

Abstract
Influenza A viruses constitute a major and ongoing global public health concern. Current antiviral strategies target viral gene products; however, the emergence of drug-resistant viruses highlights the need for novel antiviral approaches. Cleavage of the influenza virus hemagglutinin (HA) by host cell proteases is crucial for viral infectivity and therefore presents a potential drug target. Peptide-conjugated phosphorodiamidate morpholino oligomers (PPMO) are single-stranded-DNA-like antisense agents that readily enter cells and can act as antisense agents by sterically blocking cRNA. Here, we evaluated the effect of PPMO targeted to regions of the pre-mRNA or mRNA of the HA-cleaving protease TMPRSS2 on proteolytic activation and spread of influenza viruses in human Calu-3 airway epithelial cells. We found that treatment of cells with a PPMO (T-ex5) designed to interfere with TMPRSS2 pre-mRNA splicing resulted in TMPRSS2 mRNA lacking exon 5 and consequently the expression of a truncated and enzymatically inactive form of TMPRSS2. Altered splicing of TMPRSS2 mRNA by the T-ex5 PPMO prevented HA cleavage in different human seasonal and pandemic influenza A viruses and suppressed viral titers by 2 to 3 log10 units, strongly suggesting that TMPRSS2 is responsible for HA cleavage in Calu-3 airway cells. The data indicate that PPMO provide a useful reagent for investigating HA-activating proteases and may represent a promising strategy for the development of novel therapeutics to address influenza infections.
Influenza viruses are responsible for recurrent outbreaks of acute respiratory illness which affect millions of people worldwide. Of the three genera (A, B, and C) of influenza viruses, influenza A viruses represent the most serious threat to public health, causing yearly seasonal outbreaks and occasional pandemics, notably the ongoing swine-origin H1N1 outbreak. The genomes of influenza A viruses consist of eight segments of single-stranded, negative-sense RNA, which together encode 10 to 12 proteins. The virions are enveloped and contain two major spike glycoproteins, hemagglutinin (HA) and neuraminidase (NA). Based on antigenic criteria, 16 HA subtypes (H1 to H16) and 9 NA subtypes (N1 to N9) have been identified.
Influenza virus replication is initiated by the HA, which mediates entry into the target cell through virion binding to sialic-acid containing cell surface receptors and, upon endocytosis, fusion of the viral envelope with the endosomal membrane, resulting in release of viral genomic RNA into the cytoplasm. HA is synthesized as a precursor protein, HA0, and requires cleavage into the subunits HA1 and HA2 by a host cell protease to gain its fusion capacity (14, 22). Proteolytic cleavage of HA is a prerequisite for conformational changes that occur at low pH in the endosome and which expose the hydrophobic fusion peptide of the HA2 subunit and thereby enable membrane fusion (34, 37). The cleavage sites present in HA vary between viral strains, and can affect tissue tropism, virus spread, and pathogenicity. The HAs of highly pathogenic avian influenza viruses (HPAIV) of subtypes H5 and H7 contain a multibasic cleavage site (consensus sequence R-X-R/K-R) which is cleaved by ubiquitous proteases such as furin or PC5/6, supporting systemic infection with an often fatal outcome (14, 18, 38, 43). In contrast, the HAs of most other influenza viruses, including the H1, H2, and H3 subtypes typically infecting humans, contain a monobasic cleavage site, usually an arginine and infrequently a lysine, and require activation by trypsin-like proteases (3, 23). The expression of proteases capable of cleaving HA is restricted to specific tissues, thereby restricting the spread of viral infection.
Human airway trypsin-like protease (HAT) and TMPRSS2 (transmembrane protease serine S1 member 2, also known as epitheliasin) are present in human airway epithelial cells and have been shown to be capable of cleaving HAs having a monobasic cleavage site (5). In agreement with this, TMPRSS2 and the related protease TMPRSS4 have been reported to cleave the HA of the 1918 H1N1 virus at a monobasic cleavage motif (8). However, for many cell types, the protease(s) responsible for HA cleavage remains poorly defined. More extensive profiling of HA-activating proteases in various cell types is of basic research interest and may be useful in the development of novel interventional strategies to address influenza A infections.
Phosphorodiamidate morpholino oligomers (PMO) are single-stranded DNA analogs containing the DNA nucleobases A, C, G, and T and a novel backbone consisting of morpholine rings and phosphorodiamidate intersubunit linkages (40). PMO are water soluble, nuclease resistant, and typically synthesized to a length containing 20 to 25 bases (19, 40). The mechanism of antisense action of PMO is through steric blocking of cRNA (36, 39). PMO are often designed against sequences in the 5′ untranslated region (UTR) and/or the AUG translation start codon region of mRNA, for the purpose of interfering with early events in the process of translation (10, 35, 39). PMO have also been shown to be capable of interfering with spliceosome-mediated reactions of mRNA maturation (16, 24, 28, 29). To facilitate entry into cells, an arginine-rich cell-penetrating peptide (CPP) may be conjugated to PMO to produce peptide-PMO (PPMO) (25, 30, 46). PPMO have shown considerable antiviral activity against a number of positive- and negative-strand RNA viruses in both cell cultures and murine experimental systems (reviewed in reference 35). Against influenza A viruses, PPMO targeting highly conserved sequences of PB1 and NP RNAs had potent and specific antiviral activity against multiple subtypes in experimentally infected cell cultures and mice (13, 15, 26).
In this study, we investigated whether PPMO designed to target the pre-mRNAs or mRNAs of cellular HA-activating proteases could interfere with proteolytic activation of HA and thereby suppress spread of influenza viruses in Calu-3 human airway epithelial cells. We found that TMPRSS2 and TMPRSS4, but not HAT, are expressed in Calu-3 cells. Using different seasonal and pandemic human influenza A virus strains, we show that a splice-altering PPMO was able to specifically interfere with TMPRSS2 mRNA maturation, resulting in expression of enzymatically inactive TMPRSS2 and thereby inhibiting HA cleavage and influenza virus propagation.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
Calu-3 human bronchial epithelial cells were cultured in Dulbecco modified Eagle medium (DMEM)-F-12 Ham (1:1) (Gibco) supplemented with 10% fetal calf serum (FCS), penicillin, streptomycin, and glutamine, with fresh culture medium replenished every 2 to 3 days. Madin-Darby canine kidney (MDCK) cells and A549 human lung carcinoma cells were cultured in DMEM supplemented with 10% FCS, antibiotics, and glutamine. Infection experiments and PPMO treatment of cells were performed using infection medium (DMEM supplemented with 0.1% bovine serum albumin [BSA], glutamine, and antibiotics). All cell growth and incubations occurred at 37°C and 5% CO2.
The influenza viruses used in this study were A/Memphis/14/96 (H1N1) (provided by Robert Webster, St. Jude Children's Research Hospital, Memphis, TN), A/Aichi/2/68 (H3N2), A/ostrich/Italy/984/2000 (H7N1) (provided by Ilaria Capua, Istituto Zooprofilattico Sperimentale delle Venezie, V.le dell'Università, Legnaro, Italy), and pandemic H1N1 virus A/Hamburg/5/2009 (provided by Mikhail Matrosovich, Institute of Virology, Philipps University Marburg, Marburg, Germany). Human viruses were propagated in MDCK cells in infection medium containing 1 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma). A/ostrich/Italy/984/2000 (H7N1) was grown in the allantoic cavities of 11-day-old embryonated chicken eggs. Cell supernatants and allantoic fluid were cleared by low-speed centrifugation and stored at −80°C.
Expression plasmid pCAGGS-HA encoding the HA of A/HongKong/1/68 (H3N2) and plasmids pCAGGS-TMPRSS2 encoding TMPRSS2 and pCAGGS-TMPRSS2(S441A) encoding enzymatically inactive mutant S441A, each with a C-terminal FLAG epitope, have been described previously (5, 6).
Antibodies.
A monoclonal antibody against the influenza A virus nucleoprotein (NP) was provided by Alexander Klimov (Centers for Disease Control, Atlanta, GA). Rabbit serum against H1 was provided by Mikhail Matrosovich (Institute of Virology, Philipps University Marburg, Marburg, Germany). Rabbit serum against H3 was derived from rabbits immunized with A/Aichi/2/68 (H3N2). Polyclonal rabbit anti-FLAG was purchased from Sigma. Species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Dako.
PPMO.
Phosphorodiamidate morpholino oligomers (PMO) were synthesized at AVI BioPharma Inc. (Corvallis, OR) as previously described (40). The cell-penetrating peptide (CPP) (RXRRBR)2XB (R, arginine; X, 6-aminohexanoic acid; B, beta-alanine) was covalently conjugated to the 5′ end of each PMO through a noncleavable linker, to produce peptide-PMO (PPMO), by methods previously described (46).
RNA isolation and RT-PCR analysis.
Total RNA was isolated from Calu-3 cells using the RNeasy minikit (Qiagen) according to the manufacturer's protocol. Total RNA of human tracheobronchial epithelial (HTBE) cells was provided by Mikhail Matrosovich (Institute of Virology, Philipps University Marburg, Germany). For analysis of TMPRSS2 mRNA after PPMO treatment, total RNA was isolated from confluent Calu-3 cells grown in six-well plates and treated with PPMO (25 μM) in infection medium for 24 to 72 h. Reverse transcription-PCR (RT-PCR) was carried out with total RNA using the one-step RT-PCR kit (Qiagen) according to the supplier's protocol. For detection of HAT-, TMPRSS2-, and TMPRSS4-specific mRNAs in Calu-3 and HTBE cells, primers HAT-657fwd (5′ C ATG TGG ATC CTG ACA GCA GC 3′) and HAT-1113rev (5′ TGG GCC ACC AGA GTC ACC CTG GTC 3′) designed to amplify nucleotides (nt) 657 to 1113 of HAT mRNA, TMPRSS2-703fwd (5′ GC AGT GGT TTC TTT ACG CTG 3′) and TMPRSS2-1336rev (5′ C CAG AGG CCC TCC ACT GTC ACC CTG GCA A 3′) designed to amplify nt 703 to 1336 of TMPRSS2 mRNA, and TMPRSS4-111fwd (5′ C ATA GCA CTA CTG AGC C 3′) and TMPRSS4-735rev (5′ GG TAT GTT TCC TGA AGC AG 3′) designed to amplify nt 111 to 735 of TMPRSS4 mRNA (accession number NM_001083947) were used. For detection of TMPRSS2-specific mRNA after PPMO treatment, primers TMPRSS2-108fwd (5′ CTA CGA GGT GCA TCC 3′) and TMPRSS2-1336rev were used to amplify nt 108 to 1336 of TMPRSS2-mRNA. RT-PCR products were resolved on a 0.8% agarose gel stained with ethidium bromide. To determine the sequences of truncated TMPRSS2 mRNA fragments, the respective RT-PCR products were extracted from the gel and sequenced.
Cloning and transient expression of TMPRSS2 mutants Δex4/5 and Δex5.
cDNAs encoding TMPRSS2 lacking exons 4 and 5 (Δex4/5) or TMPRSS2 lacking exon 5 (Δex5) were cloned from total RNAs of T-ex4 and T-ex5 PPMO-treated Calu-3 cells, respectively, by RT-PCR (as described above) using TMPRSS2-specific primers EcoRI-for (5′CA GAA TTC ACC ATG GCT TTG AAC TCA GGG 3′) and NotI-FLAG-rev (5′ CGA TGC GGC CGC CTA CTT GTC ATC GTC ATC CTT GTA GTC TCC GCC GTC TGC CCT CAT 3′). The resulting PCR products encoding Δex4/5 or Δex5, each with a C-terminal FLAG epitope sequence, were subcloned into the mammalian expression plasmid pCAGGS using EcoRI and NotI restriction sites. To analyze expression of Δex4/5 and Δex5, the various pCAGGS expression plasmids were transiently transfected into confluent A549 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol and incubated at 37°C for 24 h. Cell lysates were subjected to SDS-PAGE and Western blot analysis using a FLAG-specific antibody as described below. To analyze the coexpression of HA with Δex4/5, Δex5, TMPRSS2, or S441A, confluent A549 monolayers were cotransfected with appropriate pCAGGS plasmids and analyzed by Western blotting as described previously (5).
Multicycle viral replication.
For analysis of multicycle replication kinetics, Calu-3 cells were seeded in six-well plates, grown to confluence, and inoculated with virus at a multiplicity of infection (MOI) of 0.0001 in infection medium for 1 h. The cells were then washed with phosphate-buffered saline (PBS) and incubated in infection medium with or without TPCK-trypsin (0.5 μg/ml) for 72 h. At 24, 48, and 72 h postinfection (p.i.), supernatants were collected and viral titers determined by plaque titration using MDCK cells. Plaque assays were performed in 24-well plates with Avicel overlay (27). Briefly, MDCK cells were inoculated with 10-fold serial dilutions of each sample for 1 h. The inoculum was then removed and replaced by Avicel overlay containing 1 μg/ml TPCK-trypsin for human viruses or no TPCK-trypsin for the avian H7N1 virus. Cells were incubated for 24 to 48 h and subsequently immunostained against the viral nucleoprotein as described previously (4). Briefly, at 24 h p.i. cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.3% Triton X-100-PBS, incubated with mouse anti-NP and HRP-conjugated secondary antibodies, and then detected using the peroxidase substrate TrueBlue (KPL). To analyze multicycle replication kinetics in PPMO-treated cells, Calu-3 cells were seeded in six-well plates, grown to confluence, and treated with 5, 10, or 25 μM PPMO in infection medium for 24 h. The cells were then inoculated with virus at an MOI of 0.0001 for 1 h in the absence of PPMO, washed with PBS, and replenished with fresh infection medium containing 5, 10, or 25 μM PPMO. At 24, 48, and 72 h p.i., supernatants were collected and viral titers determined by plaque titration as described above.
SDS-PAGE and Western blot analysis.
At the indicated time points, cells were washed with PBS, lysed in reducing SDS-PAGE sample buffer, and heated at 95°C for 5 min. Virus-containing cell supernatants were cleared of cell debris by low-speed centrifugation (4,000 rpm, 5 min), and then virus was pelleted by ultracentrifugation (28,000 rpm, 2 h, 4°C). Pellets were resuspended in reducing SDS sample buffer and heated at 95°C for 5 min. Proteins were subjected to SDS-PAGE (12% gel), transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare), and detected by incubation with primary antibodies and species-specific peroxidase-conjugated secondary antibodies, followed by incubation with ECL peroxidase substrate (Pierce) and exposure to autoradiography film (CEA).
Cytotoxicity assay.
To determine the viability of PPMO treated cells, a quantitative colorimetric 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Sigma) was used. Calu-3 cells grown in 96-well plates to about 90% confluence were treated with PBS (mock) or 5 to 25 μM PPMO in infection medium (total volume, 100 μl per well) for 30 h at 37°C. Next, 10 μl of MTT stock solution (5 mg/ml in PBS) was added to each well and the cells further incubated for 2 to 3 h at 37°C until purple formazan crystals were visible. Finally, the MTT-containing medium was removed and the formazan dissolved in 200 μl dimethyl sulfoxide (DMSO), after which the absorbance was measured at 562 nm on a microplate enzyme-linked immunosorbent assay (ELISA) reader.
RESULTS
Proteolytic activation of human influenza viruses by endogenous proteases in Calu-3 cells.
Calu-3 is an immortalized human airway epithelial cell line which is regularly used in studying respiratory pathologies. Calu-3 cells have been shown to allow multicycle replication of human H3N2 viruses in the absence of exogenous trypsin (44), suggesting that an effective HA-activating protease(s) is present in these cells. To confirm that Calu-3 cells can support proteolytic activation of influenza viruses possessing HA with a monobasic cleavage site, we analyzed the growth of human isolate Memphis/96 (H1N1) in the presence or absence of exogenous trypsin. The cells were infected at a low MOI, and at the indicated time points viral titers were determined by plaque titration. In addition, virus-containing supernatants were analyzed by SDS-PAGE and Western blotting using HA-specific antibodies. As shown in Fig. 1 A, Memphis/96 (H1N1) replicated to a slightly higher titer in trypsin-treated cells than in nontreated cells at 24 h p.i. but to similar titers at 48 h and 72 h p.i. Western blot analysis of virus-containing supernatants at 72 h postinfection showed that HA0 was cleaved in the absence of trypsin as efficiently as it was in the presence of trypsin (Fig. 1B), providing evidence of efficient endogenous HA-activating protease activity in Calu-3 cells.
FIG. 1.
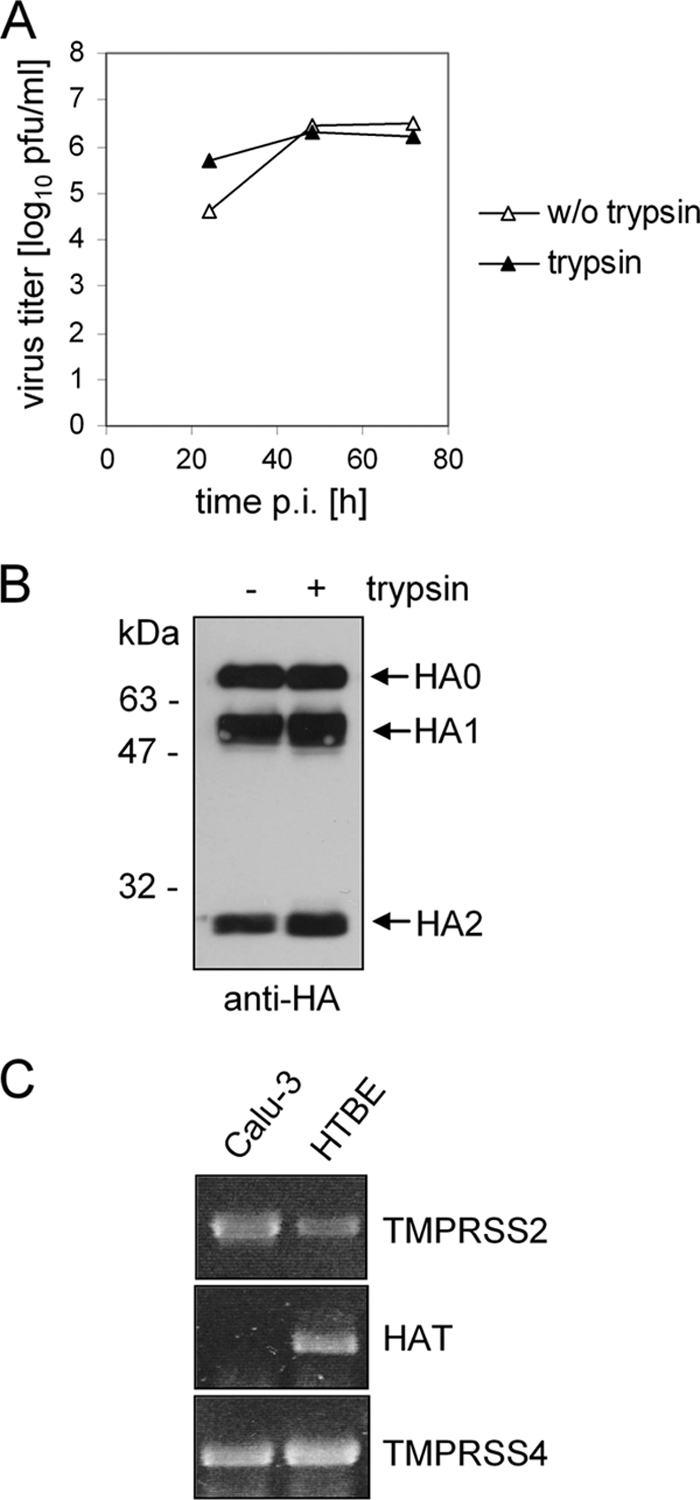
Proteolytic activation of influenza viruses in Calu-3 cells. (A) Calu-3 cells were infected with Memphis/96 (H1N1) at an MOI of 0.0001 and incubated in the absence or presence of trypsin (0.5 μg/ml). At 24, 48, and 72 h p.i., the virus titer was determined by plaque assay. (B) At 72 h p.i., virus-containing supernatants were analyzed by SDS-PAGE under reducing conditions and Western blotting using H1-specific antibodies. (C) Expression of HA-activating proteases in Calu-3 cells. Total RNA of Calu-3 cells was isolated and used as target for RT-PCR with sets of primers specific for TMPRSS2, HAT, and TMPRSS4. Total RNA of human tracheobronchial epithelial (HTBE) cells was used as control.
TMPRSS2 and TMPRSS4, but not HAT, are expressed in Calu-3 cells.
In order to identify the protease(s) responsible for HA cleavage in Calu-3 cells, we first examined the expression of TMPRSS2, TMPRSS4, and HAT, the human proteases that have been shown to be capable of cleaving HA at a monobasic cleavage site (5, 8). Total RNA was isolated from Calu-3 cells and analyzed by RT-PCR using protease-specific primers. Total RNA from human tracheobronchial epithelial (HTBE) cells, which have been previously shown to express HAT and TMPRSS2 (5), was used as a control. Using gene-specific sets of primers, PCR products for HAT, TMPRSS2 and TMPRSS4 were amplified from HTBE RNA, while only TMPRSS2- and TMPRSS4-specific PCR products were amplified from Calu-3 cells (Fig. 1C), indicating that TMPRSS2 and TMPRSS4, but not HAT, are expressed in Calu-3 cells.
PPMO design and PPMO-induced exon skipping of TMPRSS2 pre-mRNA.
To investigate the role of TMPRSS2 in proteolytic activation of HA in Calu-3 cells, we attempted to knock down TMPRSS2 expression with the use of three antisense PPMO compounds designed to target different regions of TMPRSS2 pre-mRNA or mRNA. The PPMO sequences and target locations are described in Table 1. A diagram of the PPMO target locations in the TMPRSS2 pre-mRNA and mRNA is shown in Fig. 2 A. The T-AUG PPMO was designed to interfere with translation initiation by targeting the sequence spanning the AUG translation start site. The T-ex4 and T-ex5 PPMO were designed to interfere with the correct splicing of exons 4 and 5, respectively, of TMPRSS2 pre-mRNA. As a control, a PPMO of nonsense sequence designated “scramble” was used. To directly evaluate the ability of T-ex4 and T-ex5 to alter normal TMPRSS2 splicing patterns, Calu-3 cells were treated with the various PPMO for 24 h before total RNA isolation and RT-PCR analysis with primers designed to amplify nucleotides 108 to 1336 of TMPRSS2 mRNA. A full-length TMPRSS2 PCR product of 1,228 bp was expected from the mRNA of untreated as well as scramble- and T-AUG-treated cells, whereas PCR fragments of a shorter length, resulting from the absence of the respective exons, were expected from T-ex4- and T-ex5-treated cells. Figure 2B shows that a full-length fragment of 1,228 bp was amplified from cellular RNA of mock-, scramble PPMO-, or T-AUG PPMO-treated cells. In contrast, a truncated fragment of about 1,100 bp was amplified from T-ex5 PPMO-treated cells. DNA sequencing revealed that nucleotides 382 to 501 (corresponding to the entire exon 5) were deleted without causing a frameshift in the coding sequence (Fig. 2C). In T-ex4-treated cells several truncated fragments were obtained, with the major product of about 1,020 bp having an in-frame deletion of nucleotides 295 to 501 (corresponding to exons 4 and 5) (Fig. 2C). In addition, some full-length TMPRSS2 mRNA was detected in T-ex4-treated cells (Fig. 2B). These data indicate that T-ex4 and T-ex5 were moderately and highly effective, respectively, at specifically inducing exon skipping in TMPRSS2 pre-mRNA, in a manner consistent with their design. Furthermore, the data indicate that TMPRSS2 is expressed without an LDLRA domain in T-ex5-treated cells and without both the LDLRA and transmembrane domains in T-ex4-treated cells (see Fig. 2A). Unfortunately, effective TMPRSS2-specific antibodies were not available for this study, and we were therefore unable to directly evaluate the ability of T-AUG to suppress TMPRSS2 protein production.
TABLE 1.
PPMO names, sequences, and targets
| Name | Sequence (5′ → 3′) | Target location in TMPRSS2 mRNAa |
|---|---|---|
| Scramble | TGCTCTGTCTACAGTAGTGTCA | Nonsense sequence control |
| T-AUG | CAAAGCCATCTTGCTGTTATCAAC | nt 42-65 (initiator AUG) |
| T-ex4 | TGATGCACAGTGCTTTCTTAGTCT | nt 295-318 (5′ end of exon 4, adjacent to splice site) |
| T-ex5 | CAGAGTTGGAGCACTTGCTGCCCA | nt 382-405 (5′ end of exon 5, adjacent to splice site) |
GenBank accession number NM_U75329.
FIG. 2.

TMPRSS2-specific PPMO and exon skipping. (A) Organization of TMPRSS2 pre-mRNA and mRNA and domain structure of TMPRSS2 protein. Nontranslated exons are indicated by white boxes, translated exons by gray boxes, and introns indicated by lines. TM, transmembrane domain; LDLRA, low-density lipoprotein receptor class A domain, SRCR, scavenger receptor cysteine-rich domain; HDS, serine protease domain containing the catalytic triad consisting of histidine (H), aspartate (D), and serine (S) (20). PPMO target locations in pre-mRNA and mRNA are highlighted. Numbers in the protein scheme refer to amino acids of the respective domains (32). The active-site serine (S441) and the site of zymogen activation (arrow) are indicated. The protein schemes of Δex4/5 and Δex5 represent the domain structures of truncated forms of TMPRSS2 expressed from TMPRSS2 mRNA lacking exons 4 and 5 or lacking exon 5, as produced in T-ex4 and T-ex5 PPMO-treated Calu-3 cells, respectively. Δex4/5 has a deletion of amino acids 80 to 149, and Δex5 lacks amino acids 109 to 149. (B) Analysis of TMPRSS2-specific mRNA in PPMO-treated Calu-3 cells. Calu-3 cells were treated with 25 μM PPMO for 24 h. Total RNA was isolated and analyzed by RT-PCR using TMPRSS2-specific primers to amplify a full-length fragment of 1,228 bp. Full-length and truncated PCR products (Δex4/5 and Δex5) are indicated by arrows. (C) PPMO-induced exon skipping of TMPRSS2 mRNA. Truncated DNA fragments Δex4/5 and Δex5 were extracted from the gel and analyzed by sequencing. The sequences shown represent nucleotides (nt) 288 to 512 and 375 to 512, respectively, spanning the sites of PPMO-induced deletions (indicated by a dotted line). Deletions of nt 295 to 501 and 382 to 501, respectively, are in frame and in each instance generate an isoleucine codon at the deletion site. Amino acids and nucleotides adjacent to the respective deletions are indicated. (D) Analysis of TMPRSS4-specific mRNA in PPMO-treated Calu-3 cells. Total RNA of PPMO-treated Calu-3 cells was analyzed by RT-PCR using TMPRSS4-specific primers to amplify a fragment of 624 bp.
Domain structure analysis of TMPRSS2 and TMPRSS4 indicates that they are functionally similar proteases (7), yet their nucleotide sequences show little sequence agreement, and therefore TMPRSS2-specific PPMO would not be expected to anneal to TMPRSS4 pre-mRNA or mRNA. RT-PCR analysis using TMPRSS4-specific primers showed that PPMO treatment did not affect TMPRSS4 mRNA maturation in Calu-3 cells (Fig. 2D).
TMPRSS2 mutants Δex4/5 and Δex5 lack enzymatic activity.
In order to examine the effects of T-ex4 and T-ex5 PPMO on TMPRSS2 expression and enzymatic activity in Calu-3 cells without the use of TMPRSS2-specific antibodies, we cloned the cDNAs encoding TMPRSS2 lacking exon 5 (designated Δex5) and TMPRSS2 lacking exons 4 and 5 (designated Δex4/5) from total RNAs of T-ex5 and T-ex4 PPMO-treated Calu-3 cells. The cDNAs were subcloned into the eukaryotic expression plasmid pCAGGS, and a C-terminal FLAG epitope sequence was attached to facilitate protein detection. To analyze expression of Δex4/5 and Δex5, human A549 cells, which do not support cleavage of HA with a monobasic cleavage site (5), were transfected with pCAGGS expression plasmids encoding either TMPRSS2, Δex4/5, or Δex5 and cell lysates were analyzed by Western blotting using a FLAG-specific antibody. As a control, cells were transfected with a plasmid encoding enzymatically inactive TMPRSS2 mutant S441A, in which the active-site serine is replaced by an alanine (Fig. 2A). TMPRSS2 was found to be expressed as a full-length protein (70 kDa) and a processed form (32 kDa), representing the zymogen and the cleaved catalytic domain of the mature form, respectively (Fig. 3 A). In contrast, active-site mutant S441A was expressed only as a zymogen, due to an absence of autocatalytic processing. TMPRSS2 mutants Δex4/5 and Δex5 showed lower molecular masses than full-length TMPRSS2, migrating at ∼50 kDa and ∼63 kDa, respectively, consistent with a lack of both the transmembrane and LDLRA domains or only the LDLRA domain (Fig. 3A; see also Fig. 2A). Interestingly, zymogens but no mature forms of Δex4/5 and Δex5 were detected, suggesting that Δex4/5 and Δex5 lack enzymatic activity and are therefore not capable of autocatalytic activation. The data indicate that the LDLRA domain is essential for enzymatic activity of TMPRSS2.
FIG. 3.

Expression of TMPRSS2 mutants Δex4/5 and Δex5 in A549 cells and coexpression of Δex4/5 and Δex5 with HA. (A) A549 cells were transfected with plasmids encoding either TMPRSS2, enzymatically inactive TMPRSS2 mutant S441A, Δex4/5, or Δex5, each with a C-terminal FLAG epitope. Cells transfected with empty pCAGGS were used as mock controls. At 24 h posttransfection, cell lysates were analyzed by SDS-PAGE and Western blotting using a FLAG-specific antibody. Zymogens and the mature form are indicated. A nonspecific band is indicated by an asterisk. (B) A549 cells were transfected with pCAGGS (mock) or cotransfected with pCAGGS-HA and either empty pCAGGS or pCAGGS plasmids encoding either TMPRSS2, S441A, Δex4/5, or Δex5. Cell lysates were subjected to SDS-PAGE and Western blot analysis using antibodies against HA.
In addition, Δex4/5 and Δex5 were coexpressed with HA in A549 cells and cell lysates subsequently analyzed for HA cleavage by Western blotting. As shown in Fig. 3B, HA0 was cleaved into HA1 and HA2 by coexpression of TMPRSS2, while no cleavage of HA0 was observed by coexpression of S441A, Δex4/5, or Δex5. Taken together, these data strongly suggest that T-ex4 and T-ex5 PPMO treatment of Calu-3 cells results in expression of truncated forms of TMPRSS2 which lack enzymatic activity and are incapable of supporting HA cleavage.
Inhibition of HA cleavage by T-ex5 PPMO in Calu-3 cells.
In order to examine whether TMPRSS2-directed PPMO could affect proteolytic cleavage of HA, Calu-3 cells were treated with PPMO for 24 h and then infected with Memphis/96 (H1N1) and further incubated in the presence of PPMO. At 72 h postinfection, virus-containing supernatants were analyzed by SDS-PAGE and Western blotting using HA-specific antibodies. As expected, progeny virus released from mock- and scramble PPMO-treated cells contained cleaved HA (Fig. 4 A, upper panel). In contrast, the T-ex5-treated cells released virions containing only noncleaved HA0. In addition, smaller amounts of virus were present in the supernatants of T-ex5 treated cells compared to controls, further indicating suppression of viral replication due to the inhibition of HA cleavage. Treatment with 25 μM T-ex4, however, did not affect cleavage of HA. To evaluate whether T-ex4 and T-ex5 interfered with TMPRSS2 mRNA splicing, total RNA was isolated at 72 h p.i. and analyzed by RT-PCR. A truncated PCR product containing a deletion of exon 5 was amplified from total RNA of T-ex5-treated cells, demonstrating efficient alteration of TMPRSS2 mRNA splicing (Fig. 4A, lower panel). In T-ex4-treated cells, as in the experiment with uninfected Calu-3 cells described above, a predominant PCR product of about 1,020 bp, containing a deletion of exons 4 and 5, was detected; however, a moderate amount of full-length PCR fragment was also amplified. These data show that the T-ex4 PPMO was less effective than T-ex5 at interfering with normal splicing and that a moderate quantity of full-length TMPRSS2 mRNA was sufficient to support proteolytic activation of progeny virions in these cells. Cells treated with the T-AUG PPMO showed an HA cleavage pattern and virus production similar to those of controls (Fig. 4A, upper panel). These data indicate that PPMO-induced deletion of exon 5 suppressed expression of enzymatically active TMPRSS2 in Calu-3 cells and thereby inhibited cleavage of HA. The results also indicate that TMPRSS2 is necessary for HA cleavage in Calu-3 cells.
FIG. 4.

Inhibition of HA cleavage in T-ex5-treated Calu-3 cells. (A) Confluent Calu-3 monolayers were treated with PPMO (25 μM) for 24 h prior to infection. Cells were inoculated with Memphis/96 (H1N1) at an MOI of 0.0001 for 1 h, and the inoculum was then removed and the cells incubated in the presence of PPMO (25 μM). At 72 h p.i., virus-containing supernatants were pelleted by ultracentrifugation and analyzed by SDS-PAGE and Western blotting using H1-specific antibodies (upper panel). Total cellular RNA was isolated and analyzed by RT-PCR using TMPRSS2-specific primers to amplify a full-length fragment of 1,228 bp. Full-length products and truncated PCR products are indicated by arrows (lower panel). The full-length PCR product in PPMO T-ex4-treated cells is indicated by a white arrow. (B) Dose-dependent inhibition of HA cleavage by PPMO T-ex5. Calu-3 monolayers were treated with different concentrations of PPMO (5, 10, or 25 μM) for 24 h prior to infection. Cells were inoculated at an MOI of 0.0001 for 1 h, and the inoculum was then removed and the cells further incubated in the presence of PPMO (5, 10, or 25 μM). At 72 h p.i., virus-containing supernatants were subjected to SDS-PAGE and Western blot analysis with H1-specific antibodies (upper panel). Total RNA was isolated and analyzed by RT-PCR using TMPRSS2-specific primers as described above (lower panel).
The effect of T-ex5 on TMPRSS2 mRNA splicing, HA cleavage, and virus production in Calu-3 cells was further characterized by dose-response analysis. Cells were treated with 5, 10, or 25 μM T-ex5 for 24 h, infected with Memphis/96 (H1N1), and further incubated in the presence of different PPMO concentrations for 72 h. Subsequently the effects of T-ex5 treatment on mRNA length and HA cleavage pattern were evaluated by RT-PCR and Western blotting, respectively. Examination of progeny virus released into the supernatant showed that inhibition of HA cleavage by T-ex5 treatment was dose dependent (Fig. 4B, upper panel) and consistent with the levels of truncated TMPRSS2 mRNA produced by cells under similar conditions. Infected cells treated with 25 μM T-ex5 showed complete inhibition of HA cleavage and little or no detectable full-length TMPRSS2 mRNA (Fig. 4B, lower panel).
Suppression of influenza virus propagation in Calu-3 cells by T-ex5 treatment.
To further characterize the inhibitory potential of T-ex5 on influenza virus replication in Calu-3 cells, we examined its effect on the growth of different human influenza A viruses. Cells were treated with 25 μM PPMO for 24 h prior to infection and then infected with human isolate Memphis/96 (H1N1), Hamburg/09 (H1N1), or Aichi/68 (H3N2) at a low MOI and further incubated in the presence of 25 μM PPMO for 72 h. At 24, 48, and 72 h p.i., viral growth was determined by plaque titration. As shown in Fig. 5 A, mock- and scramble PPMO-treated cells showed similar virus titers, indicating that PPMO treatment did not generically affect the propagation of any of the three viruses in Calu-3 cells. In contrast, treatment with 25 μM T-ex5 PPMO generated a 1- to 2-log10-unit reduction in viral titers of Memphis/96 (H1N1) and Hamburg/09 (H1N1) and an approximately 2.5-log10-unit reduction in viral titer of Aichi/68 (H3N2) at 24 h p.i. Further, 2- to 3-log10-unit reductions in the titers of all three virus isolates were observed in T-ex5-treated cells at 48 h and 72 h p.i., except for Aichi/68 (H3N2) at 72 h. Consistent with the dose-dependent inhibition of HA cleavage of progeny virus (Fig. 4B), the viral titer was affected in a roughly dose-responsive manner, in that 5 or 10 μM T-ex5 treatments produced less than a 1-log-unit reduction in titer of Memphis/96 (H1N1), whereas 25 μM produced a multi-log-unit reduction in titer at 48 and 72 h (Fig. 5A, upper panel).
FIG. 5.

Inhibition of cleavage of HA with a monobasic cleavage site by T-ex5 PPMO results in suppression of viral replication. (A) Confluent Calu-3 cells were pretreated with PPMO (5, 10, or 25 μM) for 24 h, inoculated at an MOI of 0.0001, and then further incubated in the presence of PPMO (5, 10, or 25 μM) for 72 h. At 24, 48, and 72 h p.i., viral titers were determined by plaque assay. (B) Rescue of virus propagation in T-ex5 PPMO-treated cells by exogenous trypsin. Calu-3 cells were pretreated with PPMO (25 μM) for 24 h, inoculated at an MOI of 0.0001, and then further incubated in the presence of PPMO (25 μM) and 0.5 μg/ml TPCK-trypsin for 72 h. Viral growth was determined by plaque titration. (C) Viral growth of influenza virus with a multibasic HA cleavage site is not affected by T-ex5 PPMO. Calu-3 cells were pretreated with PPMO (25 μM), infected at a MOI of 0.0001, and then further incubated in the presence of PPMO (25 μM) for 72 h. Viral growth was determined by plaque titration. All growth curves shown are the mean results from two independent experiments. (D) Effect of PPMO on cell viability. Calu-3 cells were treated with the indicated concentrations of PPMO for 30 h at 37°C. Cell viability compared to that of nontreated control cells was measured by MTT cell proliferation assay. The results are presented as mean values from two independent experiments.
To further confirm that the suppression of virus propagation by T-ex5 PPMO was due to inhibition of HA cleavage by TMPRSS2 and not to nonspecific effects, viral growth of Memphis/96 (H1N1) in T-ex5-PPMO treated cells was monitored in the presence of exogenous trypsin. Cells were pretreated with PPMO, infected as described above, and incubated in the presence of PPMO and trypsin for 72 h. As shown in Fig. 5B, suppression of viral growth in T-ex5-treated cells was rescued by trypsin, with Memphis/96 (H1N1) replicating to similar titers in untreated or scramble- or T-ex5-treated cells in the presence of trypsin. In addition, we investigated multicycle replication of the highly pathogenic avian virus ostrich/Italy/00 (H7N1) in mock- and PPMO-treated cells as a control. The HA of ostrich/Italy/00 (H7N1) contains a multibasic cleavage site which can be activated by ubiquitously expressed proteases such as furin (18, 38) and should therefore be unaffected by reduced TMPRSS2 expression. Calu-3 cells were treated with 25 μM PPMO for 24 h, infected with ostrich/Italy/00 (H7N1) at a low MOI, and incubated in the presence of PPMO for 72 h. Viral growth of ostrich/Italy/00 (H7N1) was not affected by T-ex5 PPMO (Fig. 5C), providing evidence for proteolytic activation of HA in those cells. These data demonstrate that inhibition of H1N1 or H3N2 influenza virus propagation in Calu-3 cells by PPMO T-ex5 was produced specifically through inhibition of proteolytic activation of HA having a monobasic cleavage site.
To evaluate the effect of PPMO on cell viability, uninfected Calu-3 cells were treated with PPMO over a concentration range of 5 to 25 μM for 30 h and cytotoxicity was determined by using a quantitative colorimetric MTT cell proliferation assay. All PPMO were found to be noncytotoxic to cells, causing less than a 10% reduction of cell viability at all concentrations tested (Fig. 5D).
Taken together, our data demonstrate that T-ex5 PPMO treatment interferes with TMPRSS2 mRNA maturation, resulting in expression of enzymatically inactive TMPRSS2 lacking the LDLRA domain. By inducing missplicing of TMPRSS2 mRNA, T-ex5 PPMO treatment prevents HA cleavage and consequently inhibits influenza virus growth and spread in Calu-3 cells.
DISCUSSION
In the present study we examined the effect of PPMO designed to interfere with the expression of influenza A virus HA-activating host cell proteases on proteolytic activation and viral spread in human Calu-3 airway epithelial cells. We aimed both to identify relevant proteases in Calu-3 cells and to explore the antiviral potential of cellular protease-targeting PPMO. Calu-3 cells are derived from the submucosal gland of the human bronchial epithelium and provide a convenient cell culture system to study diverse aspects of airway epithelial cell biology, drug delivery, and toxicology (11, 12). Furthermore, the Calu-3 cell line is one of the few cell lines that support propagation of human influenza viruses without the need of exogenous trypsin, indicating the presence of one or more relevant HA-activating proteases. RT-PCR analysis in the present study revealed that TMPRSS2 and TMPRSS4 are expressed in Calu-3 cells, while HAT is not.
Hence, in this study we tested PPMO antisense agents designed to interfere with TMPRSS2 mRNA maturation or expression. The T-ex5 PPMO, designed against sequence adjacent to a splice junction of TMPRSS2 pre-mRNA, was highly effective at altering the splicing pattern of its target, resulting in TMPRSS2 mRNA lacking exon 5 and consequently expression of TMPRSS2 protein lacking the LDLRA domain. In doing so, T-ex5 PPMO prevented cleavage of HA containing a monobasic cleavage site in Calu-3 cells and produced over 95% reductions in the titers of several different human strains of influenza A viruses at 24, 48, and 72 h p.i., at a concentration shown to be noncytotoxic. Suppression of viral growth was specifically due to inhibition of cleavage of HA with a monobasic cleavage site, since propagation of an avian H7N1 virus that possesses an HA with a multibasic cleavage motif was not affected by T-ex5 PPMO. Expression analysis in A549 cells revealed that a TMPRSS2 mutant lacking the LDLRA domain was capable of neither autocatalytic activation nor HA cleavage, indicating that the LDLRA domain is necessary for protease activity. Mutations in the LDLRA domains of type II transmembrane proteases TMPRSS3 and TMPRSS6 have previously been shown to abolish protease activity (17, 33). The data here indicate that TMPRSS2 is responsible for HA cleavage in Calu-3 cells and suggest that TMPRSS2 is likely important for HA activation in human airways in vivo.
Here, RT-PCR analysis revealed that TMPRSS4 is also expressed in Calu-3 cells, at least at the mRNA level. However, T-ex5 PPMO knockdown of TMPRSS2 expression reduced viral titers by 100- to 1,000-fold, indicating that HA is cleaved predominantly by TMPRSS2 in Calu-3 cells. It remains unknown whether the low level of residual HA activation was the result of a low level of remaining TMPRSS2 expression or of the presence of another HA-activating protease, e.g., TMPRSS4. A very recent report describing RNA interference (RNAi)-mediated knockdown of TMPRSS2 and TMPRSS4 indicates that both proteases may support HA cleavage in the human colon cancer cell line Caco-2 (2). It would be interesting to investigate the expression levels of various HA-activating proteases in various cell lines that support HA cleavage and to examine whether endogenous protease inhibitors contribute to differences in HA activation. Little knowledge exists about the expression levels and proteolytic activities of HA-activating proteases in different cell types and tissues of the human airways. We assume that different proteases are involved in HA cleavage in different cell types of the upper and lower respiratory tracts. Immunohistochemical studies revealed that HAT is expressed exclusively in ciliated cells of trachea and bronchi (41), whereas TMPRSS2 is apparently expressed in a more widespread manner in epithelial cells of the upper and lower respiratory tracts (9, 20, 42). TMPRSS4 was shown to be expressed in lung tissue and was suggested to play a role in influenza virus spread into the lung (8); however, it may be expressed in epithelial cells of the bronchi and trachea as well.
In this study, we treated the cells with PPMO once before and once after infection. The growth kinetics of Aichi/68 (H3N2) in T-ex5-treated cells (Fig. 5A) exhibit a rebound of virus production at 72 h postinfection, and Western blot analysis of progeny virions at 72 h p.i. revealed slight amounts of cleaved HA (data not shown). We did not investigate whether the diminished antiviral activity was due to PPMO degradation or its saturation with target. Treatment with T-ex4 PPMO moderately reduced the amount of correctly spliced TMPRSS2 mRNA yet had little effect on virus propagation, indicating that reduced expression of TMPRSS2 is still sufficient to support proteolytic activation of HA in Calu-3 cell cultures. We assume that repeated addition of T-ex5 PPMO at a later time point p.i. would have provided continued suppression of virus propagation.
Our results suggest that RNAs coding for human cellular proteases involved in HA cleavage may constitute productive targets for further development of PPMO-based therapeutic strategies against influenza infections. However, it has yet to be convincingly demonstrated that PPMO targeting any host factor RNA can be effective in treating a viral infection in vivo. Two studies have detailed in vivo inhibition of influenza A virus infections by PPMO targeting viral RNAs encoding the polymerase subunits PB1 and NP, administered by intranasal instillation. Although relatively high levels of PPMO were required to achieve inhibition of virus production in the cell culture experiments here, Gabriel and coworkers (13) found that a dose of only 150 ng PPMO/kg suppressed viral titers in the lungs of mice and provided protection to 50% of mice challenged with a lethal H7N7 infection. Lupfer et al. (26) showed that the peptide component of PPMO was necessary for efficacy against H3N8 influenza virus in mice, as PMO (without a peptide conjugate) were ineffective. Direct study of the antiviral efficacy of virus- compared to host-directed PPMO remains to be carried out. Future study may include evaluation of a combination of PPMO targeted against viral RNA and HA-activating cellular proteases, as such a strategy may provide an enhanced antiviral effect and reduced likelihood of the generation of escape mutants compared to the targeting of viral RNA alone.
Current anti-influenza drugs target the viral neuraminidase (NA) or the ion channel protein M2. The emergence of drug-resistant viruses, however, highlights the need for the development of novel antiviral strategies. Targeting of cellular factors is a relatively new antiviral strategy that may reduce or avoid the emergence of escape mutants. Host cell factors are infrequently considered as drug targets, however, in part because of concerns over possible toxicity. Interestingly, TMPRSS2-deficient mice have been shown to lack a discernible phenotype (21), indicating functional redundancy in the host and providing further support to the concept of targeting HA-activating proteases in general, and TMPRSS2 in particular, as a novel approach to influenza therapy.
To date, a lack of knowledge about HA-activating proteases has limited the development of antiviral drugs which target them. Recent progress in the identification and the understanding of HA-activating proteases in humans has been considerable, however, and provides an improved basis for the design of novel antiviral strategies (1, 4, 5, 6, 8, 31, 45). The present study demonstrates that PPMO constitute useful reagents to investigate HA-activating proteases and are worthy of further investigation as potential host factor-directed drugs for influenza treatment.
Acknowledgments
We thank the Chemistry Group at AVI BioPharma, Corvallis, OR, for expert production of all PPMO used in this study. We thank Mikhail Matrosovich for providing antibodies and viruses and Petra Neubauer-Rädel for technical assistance.
This work was supported by Deutsche Forschungsgemeinschaft grant SFB 593.
Footnotes
Published ahead of print on 1 December 2010.
REFERENCES
- 1.Becker, G. L., et al. 2010. Potent inhibitors of furin and furin-like proprotein convertases containing decarboxylated P1 arginine mimetics. J. Med. Chem. 53:1067-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertram, S., et al. 2010. TMPRSS2 and TMPRSS4 facilitate trypsin-independent influenza virus spread in Caco-2 cells. J. Virol. 84:10016-10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosch, F. X., W. Garten, H. D. Klenk, and R. Rott. 1981. Proteolytic cleavage of influenza virus hemagglutinins: primary structure of the connecting peptide between HA1 and HA2 determines proteolytic cleavability and pathogenicity of avian influenza viruses. Virology 113:725-735. [DOI] [PubMed] [Google Scholar]
- 4.Böttcher, E., C. Freuer, T. Steinmetzer, H. D. Klenk, and W. Garten. 2009. MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine 27:6324-6329. [DOI] [PubMed] [Google Scholar]
- 5.Böttcher, E., et al. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 80:9896-9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Böttcher-Friebertshäuser, E., et al. 2010. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 84:5605-5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bugge, T. H., T. M. Antalis, and Q. Wu. 2009. Type II transmembrane serine proteases. J. Biol. Chem. 284:23177-23181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaipan, C., et al. 2009. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 83:3200-3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donaldson, S. H., et al. 2002. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 277:8338-8345. [DOI] [PubMed] [Google Scholar]
- 10.Eisen, J. S., and J. C. Smith. 2008. Controlling morpholino experiments: don't stop making antisense. Development 135:1735-1743. [DOI] [PubMed] [Google Scholar]
- 11.Florea, B. I., M. L. Cassara, H. E. Junginger, and G. Borchard. 2003. Drug transport and metabolism characteristics of the human airway epithelial cell line Calu-3. J. Control Release 87:131-138. [DOI] [PubMed] [Google Scholar]
- 12.Forbes, B. 2000. Human airway epithelial cell lines for in vitro drug transport and metabolism studies. Pharm. Sci. Technol. Today 3:18-27. [DOI] [PubMed] [Google Scholar]
- 13.Gabriel, G., A. Nordmann, D. A. Stein, P. L. Iversen, and H. D. Klenk. 2008. Morpholino oligomers targeting the PB1 and NP genes enhance the survival of mice infected with highly pathogenic influenza A H7N7 virus. J. Gen. Virol. 89:939-948. [DOI] [PubMed] [Google Scholar]
- 14.Garten, W., and H. D. Klenk. 2008. Cleavage activation of the influenza virus hemagglutinin and its role in pathogenesis. Monographs in virology 27. Avian influenza. Karger, Basel, Switzerland.
- 15.Ge, Q., et al. 2006. Inhibition of multiple subtypes of influenza A virus in cell cultures with morpholino oligomers. Antimicrob. Agents Chemother. 50:3724-3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giles, R. V., D. G. Spiller, R. E. Clark, and D. M. Tidd. 1999. Antisense morpholino oligonucleotide analog induces missplicing of C-myc mRNA. Antisense Nucleic Acid Drug Dev. 9:213-220. [DOI] [PubMed] [Google Scholar]
- 17.Guipponi, M., S. E. Antonarakis, and H. S. Scott. 2008. TMPRSS3, a type II transmembrane serine protease mutated in non-syndromic autosomal recessive deafness. Front. Biosci. 13:1557-1567. [DOI] [PubMed] [Google Scholar]
- 18.Horimoto, T., K. Nakayama, S. P. Smeekens, and Y. Kawaoka. 1994. Proprotein-processing endoproteases PC6 and furin both activate hemagglutinin of virulent avian influenza viruses. J. Virol. 68:6074-6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hudziak, R. M., et al. 1996. Resistance of morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucleic Acid Drug Dev. 6:267-272. [DOI] [PubMed] [Google Scholar]
- 20.Jacquinet, E., et al. 2001. Cloning and characterization of the cDNA and gene for human epitheliasin. Eur. J. Biochem. 268:2687-2699. [DOI] [PubMed] [Google Scholar]
- 21.Kim, T. S., C. Heinlein, R. C. Hackman, and P. S. Nelson. 2006. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell. Biol. 26:965-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klenk, H. D., and W. Garten. 1994. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 2:39-43. [DOI] [PubMed] [Google Scholar]
- 23.Klenk, H. D., R. Rott, M. Orlich, and J. Blödorn. 1975. Activation of influenza A viruses by trypsin treatment. Virology 68:426-439. [DOI] [PubMed] [Google Scholar]
- 24.Krähling, V., D. A. Stein, M. Spiegel, F. Weber, and E. Mühlberger. 2009. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J. Virol. 83:2298-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lebleu, B., et al. 2008. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Deliv. Rev. 60:517-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lupfer, C., et al. 2008. Inhibition of influenza A H3N8 virus infections in mice by morpholino oligomers. Arch. Virol. 153:929-937. [DOI] [PubMed] [Google Scholar]
- 27.Matrosovich, M., T. Matrosovich, W. Garten, and H. D. Klenk. 2006. New low-viscosity overlay medium for viral plaque assays. Virol. J. 3:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moerdyk-Schauwecker, M., et al. 2009. Inhibition of HSV-1 ocular infection with morpholino oligomers targeting ICP0 and ICP27. Antiviral Res. 84:131-141. [DOI] [PubMed] [Google Scholar]
- 29.Moulton, H. M., and J. D. Moulton. 2010. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta 1798:2296-2303. [DOI] [PubMed] [Google Scholar]
- 30.Moulton, H. M., M. H. Nelson, S. A. Hatlevig, M. T. Reddy, and P. L. Iversen. 2004. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug. Chem. 15:290-299. [DOI] [PubMed] [Google Scholar]
- 31.Okumura, Y., et al. 2010. Novel type II transmembrane serine proteases, MSPL and TMPRSS13, proteolytically activate membrane fusion activity of the hemagglutinin of highly pathogenic avian influenza viruses and induce their multicycle replication. J. Virol. 84:5089-5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paoloni-Giacobino, A., H. Chen, M. C. Peitsch, C. Rossier, and S. E. Antonarakis. 1997. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics 44:309-320. (Erratum, 77:114, 2001.) [DOI] [PubMed] [Google Scholar]
- 33.Silvestri, L., et al. 2009. Molecular mechanisms of the defective hepcidin inhibition in TMPRSS6 mutations associated with iron-refractory iron deficiency anemia. Blood 113:5605-5608. [DOI] [PubMed] [Google Scholar]
- 34.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531-569. [DOI] [PubMed] [Google Scholar]
- 35.Stein, D. A. 2008. Inhibition of RNA virus infections with peptide-conjugated morpholino oligomers. Curr. Pharm. Des. 14:2619-2634. [DOI] [PubMed] [Google Scholar]
- 36.Stein, D., E. Foster, S. B. Huang, D. Weller, and J. Summerton. 1997. A specificity comparison of four antisense types: morpholino, 2′-O-methyl RNA, DNA, and phosphorothioate DNA. Antisense Nucleic Acid Drug Dev. 7:151-157. [DOI] [PubMed] [Google Scholar]
- 37.Steinhauer, D. A. 1999. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258:1-20. [DOI] [PubMed] [Google Scholar]
- 38.Stieneke-Gröber, A., et al. 1992. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 11:2407-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Summerton, J. 1999. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim. Biophys. Acta 1489:141-158. [DOI] [PubMed] [Google Scholar]
- 40.Summerton, J., and D. Weller. 1997. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 7:187-195. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi, M., et al. 2001. Localization of human airway trypsin-like protease in the airway: an immunohistochemical study. Histochem. Cell Biol. 115:181-187. [DOI] [PubMed] [Google Scholar]
- 42.Vaarala, M. H., K. S. Porvari, S. Kellokumpu, A. P. Kyllönen, and P. T. Vihko. 2001. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J. Pathol. 193:134-140. [DOI] [PubMed] [Google Scholar]
- 43.Vey, M., et al. 1992. Hemagglutinin activation of pathogenic avian influenza viruses of serotype H7 requires the protease recognition motif R-X-K/R-R. Virology 188:408-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng, H., et al. 2007. Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type I interferon response in polarized human bronchial epithelial cells. J. Virol. 81:12439-12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhirnov, O. P., M. R. Ikizler, and P. F. Wright. 2002. Cleavage of influenza A virus hemagglutinin in human respiratory epithelium is cell associated and sensitive to exogenous antiproteases. J. Virol. 76:8682-8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu, R. P., et al. 2007. Cell-penetrating peptides as transporters for morpholino oligomers: effects of amino acid composition on intracellular delivery and cytotoxicity. Nucleic Acids Res. 35:5182-5191. [DOI] [PMC free article] [PubMed] [Google Scholar]