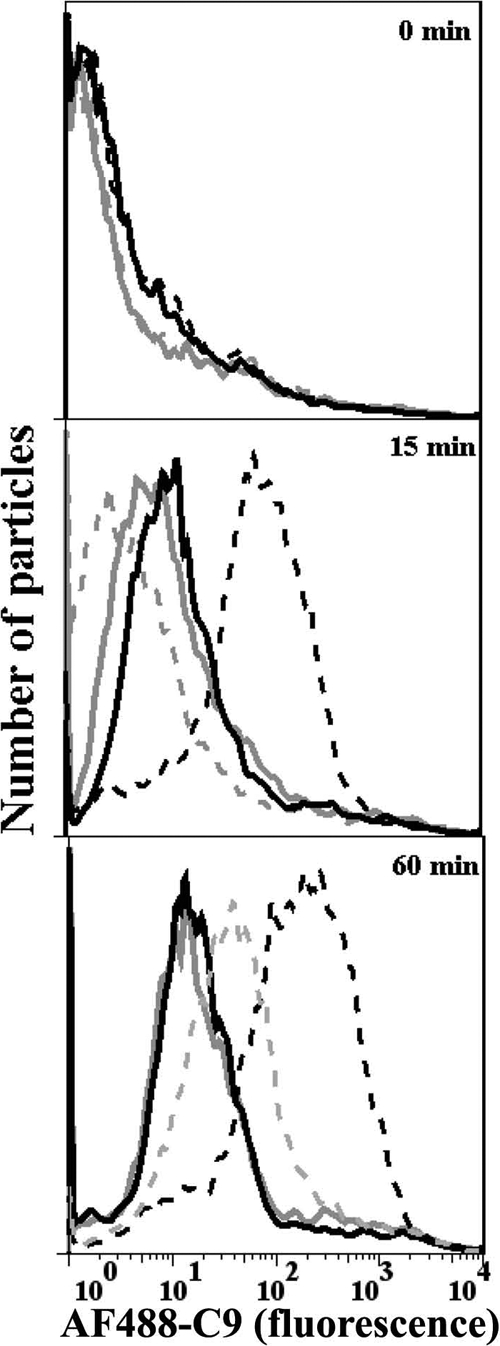
The physiological form of properdin (P3) bound to Chlamydia pneumoniae accelerates C9 deposition on EBs. C. pneumoniae bacteria (50 × 106 IFU) were incubated with (black lines) or without (gray lines) 3 μg P3 in 100 μl HBSS-0.1% BSA for 30 min at 4°C. The particles were then washed, and 15% properdin-depleted serum plus Alexa Fluor 488-labeled C9 (AF488-C9) was added in the presence of HBSS-0.1% BSA-Mg-EGTA (dashed lines) or HBSS-0.1% BSA-EDTA (solid lines) for 0, 15, and 60 min at 37°C. The particles were then washed with cold HBSS-EDTA, and Alexa Fluor 488-labeled C9 deposition was assessed by flow cytometry. The number of particles versus C9 deposition (fluorescence intensity) is shown.
The presence of properdin in serum is important in controlling C. pneumoniae infection.
Studies in C. trachomatis suggest that early but not late complement components are important to control the infection of cells (31). In order to define the role of the early complement components on C. pneumoniae infection, EBs were preincubated with normal human serum and serum deficient in C5, C8/C9, or properdin and their levels of infection were compared to those obtained with HI-NHS. Figure 6 A shows that normal human serum blocks 98.6% (±0.6%) of C. pneumoniae inclusion formation compared to HI-NHS, and serum lacking C5 or C8/C9 blocks the infection by 92.5% (±1.5%). However, the properdin-depleted serum cannot block C. pneumoniae infection effectively (30% ± 13%) compared with NHS, demonstrating that early complement activation events block most of the infectious particles. To further define the role of properdin, native properdin was added back to the properdin-depleted serum and this reduced the level of infection 3-fold (Fig. 6B and C), suggesting that properdin is important for controlling C. pneumoniae infection.
FIG. 6.

Early complement components and the presence of properdin in serum are important in controlling C. pneumoniae infection. (A) EBs were preincubated with 30% NHS or serum depleted of C5, C8/C9, or properdin for 1 h at 37°C. EBs were then used to infect HEp-2 monolayers as described in Materials and Methods, and the percentage of infection of HEp2 cells was measured relative to that with heat-inactivated serum (not shown). (B) EBs were preincubated with 30% NHS, HI-NHS, or properdin-depleted serum (P-depl), with or without P3 (15 μg/ml), in RPMI 1640 plus Ca2+/Mg2+ for 1 h at 37°C. In both panels A and B, the HEp2 cells were fixed at 48 h postinfection and stained with anti-C. pneumoniae MOMP antibody. The numbers of C. pneumoniae inclusions in each well were counted in 30 fields at 600 high-powered fields (HPF). Three separate wells were used for each condition. The statistical significances of the differences between the abilities of the HI-NHS serum and the properdin-depleted serum to control the infection (nonsignificant [n.s.]) or between the abilities of the HI-NHS serum and the properdin-depleted serum plus P3 to control the infection (P < 0.01) are included in the graph. (C) EBs were preincubated with serum as described for panel A. HEp-2 cells were infected with C. pneumoniae (multiplicity of infection of 1), and at 48 h postinfection, the cells were fixed and stained with anti-C. pneumoniae MOMP. Alexa Fluor 488 (green) and bright-field images were merged.
DISCUSSION
Our studies revealed that the predominant physiological forms of properdin (P2, P3, and P4) bind to C. pneumoniae and accelerate activation of the alternative pathway of complement, as measured by C3 and C9 deposition. This is the first description of an intracellular pathogen being targeted by properdin. We also found that properdin in serum is important for controlling infection. The data collectively suggest that properdin is a pattern recognition molecule and may play a role during Chlamydia infection.
During the infection, C. pneumoniae disseminates from lung epithelial cells, the primary target tissue of infection, to the rest of the body, including the blood vessels. During this journey, C. pneumoniae may encounter complement proteins in blood or components that are produced locally in the microenvironment, which could play a role in controlling Chlamydia infection. Thus, complement activation would be faster on surfaces of C. pneumoniae bacteria that have prebound properdin, as shown in our in vitro results (Fig. 3 and 4). This complement activation also led to faster and higher levels of C9 deposition, as a measure of MAC formation (Fig. 5), that could lyse the bacterium, as occurs with other Gram-negative bacteria (36).
Direct binding of native properdin from serum in the presence of EDTA, which inhibits complement activation, was not found on EBs (data not shown). However, when complement is allowed to activate (in the presence of Mg-EGTA), properdin binds indirectly (data not shown), due to its role as a stabilizer of the C3 and C5 convertases of the alternative pathway. This suggests that an inhibitor in serum may interfere with the ability of properdin to bind to surfaces. Recently, serum amyloid P has been reported to inhibit the capacity of properdin to initiate complement activation (34). On the other hand, it has been shown that properdin produced locally by neutrophils is not inhibited and may be able to bind directly to certain cells (25, 26). In addition, other cells, such as monocytes, macrophages, T cells, mast cells, and endothelial cells, increase the production of properdin upon activation with cytokines and shear stress in vitro (6, 45, 46, 49, 53) or in vivo when the classical pathway is activated in the absence of factor B, an essential alternative pathway protein (55). Moreover, it has been shown that the main source of properdin in blood is derived from myeloid lineage (CD11b+) cells (28) and that properdin released by activated neutrophils, CD11b+ cells, can remain bound to the cell (7). Thus, properdin, which is produced locally by cells that are typically infected by EBs, would be in the microenvironment surrounding released EBs and may interact directly with C. pneumoniae.
Studies to define the role of complement in controlling Chlamydia infection are limited. For instance, studies with C. trachomatis suggested that early- but not late-stage complement components (C5 and C8) were important for controlling infection in vitro on murine McCoy cells (31). In agreement with this study, we found that with C. pneumoniae, most of the infectious particles (70% ± 13%) were controlled by the deposition of early complement components since the absence of late complement components (C5 and C8/C9) did not reduce the infection of human HEp-2 cells (Fig. 6A). Although the contribution of late complement components during the infection of HEp-2 cells with C. pneumoniae is rather low (Fig. 6A), the effect of subproducts, such as C5a and C3a, has not been examined in C. pneumoniae infection in vitro or in vivo. Studies performed in C5-deficient mice infected with Chlamydia trachomatis mouse pneumonitis (MoPn) or mice treated with cobra venom factor, used to deplete C3 and C5, had no effect on the infection burden (52). However, it has been shown that complement activation and generation of potent chemotactic C5a in C. trachomatis (a human Chlamydia species) may play a role in attracting PMN in vitro (33, 41), which agrees with the general role of C5a in inflammation in vivo (18) and with the role of an alternative pathway in the activation of neutrophils and C5a release (7, 19, 23). Although Chlamydia trachomatis has been shown to infect neutrophils in the absence of complement (41), the presence of serum (negative for antibodies against Chlamydia) may contribute to the inhibition of the formation of inclusions in these cells (57). In addition, C. trachomatis encephalitis was found in a patient with a defect in the alternative pathway but not in the classical pathway (4). Whether these discrepancies in the role of complement in Chlamydia infection are due to the species of Chlamydia or to the host (human or murine) used in these studies remains to be determined.
Despite the fact that the classical and lectin pathways are still intact, depletion of properdin from serum drastically increased the level of infection, which is largely restored when properdin is added back to the properdin-depleted serum (Fig. 6). Because depletion of late complement components (C5 and C8/C9) did not affect the ability of the serum to control the infection, it is possible that properdin bound to C. pneumoniae drives the EBs to endocytic compartments, such as lysosomes, in which Chlamydiae are killed, thus controlling the rate of inclusion formation. On the other hand, properdin either bound to C. pneumoniae directly or as a stabilizer of the C3 and C5 convertases of the alternative pathway, or by amplifying deposition of C3b after initial complement activation by all pathways (22), may induce higher levels of C3b subproducts on EBs. These subproducts (iC3b or C3dg) on C. pneumoniae bound to properdin, compared with those on C. pneumoniae alone, could influence the clearance of EBs through complement receptors. Studies aimed at determining the effect of EB-bound properdin or C3b subproducts on EB survival are warranted.
Properdin-deficient individuals have recurrent pneumonia (44) and are more susceptible to Neisseria infections (14) due to a defective alternative pathway. Moreover, it has been shown that properdin is found in atherosclerotic plaques (43), suggesting that properdin may play a role in the pathology of atherosclerosis. On the other hand, C. pneumoniae infection has been associated with atherosclerosis and pneumonia (3, 8). Once C. pneumoniae infects vascular tissues, EBs released subsequently after disruption of infected cells may lead to properdin-mediated complement activation and vascular tissue damage. Based on the abilities of properdin to bind to C. pneumoniae (Fig. 2), to accelerate complement activation (Fig. 3 and 4), to control C. pneumoniae infection (Fig. 6), and to play a role in tissue injury (15, 27), there may be a link between complement and Chlamydia in the pathogenesis of atherosclerosis and pneumonia.
Properdin was suggested to be a pattern recognition molecule more than 50 years ago by Pillemer et al. (38). However, these results were discarded when it was demonstrated that properdin acted through stabilizing the C3bBb convertase. Recent reports have reopened the controversy surrounding the basic functions of this protein, suggesting that properdin binds directly to a variety of surfaces, including live, apoptotic, and necrotic cells, and to N. gonorrhoeae (26, 48, 56). We have recently shown (1, 12) that although properdin can function as a pattern recognition molecule of the alternative pathway, it is more selective than originally proposed. In order to effectively study the selective interaction of properdin with surfaces, the physiological forms (P2, P3, and P4) need to be separated from the nonphysiological aggregates, which have the abnormal property to consume complement in fluid phase (37), and need to bind nonspecifically to several surfaces, including live cells (12) and Neisseria (1). As shown here, we have used the physiological forms of properdin (P2, P3, and P4) and demonstrated that they can bind to C. pneumoniae and promote complement activation. Although it has been shown that properdin may bind to certain molecular structures, such as cell surface glycosaminoglycans (GAGs), DNA, lipopolysaccharide (LPS), and nonsulfated glycoconjugates (24, 26, 56), the specific ligand on EBs has not been determined. To our knowledge, this is the first study that uses native human properdin to demonstrate direct and specific interaction with a pathogen resulting in impaired infection.
Acknowledgments
This work has been supported by NIH grants DK-35081 (M.K.P.), 1P30HL101317-01 (V.P.F.), and AHA 0735101N (V.P.F.).
We thank Benjamin Wizel for providing us with the C. pneumoniae Kajaani 6 strain and Staci Snyder, Heather N. Emch, and Laci J. Bloomfield for excellent technical assistance.
M. K. Pangburn is an officer of and has a financial interest in Complement Technology, Inc., a supplier of complement reagents.
Editor: R. P. Morrison
Footnotes
Published ahead of print on 6 December 2010.
REFERENCES
- 1.Agarwal, S., et al. 2010. An evaluation of the role of properdin in alternative pathway activation on Neisseria meningitidis and Neisseria gonorrhoeae. J. Immunol. 185:507-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belland, R., D. M. Ojcius, and G. I. Byrne. 2004. Chlamydia. Nat. Rev. Microbiol. 2:530-531. [DOI] [PubMed] [Google Scholar]
- 3.Belland, R. J., S. P. Ouellette, J. Gieffers, and G. I. Byrne. 2004. Chlamydia pneumoniae and atherosclerosis. Cell. Microbiol. 6:117-127. [DOI] [PubMed] [Google Scholar]
- 4.Bertsche, A., M. H. Wagner, R. Bollmann, M. Obladen, and U. Felderhoff-Mueser. 2008. An unusual manifestation of a neonatal Chlamydia infection. J. Child Neurol. 23:948-949. [DOI] [PubMed] [Google Scholar]
- 5.Biesecker, G., and H. J. Muller-Eberhard. 1980. The ninth component of human complement: purification and physicochemical characterization. J. Immunol. 124:1291-1296. [PubMed] [Google Scholar]
- 6.Bongrazio, M., A. R. Pries, and A. Zakrzewicz. 2003. The endothelium as physiological source of properdin: role of wall shear stress. Mol. Immunol. 39:669-675. [DOI] [PubMed] [Google Scholar]
- 7.Camous, L., et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood, in press. [DOI] [PubMed]
- 8.Campbell, L. A., and C. C. Kuo. 2004. Chlamydia pneumoniae: an infectious risk factor for atherosclerosis? Nat. Rev. Microbiol. 2:23-32. [DOI] [PubMed] [Google Scholar]
- 9.Cortes, C., K. A. Rzomp, A. Tvinnereim, M. A. Scidmore, and B. Wizel. 2007. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infect. Immun. 75:5586-5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farries, T. C., J. T. Finch, P. J. Lachmann, and R. A. Harrison. 1987. Resolution and analysis of ‘native’ and ‘activated’ properdin. Biochem. J. 243:507-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fearon, D. T., and K. F. Austen. 1975. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J. Exp. Med. 142:856-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferreira, V. P., C. Cortes, and M. K. Pangburn. 2010. Native polymeric forms of properdin selectively bind to targets and promote activation of the alternative pathway of complement. Immunobiology 215:932-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fields, K. A., and T. Hackstadt. 2002. The chlamydial inclusion: escape from the endocytic pathway. Annu. Rev. Cell Dev. Biol. 18:221-245. [DOI] [PubMed] [Google Scholar]
- 14.Fijen, C. A., et al. 1999. Properdin deficiency: molecular basis and disease association. Mol. Immunol. 36:863-867. [DOI] [PubMed] [Google Scholar]
- 15.Gaarkeuken, H., et al. 2008. Complement activation by tubular cells is mediated by properdin binding. Am. J. Physiol. Renal Physiol. 295:F1397-F1403. [DOI] [PubMed] [Google Scholar]
- 16.Gieffers, J., et al. 2004. Phagocytes transmit Chlamydia pneumoniae from the lungs to the vasculature. Eur. Respir. J. 23:506-510. [DOI] [PubMed] [Google Scholar]
- 17.Reference deleted.
- 18.Guo, R. F., and P. A. Ward. 2005. Role of C5a in inflammatory responses. Annu. Rev. Immunol. 23:821-852. [DOI] [PubMed] [Google Scholar]
- 19.Gupta-Bansal, R., J. B. Parent, and K. R. Brunden. 2000. Inhibition of complement alternative pathway function with anti-properdin monoclonal antibodies. Mol. Immunol. 37:191-201. [DOI] [PubMed] [Google Scholar]
- 20.Hahn, D. L. 1999. Chlamydia pneumoniae, asthma, and COPD: what is the evidence? Ann. Allergy Asthma Immunol. 83:271-. 88:291. [DOI] [PubMed] [Google Scholar]
- 21.Hall, R. T., T. Strugnell, X. Wu, D. V. Devine, and H. G. Stiver. 1993. Characterization of kinetics and target proteins for binding of human complement component C3 to the surface-exposed outer membrane of Chlamydia trachomatis serovar L2. Infect. Immun. 61:1829-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harboe, M., and T. E. Mollnes. 2008. The alternative complement pathway revisited. J. Cell. Mol. Med. 12:1074-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harboe, M., G. Ulvund, L. Vien, M. Fung, and T. E. Mollnes. 2004. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 138:439-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holt, G. D., M. K. Pangburn, and V. Ginsburg. 1990. Properdin binds to sulfatide [Gal(3-SO4)beta 1-1Cer] and has a sequence homology with other proteins that bind sulfated glycoconjugates. J. Biol. Chem. 265:2852-2855. [PubMed] [Google Scholar]
- 25.Kemper, C., J. P. Atkinson, and D. E. Hourcade. 2010. Properdin: emerging roles of a pattern-recognition molecule. Annu. Rev. Immunol. 28:131-155. [DOI] [PubMed] [Google Scholar]
- 26.Kemper, C., L. M. Mitchell, L. Zhang, and D. E. Hourcade. 2008. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc. Natl. Acad. Sci. U. S. A. 105:9023-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keslar, K., E. R. Rodriguez, C. D. Tan, R. C. Starling, and P. S. Heeger. 2008. Complement gene expression in human cardiac allograft biopsies as a correlate of histologic grade of injury. Transplantation 86:1319-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimura, Y., L. Zhou, T. Miwa, and W. C. Song. 2010. Genetic and therapeutic targeting of properdin in mice prevents complement-mediated tissue injury. J. Clin. Invest. 120:3545-3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo, C. C., and J. T. Grayston. 1990. A sensitive cell line, HL cells, for isolation and propagation of Chlamydia pneumoniae strain TWAR. J. Infect. Dis. 162:755-758. [DOI] [PubMed] [Google Scholar]
- 30.Lambris, J. D., D. Ricklin, and B. V. Geisbrecht. 2008. Complement evasion by human pathogens. Nat. Rev. Microbiol. 6:132-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin, J. S., L. L. Yan, Y. Ho, and P. A. Rice. 1992. Early complement components enhance neutralization of Chlamydia trachomatis infectivity by human sera. Infect. Immun. 60:2547-2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mastellos, D., et al. 2003. Complement: structure, functions, evolution, and viral molecular mimicry. Immunol. Res. 27:367-386. [DOI] [PubMed] [Google Scholar]
- 33.Megran, D. W., H. G. Stiver, and W. R. Bowie. 1985. Complement activation and stimulation of chemotaxis by Chlamydia trachomatis. Infect. Immun. 49:670-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitchell, L., and D. Hourcade. 2008. Inhibition of properdin-directed complement activation by serum amyloid P component. Mol. Immunol. 45:4103-4104. [Google Scholar]
- 35.Moazed, T. C., C. C. Kuo, J. T. Grayston, and L. A. Campbell. 1998. Evidence of systemic dissemination of Chlamydia pneumoniae via macrophages in the mouse. J. Infect. Dis. 177:1322-1325. [DOI] [PubMed] [Google Scholar]
- 36.O'Hara, A. M., A. P. Moran, R. Wurzner, and A. Orren. 2001. Complement-mediated lipopolysaccharide release and outer membrane damage in Escherichia coli J5: requirement for C9. Immunology 102:365-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pangburn, M. K. 1989. Analysis of the natural polymeric forms of human properdin and their functions in complement activation. J. Immunol. 142:202-207. [PubMed] [Google Scholar]
- 38.Pillemer, L., et al. 1954. The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science 120:279-285. [DOI] [PubMed] [Google Scholar]
- 39.Puolakkainen, M., J. Parker, C. C. Kuo, J. T. Grayston, and L. A. Campbell. 1995. Further characterization of Chlamydia pneumoniae specific monoclonal antibodies. Microbiol. Immunol. 39:551-554. [DOI] [PubMed] [Google Scholar]
- 40.Redecke, V., K. Dalhoff, S. Bohnet, J. Braun, and M. Maass. 1998. Interaction of Chlamydia pneumoniae and human alveolar macrophages: infection and inflammatory response. Am. J. Respir. Cell Mol. Biol. 19:721-727. [DOI] [PubMed] [Google Scholar]
- 41.Register, K. B., P. A. Morgan, and P. B. Wyrick. 1986. Interaction between Chlamydia spp. and human polymorphonuclear leukocytes in vitro. Infect. Immun. 52:664-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez, N., et al. 2005. Polymorphonuclear neutrophils improve replication of Chlamydia pneumoniae in vivo upon MyD88-dependent attraction. J. Immunol. 174:4836-4844. [DOI] [PubMed] [Google Scholar]
- 43.Ruef, J., P. Kuehnl, T. Meinertz, and M. Merten. 2008. The complement factor properdin induces formation of platelet-leukocyte aggregates via leukocyte activation. Platelets 19:359-364. [DOI] [PubMed] [Google Scholar]
- 44.Schejbel, L., V. Rosenfeldt, H. Marquart, N. H. Valerius, and P. Garred. 2009. Properdin deficiency associated with recurrent otitis media and pneumonia, and identification of male carrier with Klinefelter syndrome. Clin. Immunol. 131:456-462. [DOI] [PubMed] [Google Scholar]
- 45.Schwaeble, W., et al. 1993. Properdin, a positive regulator of complement activation, is expressed in human T cell lines and peripheral blood T cells. J. Immunol. 151:2521-2528. [PubMed] [Google Scholar]
- 46.Schwaeble, W., et al. 1994. Expression of properdin in human monocytes. Eur. J. Biochem. 219:759-764. [DOI] [PubMed] [Google Scholar]
- 47.Smith, C. A., M. K. Pangburn, C.-W. Vogel, and H. J. Muller-Eberhard. 1984. Molecular architecture of human properdin, a positive regulator of the alternative pathway of complement. J. Biol. Chem. 259:4582-4588. [PubMed] [Google Scholar]
- 48.Spitzer, D., L. M. Mitchell, J. P. Atkinson, and D. E. Hourcade. 2007. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J. Immunol. 179:2600-2608. [DOI] [PubMed] [Google Scholar]
- 49.Stover, C. M., et al. 2008. Properdin plays a protective role in polymicrobial septic peritonitis. J. Immunol. 180:3313-3318. [DOI] [PubMed] [Google Scholar]
- 50.van Zandbergen, G., et al. 2004. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J. Immunol. 172:1768-1776. [DOI] [PubMed] [Google Scholar]
- 51.Ward, P. A. 2009. Functions of C5a receptors. J. Mol. Med. 87:375-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams, D. M., J. Schachter, M. H. Weiner, and B. Grubbs. 1984. Antibody in host defense against mouse pneumonitis agent (murine Chlamydia trachomatis). Infect. Immun. 45:674-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wirthmueller, U., et al. 1997. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J. Immunol. 158:4444-4451. [PubMed] [Google Scholar]
- 54.Wizel, B., et al. 2002. Multiple Chlamydia pneumoniae antigens prime CD8+ Tc1 responses that inhibit intracellular growth of this vacuolar pathogen. J. Immunol. 169:2524-2535. [DOI] [PubMed] [Google Scholar]
- 55.Wu, X., T. Q. Xu, and J. P. Atkinson. 2010. Properdin homeostasis requires turnover of the alternative complement pathway. Proc. Natl. Acad. Sci. U. S. A. 107:19444-19448. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Xu, W., et al. 2008. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J. Immunol. 180:7613-7621. [DOI] [PubMed] [Google Scholar]
- 57.Yong, E. C., S. J. Klebanoff, and C. C. Kuo. 1982. Toxic effect of human polymorphonuclear leukocytes on Chlamydia trachomatis. Infect. Immun. 37:422-426. [DOI] [PMC free article] [PubMed] [Google Scholar]