

Abstract
Capsular polysaccharides are important virulence factors of invasive bacterial pathogens. Here we studied the role of the virulence (Vi) capsular polysaccharide of Salmonella enterica serotype Typhi (S. Typhi) in preventing innate immune recognition by complement. Comparison of capsulated S. Typhi with a noncapsulated mutant (ΔtviBCDE vexABCDE mutant) revealed that the Vi capsule interfered with complement component 3 (C3) deposition. Decreased complement fixation resulted in reduced bacterial binding to complement receptor 3 (CR3) on the surface of murine macrophages in vitro and decreased CR3-dependent clearance of Vi capsulated S. Typhi from the livers and spleens of mice. Opsonization of bacteria with immune serum prior to intraperitoneal infection increased clearance of capsulated S. Typhi from the liver. Our data suggest that the Vi capsule prevents CR3-dependent clearance, which can be overcome in part by a specific antibody response.
Salmonella enterica serotype Typhi (S. Typhi) causes an estimated 21 million annual cases of typhoid fever, a severe systemic infection resulting in 200,000 to 600,000 fatalities per year (5, 37). After ingestion, S. Typhi invades the intestinal mucosa, but symptoms develop only after an average incubation period of 2 weeks (23). The relatively long incubation period of typhoid fever suggests that S. Typhi can evade or suppress detection by the innate immune system during the initial stages of infection (26, 28, 33). However, the virulence mechanisms that enable S. Typhi to evade components of the innate immune system early after infection have long remained elusive.
One arm of the innate immune system involved in detection of invasive microbes is the complement system (9, 38). Complement deposition on the bacterial cell surface and opsonophagocytosis can be prevented by capsular polysaccharides of invasive Gram-negative pathogens, including Neisseria meningitidis, Klebsiella pneumoniae, and Escherichia coli isolates associated with extraintestinal infections (1, 15, 16, 35). S. Typhi produces the virulence (Vi) capsular polysaccharide (8), which is encoded by the viaB locus (17). The viaB locus is a 14-kb DNA region containing genes required for the regulation (tviA), the biosynthesis (tviBCDE), and the export (vexABCDE) of the Vi capsular polysaccharide (36). In S. Typhi, the role of the Vi capsular polysaccharide in reducing complement deposition has not been convincingly demonstrated. The viaB locus of S. Typhi is located on a 134-kb DNA region, termed Salmonella pathogenicity island 7 (SPI-7) (24). SPI-7 is genetically unstable and can be lost upon laboratory passage (3, 22). Clinical S. Typhi isolates expressing the Vi capsular polysaccharide tend to bind less complement on their surface in vitro than clinical isolates lacking capsule expression (20). While this report concludes that the Vi capsular polysaccharide inhibits opsonophagocytosis, the evidence is not conclusive, because it is based on comparison of nonisogenic, clinical S. Typhi isolates. Genetic differences between these clinical isolates were not defined but likely included the entire SPI-7 region. Additionally, the in vivo relevance of phenotypes attributed to the Vi capsular polysaccharide remains to be established using animal models.
With the exception of higher primates, vertebrate hosts are resistant to infection with the human-adapted S. Typhi, which has prevented the use of animal models to investigate the in vivo relevance of results obtained using tissue culture. While mice orally inoculated with S. Typhi are not suited to study the development of typhoid fever, we reasoned that this animal model could be used for studying isolated steps during infection, provided that the relevant interactions were not species specific. Here we investigated the role of the Vi capsular polysaccharide on complement-mediated phagocytosis and its impact on bacterial clearance during an infection.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Vi-positive (Vi+) S. Typhi isolate Ty2 (ATCC 19430) and E. coli strain W3110 (ATCC 39936) were obtained from the American Type Culture Collection. Plasmid pDC5 (27) and strain SW74 (39) have been described previously.
Bacteria were routinely cultured in LB broth (10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter NaCl) or on LB agar (15 g/liter agar) plates. To induce expression of the Vi capsular polysaccharide for experiments, strains were statically cultured overnight at 37°C in superoptimal broth (SOB) medium supplemented with magnesium salts (20 g/liter tryptone, 5 g/liter yeast extract, 10 mM NaCl, 10 mM KCl, 10 mM MgCl2, 10 mM MgSO4). When appropriate, the following antibiotics were added at the indicated concentrations: nalidixic acid, 0.05 mg/ml; chloramphenicol (Cm), 0.03 mg/ml; kanamycin (Kan), 0.05 mg/ml; and carbenicillin, 0.1 mg/ml.
Mutant construction.
Regions upstream (flanking region 1) and downstream (flanking region 2) of the vexE gene were amplified (PCR Supermix Hi-Fi; Invitrogen) from strain Ty2 using primer pairs 123/124 and 125/126 (Table 1), respectively. Flanking regions 1 and 2 were digested with XbaI, ligated together with T4 DNA ligase, and PCR amplified with primers 123 and 126. The resulting fragment was gel purified and cloned into the SalI site of the suicide plasmid pRDH10, yielding pSPN55. The KSAC kanamycin resistance cassette of pBS34 was cloned into the XbaI site of pSPN55, generating pSPN58. Plasmid pSPN58 was introduced into S. Typhi (Ty2) by conjugation (S17-1 λpir), and exconjugants were selected on Simmons citrate agar (BD) supplemented with 40 mM histidine, 40 mM tryptophan, and Kan. Kanr Cms colonies were screened by PCR with combinations of primers 127 and 128 as well as primers 34 and 35. A positive mutant was dubbed SPN459 [Ty2 ΔvexE(−15 to +1968)::KSAC] and confirmed to be Vi negative by slide agglutination with anti-Vi serum (BD).
TABLE 1.
Primers for cloning experiments
| Primer no. | Sequence (5′ to 3′)a |
|---|---|
| 34 | GGCATAAATTCCGTCAGC |
| 35 | TGATGACGAGCGTAATGG |
| 123 | TTCACCGTCGACAGCCAAGCAATCGCTACG |
| 124 | TTCACCTCTAGAAGCCTTATTCACGCATCC |
| 125 | TTCACCTCTAGATAATAAGCGATTTAATTGCGGTAG |
| 126 | TACGCCGTCGACCATTCATAACCCGTTCACG |
| 127 | TGATTCTGTCCGTAGAGC |
| 128 | TCAGCGAAAGCGAACACC |
| 129 | AAGGAACGGATTTTGTGG |
| 130 | TAGCCTCTTTTGACGAGC |
| AS1 | GAATTCTGCGTGAATAAGGCTGAGTAAGG |
| AS2 | TCTAGATTAACTATCCCTACGTATAATGTTTCG |
Boldface, added 5′ DNA; underlined, added restriction site.
For complementation, the vexE gene from S. Typhi strain Ty2 was PCR amplified using primers AS1 and AS2 and was cloned into pCR2.1 using the TOPO TA cloning kit (Invitrogen). The XbaI- and EcoRI-digested insert was gel purified and ligated into the XbaI- and EcoRI-digested vector pWSK29. The resulting plasmid was named pAS1.
Analyses of Vi expression.
Detection of Vi expression by flow cytometry was performed as described previously (11) using the DNA-specific stain propidium iodide, rabbit anti-Vi serum (1:250 dilution; BD), and goat anti-rabbit fluorescein isothiocyanate (FITC) conjugate (1:250 dilution; Jackson ImmunoResearch).
For agglutination assays, overnight cultures were prepared as described above and 1 ml was harvested by centrifugation at 15,000 × g for 5 min. Cells were resuspended in 10 μl of rabbit anti-Vi serum (BD). This suspension was placed on a glass slide, and agglutination was determined after 5 min.
C3 and IgG binding assays.
To determine murine complement component 3 (C3) binding, 1 ml of a bacterial overnight culture was washed with phosphate-buffered saline (PBS) and then resuspended in 0.1 ml of 10% mouse serum diluted in PBS. Samples were then placed at 37°C for 30 min. The bacteria were washed in PBS and killed by incubation in PBS containing 0.1% (wt/vol) sodium azide for 20 min. Samples were washed with PBS, and bacteria were stained with an FITC-conjugated goat anti-murine C3b monoclonal antibody (1:250 dilution; MP Biomedicals) for 1 h in the dark at room temperature. The samples were then washed three times with PBS and analyzed by flow cytometry (LSRII; Becton Dickinson).
To determine murine IgG binding, the protocol described above was performed using a 1:250 dilution of an FITC-conjugated goat anti-murine IgG monoclonal antibody (Sigma).
To determine human C3 binding, bacteria were incubated with either complement component 5 (C5)-depleted human serum (Quidel) or C3-depleted human serum (Quidel), and the bacteria were stained with an FITC-conjugated goat anti-human C3b monoclonal antibody (1:250 dilution; MP Biomedicals).
Serum sensitivity.
In serum sensitivity assays, bacterial cells (1 × 107 CFU of S. Typhi strains or 5 × 108 CFU of E. coli strains) were incubated at 37°C in 10% human serum complement (Quidel) diluted in PBS. At the indicated time points, bacterial survival was quantified by spreading serial 10-fold dilutions on LB agar plates containing the appropriate antibiotics.
Macrophage assays.
Bone marrow-derived macrophages (BMDMs) were isolated as described previously (30). Cells were seeded in 24-well plates at a density of 5 × 105 cells per well and incubated overnight in complete RPMI medium without antibiotics. THP-1 cells were obtained from the American Type Culture Collection and were differentiated with phorbol 12-myristate 13-acetate as described previously (34).
To determine bacterial attachment, BMDMs or differentiated THP-1 cells were incubated for 1 h at 4°C. Bacteria were added to cells for 1 h at 4°C at a multiplicity of infection (MOI) of 2 bacteria per macrophage. Cells were washed three times with 0.5 ml PBS, and the numbers of cell-associated bacteria were determined by spreading serial 10-fold dilutions on LB agar plates.
To determine bacterial uptake, BMDMs or differentiated THP-1 cells were infected with the indicated strains for 1 h at 37°C at an MOI of 2 bacteria per macrophage-like cell. Cells were then washed three times with 0.5 ml PBS. RPMI medium (0.5 ml) containing 0.1 mg/ml gentamicin (Gibco) was then added to the cells for 90 min. Macrophage-like cells were then washed 3 times with 0.5 ml PBS and lysed in 0.5 ml of sterile water. The recovery of bacteria from macrophages was quantified by spreading serial 10-fold dilutions on LB agar plates containing the appropriate antibiotics. When indicated, complement receptor 3 (CR3)-mediated uptake in THP-1 cells was blocked by adding 2.5 μg of anti-CD11b (clone CBRM1/5; BioLegend) per well 90 min prior to infection with bacterial strains.
Animal experiments.
Experimental procedures were approved by the University of California, Davis, Institutional Animal Care and Use Committee. Wild-type (C57BL/6J) mice and congenic CR3-deficient (B6.129S4-Itgamtm1Myd/J) mice, generated by disrupting the gene encoding CD11b, were obtained from Jackson Laboratories. For bacterial infection experiments, 6- to 8 week-old female wild-type and CR3-deficient mice were injected intraperitoneally with 1 × 108 CFU. At 4 h after infection, groups of four mice were euthanized and liver and spleen samples were collected and processed.
To generate immune serum, four wild-type mice were injected twice a month for 3 months with 1 × 105 CFU of the capsulated S. Typhi strain (Ty2). After 3 months, the serum from four immunized mice was collected and pooled. For mouse experiments using opsonized bacteria, 1 ml of a bacterial overnight culture was washed with PBS and then resuspended in 0.1 ml of 10% immune mouse serum diluted in PBS. Samples were then placed at 37°C for 30 min. The bacteria were then washed in PBS to remove excess serum that was not associated with bacteria and resuspended to the appropriate volume in PBS. Next, 6- to 8-week-old female naïve wild-type mice were injected intraperitoneally with 1 × 108 CFU each. At 4 h after infection, groups of four mice were euthanized and the livers and spleens were collected. To determine Salmonella numbers in the livers and spleens, tissue samples were homogenized in PBS and serial 10-fold dilutions were spread onto LB agar plates containing the appropriate antibiotics.
Statistical analysis.
For statistical analysis, raw data underwent logarithmic transformation. Statistical analysis of the data was performed by an analysis of variance test, followed by either an unpaired Student's t test (animal experiments and serum resistance assays) or a paired Student's t test (for tissue culture experiments).
RESULTS
The Vi capsule reduces C3 deposition and increases serum resistance.
Detection of the lipopolysaccharide (LPS) O antigen of Gram-negative bacteria by C3 is a pattern recognition event that initiates the alternative pathway of complement activation (9, 38). During the initial steps of this pathway, C3b, a cleavage products of C3, binds covalently to hydroxyl groups present in LPS, a process known as C3 fixation. Vi capsule-positive S. Typhi isolates fix less complement to their surface than nonisogenic S. Typhi isolates which are noncapsulated (20). To investigate whether this phenotype could be solely attributed to the expression of the Vi capsular polysaccharide, we compared C3 fixation on the surface of a capsulated S. Typhi isolate (Ty2) with that on the surface of an isogenic mutant carrying a precise deletion of the tviBCDE vexABCDE capsule biosynthesis genes (ΔtviB-vexE mutant, SW74). S. Typhi strain Ty2, but not the isogenic ΔtviB-vexE mutant (SW74), expressed the Vi capsule, as shown by flow cytometry (Fig. 1A and B) and slide agglutination with rabbit anti-Vi serum (data not shown). Comparison of the capsulated S. Typhi strain Ty2 with its isogenic ΔtviB-vexE mutant was thus well suited to specifically investigate the role of the Vi capsule on C3 deposition.
FIG. 1.

Vi capsule expression detected by flow cytometry. Cells of the S. Typhi wild type (A), an S. Typhi ΔtviB-vexE mutant (B), E. coli strain W3110 (C), E. coli strain W3110 carrying the cloned viaB locus (pDC5) (D), an S. Typhi vexE mutant (E), or an S. Typhi vexE mutant complemented with the cloned vexE gene (pAS1) (F) were labeled with rabbit anti-Vi serum/goat-anti rabbit FITC conjugate (Vi expression, y axis), and fluorescence intensities were determined for 10,000 particles. Each experiment was repeated three times independently with similar outcomes, and a representative example is shown.
C3 fixation on the bacterial surface was monitored by incubating bacteria in 10% normal human serum depleted for C5, a component of the membrane attack complex, to avoid bacterial killing during the assay. Compared to capsulated S. Typhi strain Ty2, the isogenic ΔtviB-vexE mutant (SW74) deposited more C3 on its surface, as indicated by an increased peak fluorescence intensity detected by flow cytometry with an anti-C3 FITC conjugate (Fig. 2A). No C3 fixation was observed in a control experiment with C3-depleted human serum (Fig. 2B). These data suggested that expression of the Vi capsule reduced C3 deposition on the bacterial surface.
FIG. 2.

The Vi capsule reduces C3 fixation and increases complement resistance. (A and B) Fixation of C3 after incubation of capsulated (wild-type) and noncapsulated (ΔtviB-vexE) S. Typhi strains in C5-depleted human serum (A) or in C3-depleted human serum (B) detected by flow cytometry using an anti-human C3 FITC conjugate. (C and D) Survival of capsulated and noncapsulated S. Typhi strains (C) or capsulated (viaB on plasmid pDC5) and noncapsulated (wild-type [W3110]) E. coli strains (D) in normal human serum. The experiment was repeated three times independently, and data points represent averages ± standard deviations. (E) Fixation of C3 after incubation of capsulated and noncapsulated S. Typhi strains in murine serum detected by flow cytometry using an anti-murine C3 FITC conjugate. (F) Fixation of C3 after incubation of capsulated (vexE mutant complemented with pAS1) and noncapsulated (vexE mutant) S. Typhi strains in murine serum detected by flow cytometry using an anti-murine C3 FITC conjugate. The experiments whose results are presented in panels A, B, E, and F were repeated three times independently with similar outcomes each time, and a representative examples are shown.
C3 fixation on the cell surface activates the alternative complement pathway, which can result in bacterial killing through formation of the membrane attack complex. To investigate whether inhibition of C3 deposition by the Vi capsule contributes to serum resistance, capsulated S. Typhi (Ty2) and its isogenic ΔtviB-vexE mutant (SW74) were incubated in 10% normal human serum and survival was monitored over time. While capsulated S. Typhi (Ty2) was serum resistant, the ΔtviB-vexE mutant (SW74) was killed over time. One hour after addition of serum, the ΔtviB-vexE mutant (SW74) was recovered in significantly lower numbers than the wild-type strain (P < 0.05; Fig. 2C). These data were consistent with those from previous reports showing that Vi capsule-negative S. Typhi isolates exhibit increased serum sensitivity (14). Introduction of the cloned tviABCDE vexABCDE genes (pDC5) into E. coli strain W3110 resulted in expression of the Vi capsular polysaccharide (Fig. 1C and D) and was sufficient to increase serum resistance (P < 0.05: Fig. 2D).
Murine complement components forming the membrane attack complex are labile in vitro, rendering serum from this species unsuitable for studying serum resistance in the test tube. We thus determined whether inhibition of C3 fixation by the Vi capsule could be reproduced with murine complement. After incubation in 10% murine serum, the ΔtviB-vexE mutant (SW74) exhibited increased C3 deposition compared to the capsulated wild type (Ty2) (Fig. 2E). These data suggested that the Vi capsule reduced fixation of both murine and human complement.
The Vi capsule reduces binding by CR3.
C3 fragments (C3b and iC3b) bound to the bacterial surface act as important opsonins for efficient phagocytosis. This process requires binding of C3 fragments deposited on the bacterial surface to their specific complement receptor, CR3 (also known as CD11b/CD18), on the surface of phagocytes. Similar to previous studies with capsulated and noncapsulated S. Typhi isolates (20), we found that the capsulated S. Typhi wild type (Ty2) bound in lower numbers to human macrophage-like (THP-1) cells than the noncapsulated ΔtviB-vexE mutant (SW74) (Fig. 3A), which resulted in decreased uptake, as determined by gentamicin protection assays (Fig. 3B). This capsule-mediated inhibition of phagocytosis was dependent on CR3, because treatment of THP-1 cells with blocking anti-CD11b antibodies reduced uptake of the noncapsulated ΔtviB-vexE mutant (SW74) to the levels of the capsulated S. Typhi wild type (Ty2) (Fig. 3B).
FIG. 3.

The Vi capsule reduces CR3-mediated interaction with macrophages. (A and B) Binding to (after incubation at 4°C) (A) and phagocytosis of (gentamicin protection assay) (B) capsulated bacteria (the S. Typhi wild type or the S. Typhi vexE mutant complemented with pAS1) and noncapsulated bacteria (an S. Typhi ΔtviB-vexE mutant or an S. Typhi vexE mutant) to human macrophage-like THP-1 cells in the absence (black bars) or presence (open bars) of blocking anti-human CD11b antibodies. (C and D) Binding to (after incubation at 4°C) (C) and phagocytosis of (gentamicin protection assay) (D) the indicated bacterial strains by murine BMDMs from wild-type mice (C57BL/6, black bars) or CR3-deficient mice (open bars). Each experiment was repeated at least three times independently, and data are shown as averages ± standard errors. The statistical significance of the differences is indicated above. NS, not significant.
To interpret results from murine challenge studies, it was relevant to determine whether reduced binding of capsulated S. Typhi could also be observed with murine phagocytes. As an additional control, we constructed a ΔvexE mutant in S. Typhi strain Ty2 (SPN459) by allelic exchange. The VexE protein is thought to transfer an unidentified acyl moiety, which anchors the Vi capsular polysaccharide in the bacterial outer membrane. Inactivation of the vexE gene results in release of soluble Vi capsular polysaccharide, while the bacterial cell surface remains noncapsulated (36). Consistent with this previous report, the Vi capsular polysaccharide was no longer detectable on the surface of the vexE mutant by flow cytometry (Fig. 1E). Introduction of the cloned vexE gene (pAS1) restored Vi capsule expression (Fig. 1F) and reduced murine C3 deposition on the surface of the vexE mutant (Fig. 2F). The ΔtviB-vexE mutant (SW74) and the vexE mutant (SPN459) were bound by BMDMs in significantly higher numbers (P < 0.05) than their capsulated parent (Ty2) or the complemented vexE mutant [SPN459(pAS1)]) (Fig. 3C). Importantly, no differences between the binding of capsulated or noncapsulated S. Typhi strains were observed when the experiment was repeated with BMDMs from CR3-deficient mice. Thus, the Vi capsule reduced CR3-mediated binding of S. Typhi to murine macrophages.
We next measured bacterial uptake by BMDMs from wild-type mice (C57BL/6) and CR3-deficient mice using a gentamicin protection assay. The ΔtviB-vexE mutant (SW74) was phagocytosed in significantly higher numbers by BMDMs of wild-type mice (P < 0.05) than by those of its capsulated parent (Ty2) (Fig. 3D). In contrast, no differences between phagocytosis of capsulated or noncapsulated S. Typhi strains were observed with BMDMs from CR3-deficient mice.
Collectively, results from in vitro studies with murine serum (Fig. 2E and F) and murine macrophages (Fig. 3C and D) supported the idea that consequences of a Vi capsule-mediated inhibition of C3 fixation could be studied in a mouse model.
The Vi capsule reduces CR3-mediated clearance of S. Typhi.
The role of the Vi capsule-mediated inhibition of opsonophagocytosis was investigated at an early time point (4 h) after intraperitoneal infection of mice, because human-adapted S. Typhi does not survive for extended periods of time in murine tissue. Capsulated S. Typhi (Ty2) was recovered in higher numbers than the ΔtviB-vexE mutant (SW74) from the blood, livers, and spleens of mice (Fig. 4). Possible reasons for the higher recovery of Vi capsulated S. Typhi included a capsule-mediated reduction of clearance through CR3 and/or an increased serum sensitivity of noncapsulated bacteria. The experiment was repeated using CR3-deficient mice (i.e., CD11b-deficient mice) to investigate the contribution of CR3-mediated opsonophagocytosis to clearance. In the livers and spleens of CR3-deficient mice, the capsulated S. Typhi strain (Ty2) and the ΔtviB-vexE mutant (SW74) were recovered in similar numbers (Fig. 4A and B), suggesting that CR3-mediated phagocytosis was primarily responsible for an enhanced clearance of noncapsulated bacteria from these organs in wild-type mice. In the blood, clearance of the ΔtviB-vexE mutant (SW74) was significantly reduced in CR3-deficient mice compared to that in wild-type mice. However, the capsulated S. Typhi strain (Ty2) was still recovered in significantly higher numbers than the ΔtviB-vexE mutant (SW74) from the blood of CR3-deficient mice (Fig. 4C). Thus, in blood the capsule contributed to survival by inhibiting opsonophagocytosis and by a second mechanism, which likely represents capsule-mediated serum resistance. Since clearance from organs (i.e., liver and spleen) was largely due to a single mechanism (i.e., CR3-mediated phagocytosis), we focused in subsequent experiments on these sites to study phagocytic clearance.
FIG. 4.
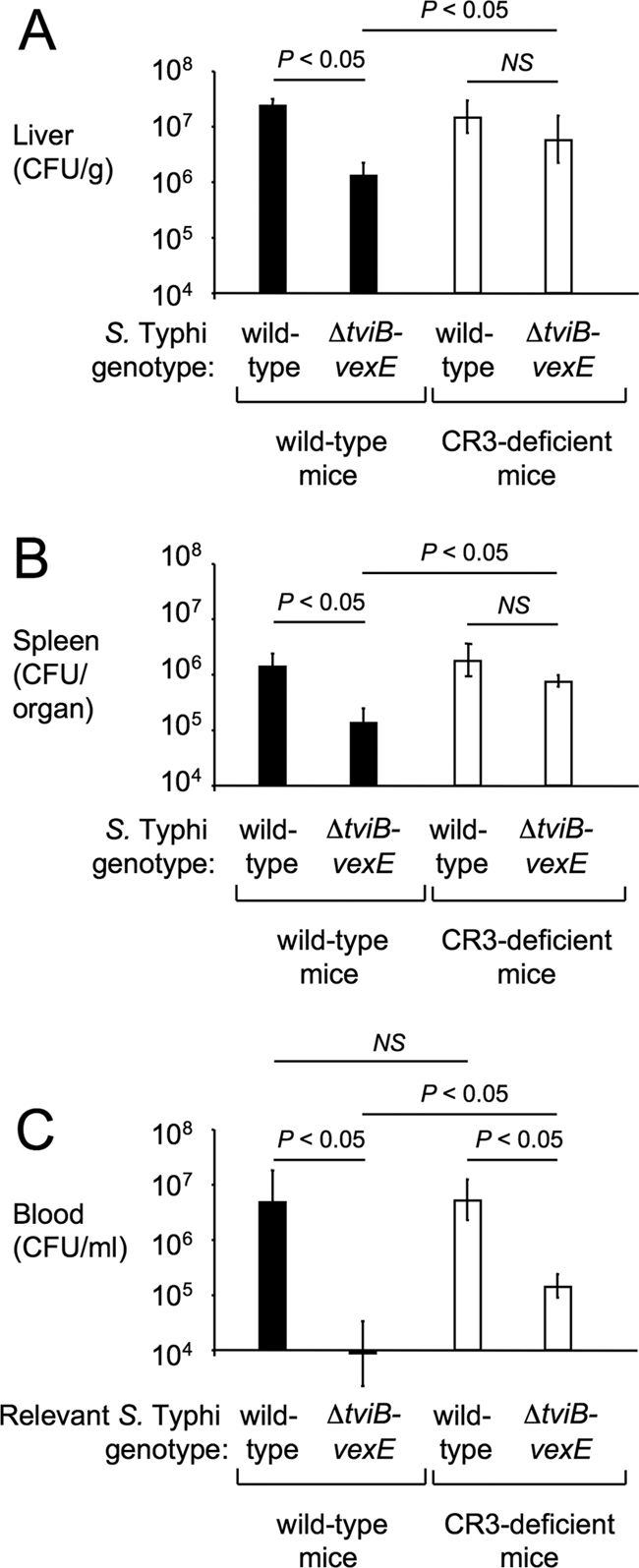
The Vi capsule reduces CR3-dependent clearance of S. Typhi from organs of mice. Recovery of capsulated (wild-type) and noncapsulated (ΔtviB-vexE) S. Typhi strains from the livers (A), spleens (B), or blood (C) of wild-type mice (C57BL/6, black bars) or CR3-deficient mice (open bars) 4 h after intraperitoneal infection. Bars represent averages ± standard deviations from four animals. The statistical significance of the differences is indicated above each graph. NS, not significant.
Antibody-mediated opsonization promotes clearance of Vi capsulated bacteria from the livers but not the spleens of mice.
We next investigated whether opsonization with specific antibodies would promote clearance of Vi capsulated S. Typhi from organs of mice. Serum from naïve mice or mice immunized with the S. Typhi wild type (immune serum) was used to opsonize bacterial strains, which were subsequently analyzed by flow cytometry or injected intraperitoneally into mice. Opsonization with immune serum resulted in equal deposition of IgG on the surfaces of the S. Typhi wild type (Ty2) and the ΔtviB-vexE mutant (SW74) (Fig. 5A), while more C3 was deposited on the surface of the ΔtviB-vexE mutant (SW74) (Fig. 5B). Opsonization with naïve serum did not result in deposition of IgG on the surface of S. Typhi strains (Fig. 5C). Opsonization with immune serum did not significantly alter bacterial recovery from the spleens of mice (Fig. 5D). However, opsonization with immune serum reduced (P < 0.05) the numbers of the Vi capsulated S. Typhi wild type recovered from the liver to the levels of the noncapsulated ΔtviB-vexE mutant (SW74) (Fig. 5E). These data suggested that opsonization with specific antibodies enhanced clearance of capsulated bacteria in the liver, but not the spleen, thereby in part overcoming the antiphagocytic properties of the Vi capsule in vivo.
FIG. 5.

Opsonization with immune serum increases clearance of capsulated S. Typhi from the liver. (A to C) Deposition of IgG (A and C) or C3 (B) on the surface of the capsulated S. Typhi wild type (Ty2) or a noncapsulated S. Typhi strain (ΔtviB-vexE mutant) after incubation in naïve mouse serum (C) or immune mouse serum (A and B) detected by flow cytometry using an anti-mouse C3 FITC conjugate (B) or anti-mouse IgG FITC conjugate (A and C). (D and E) The indicated bacterial strains were opsonized in naïve mouse serum (black bars) or immune mouse serum (open bars) and then injected intraperitoneally into mice (C57BL/6). Bars represent average numbers of bacteria recovered from the spleens (D) or the livers (E) of four animals collected 4 h after infection ± standard deviations. The statistical significance of the differences is indicated above each graph. NS, not significant.
DISCUSSION
Complement, specifically C3, senses conserved microbial structures, such as LPS of Gram-negative bacteria, which represents an important pattern recognition event in the detection of microbes by the innate immune system (9, 38). Complement activation results in the fixation of C3b on the bacterial surface, because this C3 cleavage product contains a reactive thioester group which forms esters with free hydroxyl groups of sugars (31). The O-antigen repeat unit of S. Typhi LPS contains multiple hydroxyl groups and promotes complement fixation (Fig. 6A). In contrast, the Vi capsular polysaccharide, a homopolymer of (1→4)-2-acetamido-3-O-acetyl-2-deoxy-α-d-galacturonic acid (13), does not contain free hydroxyl groups that could bind C3b covalently (Fig. 6B). The lack in the Vi polysaccharide of free hydroxyl groups available for complement fixation may explain why capsule expression reduced complement deposition. The finding that the Vi capsule prevents complement deposition reinforces the concept that inhibition of C3 fixation is a virulence strategy shared by a number of invasive capsulated bacterial pathogens, including extraintestinal pathogenic Escherichia coli (15, 35), Klebsiella pneumoniae (1), Neisseria meningitidis (16), Staphylococcus aureus (6), Streptococcus pneumoniae (2), and group B Streptococcus (21).
FIG. 6.

Repeat unit structure of the Vi capsule and the S. Typhi O antigen. Structures of the O-antigen repeat unit in S. Typhi LPS (A) (18) and the repeat unit of the Vi capsular polysaccharide (B) (13) according to previous reports. Note the presence in the O antigen and the absence in the Vi capsule of free hydroxyl groups available for covalent binding of C3b.
One of the consequences of pattern recognition of LPS by C3 is that the resulting complement fixation (opsonization) enables macrophages to phagocytose bacteria using CR3 (9). Consistent with this concept, inhibition of complement fixation by the Vi capsule reduced CR3-dependent binding to macrophages and impaired CR3-dependent clearance of S. Typhi from organs of mice. An inhibition of complement-mediated phagocytosis is a virulence property conferred by the capsular polysaccharides of other invasive bacterial pathogens, including K. pneumoniae, N. meningitidis, S. aureus, S. pneumoniae, and E. coli isolates associated with extraintestinal infections (2, 7, 10, 15, 35). Vaccination with purified Vi capsular polysaccharide confers protection against typhoid fever, suggesting that dissemination of S. Typhi involves an extracellular phase that renders the pathogen susceptible to Fc receptor-mediated phagocytosis and/or serum-mediated killing (19, 29, 32). The nature of this extracellular phase remains poorly characterized. A possible scenario has been suggested for S. enterica serotype Dublin (S. Dublin), an invasive pathogen of cattle, which can express the Vi capsular polysaccharide (12). After invading the intestinal mucosa and reaching the mesenteric lymph nodes, S. Dublin exits extracellularly into the efferent lymphatics (25), from where it disseminates throughout the body. Our data suggest that specific antibodies can enhance elimination of circulating capsulated bacteria, because opsonization with immune serum prior to infection resulted in increased clearance of S. Typhi from the liver. However, this mechanism was not equally effective for enhancing clearance of capsulated S. Typhi from the spleen. It may be relevant mentioning in this context that adaptive immune responses are not always sufficient to control S. Typhi infection, as suggested by the clinical observation that relapses can occur after a primary episode of typhoid fever (4).
Acknowledgments
This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program grant number C06 RR12088-01 from the National Center for Research Resources, National Institutes of Health. C.T. is supported by Scientist Development grant 0835248N from the American Heart Association. S.-P.N. was supported by the Floyd and Mary Schwall Fellowship in Medical Research and Public Health Service grant AI060555. Work in A.J.B.'s laboratory is supported by Public Health Service grants AI040124, AI044170, AI073120, AI076246, and AI088122.
Editor: A. Camilli
Footnotes
Published ahead of print on 22 November 2010.
REFERENCES
- 1.Alvarez, D., S. Merino, J. M. Tomas, V. J. Benedi, and S. Alberti. 2000. Capsular polysaccharide is a major complement resistance factor in lipopolysaccharide O side chain-deficient Klebsiella pneumoniae clinical isolates. Infect. Immun. 68:953-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown, E. J., K. A. Joiner, R. M. Cole, and M. Berger. 1983. Localization of complement component 3 on Streptococcus pneumoniae: anti-capsular antibody causes complement deposition on the pneumococcal capsule. Infect. Immun. 39:403-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bueno, S. M., et al. 2004. Precise excision of the large pathogenicity island, SPI7, in Salmonella enterica serovar Typhi. J. Bacteriol. 186:3202-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark, T. W., C. Daneshvar, M. Pareek, N. Perera, and I. Stephenson. 2010. Enteric fever in a UK regional infectious diseases unit: a 10 year retrospective review. J. Infect. 60:91-98. [DOI] [PubMed] [Google Scholar]
- 5.Crump, J. A., S. P. Luby, and E. D. Mintz. 2004. The global burden of typhoid fever. Bull. World Health Organ. 82:346-353. [PMC free article] [PubMed] [Google Scholar]
- 6.Cunnion, K. M., J. C. Lee, and M. M. Frank. 2001. Capsule production and growth phase influence binding of complement to Staphylococcus aureus. Infect. Immun. 69:6796-6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunnion, K. M., H. M. Zhang, and M. M. Frank. 2003. Availability of complement bound to Staphylococcus aureus to interact with membrane complement receptors influences efficiency of phagocytosis. Infect. Immun. 71:656-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Felix, A., and R. M. Pitt. 1934. A new antigen of B. typhosus. Lancet 227:186-191. [Google Scholar]
- 9.Gasque, P. 2004. Complement: a unique innate immune sensor for danger signals. Mol. Immunol. 41:1089-1098. [DOI] [PubMed] [Google Scholar]
- 10.Glynn, A. A., and C. J. Howard. 1970. The sensitivity to complement of strains of Escherichia coli related to their K antigens. Immunology 18:331-346. [PMC free article] [PubMed] [Google Scholar]
- 11.Haneda, T., et al. 2009. The capsule-encoding viaB locus reduces intestinal inflammation by a Salmonella pathogenicity island 1-independent mechanism. Infect. Immun. 77:2932-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashimoto, Y., and A. Q. Khan. 1997. Comparison of ViaB regions of Vi-positive organisms. FEMS Microbiol. Lett. 157:55-57. [DOI] [PubMed] [Google Scholar]
- 13.Heyns, K., and G. Kiessling. 1967. Strukturaufklarung des Vi-antigens aus Citrobacter freundii (E. coli) 5396/38. Carbohydr. Res. 3:340-353. [Google Scholar]
- 14.Hone, D. M., A. M. Harris, V. Lim, and M. M. Levine. 1994. Construction and characterization of isogenic O-antigen variants of Salmonella typhi. Mol. Microbiol. 13:525-530. [DOI] [PubMed] [Google Scholar]
- 15.Horwitz, M. A., and S. C. Silverstein. 1980. Influence of the Escherichia coli capsule on complement fixation and on phagocytosis and killing by human phagocytes. J. Clin. Invest. 65:82-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarvis, G. A., and N. A. Vedros. 1987. Sialic acid of group B Neisseria meningitidis regulates alternative complement pathway activation. Infect. Immun. 55:174-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson, E. M., B. Krauskopf, and L. S. Baron. 1965. Genetic mapping of Vi and somatic antigenic determinants in Salmonella. J. Bacteriol. 90:302-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kingsley, R. A., and A. J. Bäumler. 2000. Host adaptation and the emergence of infectious disease: the salmonella paradigm. Mol. Microbiol. 36:1006-1014. [DOI] [PubMed] [Google Scholar]
- 19.Klugman, K. P., et al. 1987. Protective activity of Vi capsular polysaccharide vaccine against typhoid fever. Lancet ii:1165-1169. [DOI] [PubMed]
- 20.Looney, R. J., and R. T. Steigbigel. 1986. Role of the Vi antigen of Salmonella typhi in resistance to host defense in vitro. J. Lab. Clin. Med. 108:506-516. [PubMed] [Google Scholar]
- 21.Marques, M. B., D. L. Kasper, M. K. Pangburn, and M. R. Wessels. 1992. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect. Immun. 60:3986-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nair, S., et al. 2004. Salmonella enterica serovar Typhi strains from which SPI7, a 134-kilobase island with genes for Vi exopolysaccharide and other functions, has been deleted. J. Bacteriol. 186:3214-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olsen, S. J., et al. 2003. Outbreaks of typhoid fever in the United States, 1960-99. Epidemiol. Infect. 130:13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parkhill, J., et al. 2001. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413:848-852. [DOI] [PubMed] [Google Scholar]
- 25.Pullinger, G. D., et al. 2007. Systemic translocation of Salmonella enterica serovar Dublin in cattle occurs predominantly via efferent lymphatics in a cell-free niche and requires type III secretion system 1 (T3SS-1) but not T3SS-2. Infect. Immun. 75:5191-5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raffatellu, M., et al. 2006. Capsule-mediated immune evasion: a new hypothesis explaining aspects of typhoid fever pathogenesis. Infect. Immun. 74:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raffatellu, M., et al. 2007. The capsule encoding viaB locus reduces interleukin-17 expression and mucosal innate responses in the bovine intestinal mucosa during infection with Salmonella enterica serotype Typhi. Infect. Immun. 75:4342-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raffatellu, M., R. P. Wilson, S. E. Winter, and A. J. Bäumler. 2008. Clinical pathogenesis of typhoid fever. J. Infect. Dev. Ctries. 2:260-266. [DOI] [PubMed] [Google Scholar]
- 29.Robbins, J. D., and J. B. Robbins. 1984. Reexamination of the protective role of the capsular polysaccharide (Vi antigen) of Salmonella typhi. J. Infect. Dis. 150:436-449. [DOI] [PubMed] [Google Scholar]
- 30.Rolan, H. G., and R. M. Tsolis. 2007. Mice lacking components of adaptive immunity show increased Brucella abortus virB mutant colonization. Infect. Immun. 75:2965-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sahu, A., T. R. Kozel, and M. K. Pangburn. 1994. Specificity of the thioester-containing reactive site of human C3 and its significance to complement activation. Biochem. J. 302(Pt 2):429-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tacket, C. O., et al. 1986. Safety and immunogenicity of two Salmonella typhi Vi capsular polysaccharide vaccines. J. Infect. Dis. 154:342-345. [DOI] [PubMed] [Google Scholar]
- 33.Tsolis, R. M., G. M. Young, J. V. Solnick, and A. J. Bäumler. 2008. From bench to bedside: stealth of enteroinvasive pathogens. Nat. Rev. Microbiol. 6:883-892. [DOI] [PubMed] [Google Scholar]
- 34.Tükel, C., et al. 2005. CsgA is a pathogen-associated molecular pattern of Salmonella enterica serotype Typhimurium that is recognized by Toll-like receptor 2. Mol. Microbiol. 58:289-304. [DOI] [PubMed] [Google Scholar]
- 35.Van Dijk, W. C., H. A. Verbrugh, M. E. van der Tol, R. Peters, and J. Verhoef. 1979. Role of Escherichia coli K capsular antigens during complement activation, C3 fixation, and opsonization. Infect. Immun. 25:603-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Virlogeux, I., H. Waxin, C. Ecobichon, and M. Y. Popoff. 1995. Role of the viaB locus in synthesis, transport and expression of Salmonella typhi Vi antigen. Microbiology 141(Pt 12):3039-3047. [DOI] [PubMed] [Google Scholar]
- 37.WHO. 1997. The world health report 1996—fighting disease, fostering development. World Health Forum 18:1-8. [PubMed] [Google Scholar]
- 38.Winter, S. E., A. M. Keestra, R. M. Tsolis, and A. J. Bäumler. 2010. The blessings and curses of intestinal inflammation. Cell Host Microbe 8:36-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winter, S. E., M. Raffatellu, R. P. Wilson, H. Russmann, and A. J. Bäumler. 2008. The Salmonella enterica serotype Typhi regulator TviA reduces interleukin-8 production in intestinal epithelial cells by repressing flagellin secretion. Cell. Microbiol. 10:247-261. [DOI] [PubMed] [Google Scholar]