

Abstract
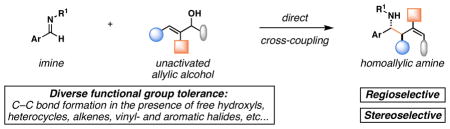
We report a reaction for the convergent coupling of allylic alcohols with imines that delivers stereodefined homoallylic amines. The process proceeds with net allylic transposition, without the intermediacy of allylic organometallic reagents, and forges two stereodefined centers and a geometrically defined di- or trisubstituted alkene with very high levels of selectivity.
Keywords: stereoselective synthesis, homoallylic amines, titanium, metallacycles, allylic alcohols
Since the birth of the field, convergent C–C bond forming reactions have defined the backbone of organic synthesis.[1] While significant advances in reaction development have recently been made in the area of catalysis,[2] contributions that describe novel bimolecular C–C bond construction remain central to the evolution of organic synthesis. Such contributions provide new paradigms for molecular assembly, greatly facilitating the manner in which complex molecules are made. Here, we describe a convergent coupling reaction between allylic alcohols and imines that delivers complex homoallylic amines with high levels of regio- and stereoselectivity by a pathway that proceeds without the intermediacy of allylic organometallic reagents (Figure 1, eq 1).
Figure 1.

Allylation for convergent C–C bond formation.
Over the last thirty years, allylation has emerged as a particularly powerful bimolecular C–C bond forming process, with current examples demonstrating the ability to achieve enantio- and diastereoselective allyl-, crotyl- and prenyl-transfer.[3] While powerful, the typical dependence on allylic organometallic reagents often restricts the utility of these processes, limiting them to the addition of these simple hydrocarbon fragments.[4] The synthesis and application of more functionalized allylic organometallic reagents for convergent coupling is complicated and typically unwieldy owing to: 1) Functional group tolerance in the preparation of the allylic organometallic reagent, 2) challenges associated with the control of site selectivity in the metalation step, 3) difficulties in controlling site-selective C–C bond formation (due to a competition between α- vs γ- attack, and the known propensity for allylic isomerization of the intermediate organometallic reagent), and 4) problems associated with attaining selectivity in both the establishment of a stereodefined alkene and the tetrahedral stereochemistry at the allylic and homoallylic positions (Figure 1, eq 2). In cases where the allylic metal reagent is generated in a catalytic fashion, functional group tolerance is often enhanced, but complexities still remain due to competing isomerization (of the allylic metal species), as well as the previously mentioned issues with regio- and stereoselection in the C–C bond forming event.[5] As such, the potential impact of the bond constructions made possible with allylic organometallic reagents (independent of whether such processes are rendered catalytic in the metal) remains limited.
Recent contributions from our laboratory have focused on harnessing the power of metallacycle-mediated C–C bond formation for new convergent coupling reactions in organic chemistry.[6] These accomplishments have derived from the development of general strategies to control the reactivity of metal–π complexes. In particular, association of neighboring alkoxides with the metal center has played a central role in these processes; functional groups that often complicate other C–C bond forming reactions.[7] One powerful mode of control is reaction via formal metallo-[3,3] rearrangement.[8] Here, we describe a new stereoselective convergent coupling reaction by formal metallo-[3,3] rearrangement that addresses long-standing problems in allyl-transfer chemistry and defines a pathway for complex allylation of imines that:
Proceeds via the direct coupling of allylic alcohols, thereby eliminating the need for preformed allylic organometallic reagents,
occurs with diverse functional group tolerance,
progresses in a highly regioselective manner with net allylic transposition,
delivers homoallylic amines with high anti- selectivity, and
establishes a stereodefined di- or tri-substituted alkene in concert with C–C bond formation (Figure 1, eq 1).
Our generic design for an allylic alcohol–imine cross-coupling process is outlined in Figure 2. Treatment of an imine (A) with a low-valent metal (B) was anticipated to result in the formation of an intermediate azametallacyclopropane (C). Addition of an allylic alkoxide (D) to this preformed complex, was expected to result in rapid and reversible ligand exchange to deliver E. Rearrangement by way of F results in the formation of a C–C bond, two-stereogenic centers and one geometrically defined substituted alkene and delivers homoallylic metallated amine G. From G, simple hydrolysis provides the complex homoallylic amine product H. Alternatively, depending on the metal employed, we envisioned a potential pathway for capturing the precious metal intermediate G via net reduction, and epimetalation with imine A. Defining this portion of the reaction would render the process catalytic in the metal component, but was thought to be necessary only if:
Figure 2.

Reaction design.
The reaction requires a complex ligand for control of selectivity (enantio-, diastereo-, or regioselectivity), or
if the metal employed is rare, expensive or toxic.
The metal (B) selected for this process was a readily available titanium alkoxide, and the control of the coupling reaction was anticipated to follow from the geometrical constraints imposed by reaction through a formal metallo-[3,3] rearrangement. As such, the primary goal of our studies was to investigate the possibility of this stereoselective new bond construction, without concern for turning over the non-toxic and readily available titanium alkoxide reagent.
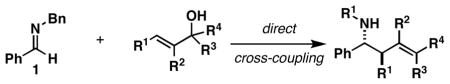
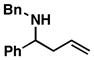
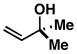
As illustrated in Table 1, coupling of allyl alcohol (2) with imine 1 delivers the simple homoallylic amine 3 in 70% yield (entry 1).[9] With more substituted allylic alcohols (4 and 6), coupling provides homoallylic amines bearing proximal tri- and tetrasubstituted alkenes (entries 2 and 3); in one case defining a useful reaction for the prenylation of aromatic imines (4→5).[10] Interestingly, coupling of allylic alcohol 8 with imine 1 proceeds with both high regio- and stereoselectivity delivering homoallylic amine 9 in 87% yield as a single geometrical isomer (Z:E ≥ 20:1; entry 4).
Table 1.
 | |||||
|---|---|---|---|---|---|
| entry | allylic alcohol | yield (%)[a] | (Z:E) | dr | product |
| 1 |
2 |
70 | – | – |
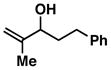
 3 |
| 2 |
 4 |
83 | – | – |
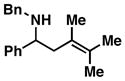
 5 |
| 3 |
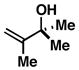 6 |
80 | – | – |
 7 |
| 4 |
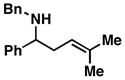
 8 |
87 | ≥ 20:1 | – |
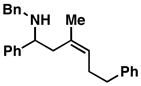 9 |
| 5 |
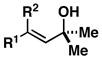 10; R1 = Me; R2 = H |
81 | – | ≥ 20:1 |
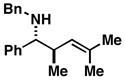 11 |
| 6 | 12; R1 = H; R2 = Me 11 | 68 | – | ≥ 20:1 | |
| 7 |
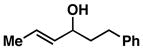 13 |
92 | 1.6:1 | ≥ 20:1[b] |
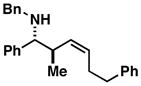 14 |
| 8 |
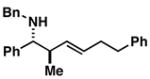
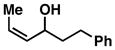 15 |
57 | ≤ 1:20 | ≥ 20:1 |
 16 |
| 9 |
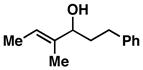 17 |
54 | ≥ 20:1 | ≥ 20:1 |
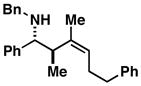 18 |
Reaction conditions: imine (1 eq), Ti(Oi-Pr)4 (1.5 eq), c-C5H9MgCl (3 eq) (−78 to −40 °C), then add lithium alkoxide of allylic alcohol (1.5 eq) in THF (−40 to 0 °C; or −40 °C to rt - entries 6 and 8), then quench with H2O.
Each alkene isomer (Z and E) was identified as the anti-diastereomer (dr ≥ 20:1).
When employing terminally substituted allylic alcohols, this C–C bond forming process proceeds in a highly anti-selective manner. For example, coupling of allylic alcohol 10 or 12 with 1 provides the stereodefined product 11 in 81 and 68% yield, respectively (dr ≥ 20:1 in both cases). With secondary allylic alcohols, bearing geometrically defined alkenes, the control of stereochemistry is more complex, as the challenge of establishing allylic and homoallylic stereochemistry is coupled to the construction of a stereodefined alkene. Nevertheless, reaction of allylic alcohol 13 with 1 provides 14 in 92% yield (anti:syn ≥ 20:1). While this coupling reaction does not deliver the stereodefined alkene with high levels of selectivity (Z:E = 1.6:1), coupling of the isomeric allylic alcohol 15 with 1 delivers 16 with much higher levels of selectivity, favoring the anti-product with a proximal (E)-disubstituted alkene (dr ≥ 20:1; E:Z ≥ 20:1; entry 8). Finally, as depicted in entry 9, coupling of the (E)-trisubstituted allylic alcohol 17 with 1 provides homoallylic amine 18 in 54% yield, in this case delivering an anti-product with a central (Z)-trisubstituted alkene (dr ≥ 20:1; Z:E ≥ 20:1).
While this convergent coupling reaction affords complex homoallylic amines that are otherwise difficult to prepare, it is also compatible with vinyl halides – a feature that further defines a rather unique stereoselective bond construction for complex molecule synthesis (Table 2). Entries 1–3 demonstrate that 2-halo-allylic alcohols (19–21) are suitable substrates for coupling with 1. As depicted in entries 4 and 5, more complex bond constructions are possible in this series. Here, high anti- selectivity is coupled to the generation of geometrically defined vinyl halides (dr ≥ 20:1; E:Z ≥ 20:1). Finally, allylic alcohols bearing carbocyclic vinyl halides are also viable partners in this coupling reaction. As illustrated in entry 6, coupling of imine 29 with 30 provides the functionalized cyclohexene 31 in 53% yield (dr ≥ 20:1).
Table 2.
 | ||||||
|---|---|---|---|---|---|---|
| entry | imine | allylic alcohol | yield (%)[a] | (E:Z)[b] | dr[b] | product |
| 1 |
 1 |
 19; X = Cl |
59 | – | – |
 22; X = Cl |
| 2 | 1 | 20; X = Br | 61 | – | – | 23; X = Br |
| 3 | 1 | 21; X = I | 60 | – | – | 24; X = I |
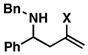
| 4 | 1 |
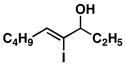 25 |
53 | ≥ 20:1 | ≥ 20:1 |
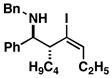 26 |
| 5 | 1 |
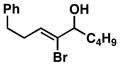 27 |
56 | ≥ 20:1 | ≥ 20:1 |
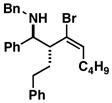 28 |
| 6 |
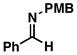 29 |
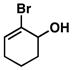 30 |
53 | – | ≥ 20:1 |
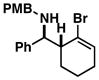 31 |
Reaction conditions: See supporting information for details.
No evidence was found for the production of stereoisomeric products.
This stereoselective convergent coupling reaction is compatible with a variety of aromatic imines and substituted allylic alcohols. Table 3 highlights the use of this reaction for the synthesis of homoallylic amines bearing heteroaromatics (33 and 38), tetrasubstituted vinyl halides (36), aromatic halides (40, 42 and 44), additional alkenes (42 and 44) as well as a trifluoromethyl substituted aromatic (40). As depicted in entry 7, this reaction can also be extended to ketimines, in this case providing the 3° carbinolamine 46 in 83% yield.
Table 3.
 | ||||
|---|---|---|---|---|
| entry | imine | allylic alcohol | yield (%)[a],[b] | product |
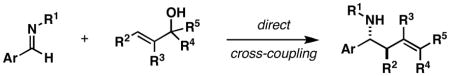
| 1 |
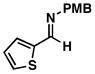 32 |
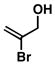 20 |
53 |
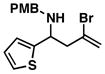 33 |
| 2 |
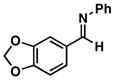 34 |
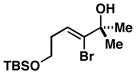 35 |
52 |
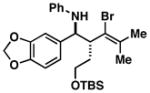 36 |
| 3 |
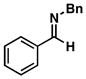 1 |
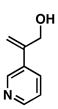 37 |
55 |
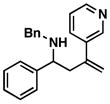 38 |
| 4 |
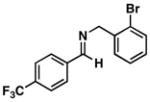 39 |
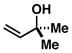 4 |
67 |
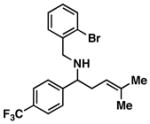 40 |
| 5 |
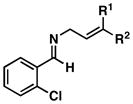 41; R1, R2 = Me |
4 | 79 |
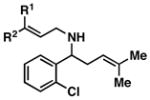 42; R1, R2 = Me |
| 6 | 43[c]; R1 = H, R2 = TMS | 4 | 69 | 44; R1 = H, R2 = TMS |
| 7 |
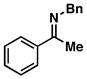 45 |
4 | 83 |
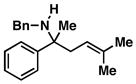 46 |
| 8 | 1 |
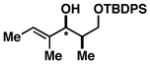 47 |
72 |
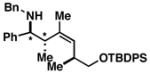 48 dr ≥ 20:1; (Z):(E) ≥ 20:1 |
Reaction conditions: See supporting information for details.
No evidence was found for the production of stereoisomeric products.
Compound 43 was used as a mixture of alkene isomers (E:Z = 4:1).
Finally, the absolute stereochemistry of this reaction can be controlled in a substrate-directed manner. As depicted in entry 8 of Table 3, coupling of the stereodefined allylic alcohol 47 with imine 1 provides the chiral stereodefined product 48 in 72% yield, as a single isomer.[11]
The regiochemical course of this coupling reaction is consistent with an empirical model based on a formal metallo-[3,3] rearrangement via the intermediacy of a mixed titanate ester (Figure 3). The stereochemical control observed is consistent with reaction through a conformation where σC–M is aligned with πC=C, while minimizing allylic strain (A-1,2/A-1,3) and developing 1,2-non bonded steric interactions (A and B; Figure 3).[12]
Figure 3.

Empirical model for regio- and stereoselection.
In conclusion, we describe a new reaction design to accomplish complex convergent coupling via formal allyl-transfer that proceeds without the requirement of allylic organometallic reagents. This process, while not yet rendered catalytic in the metal (Ti or Mg), defines a unique and powerful convergent bond construction. Due to the low cost of the metal-containing reagents, benign nature of the byproducts (TiO2 and magnesium (II) salts), and substrate-controlled stereoselection, this type of process in its current form should be of great utility in organic chemistry.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work by the American Cancer Society (RSG-06-117-01), the Arnold and Mabel Beckman Foundation, Boehringer Ingelheim, Eli Lilly & Co. and the National Institutes of Health–NIGMS (GM80266). The authors also thank the W. M. Keck Foundation Biotechnology and Resource Laboratory at Yale University for high resolution mass spectrometry and Mr. Filip Kolundzic for an early demonstration of this imine–allylic alcohol reductive cross-coupling reaction.
Footnotes
We gratefully acknowledge financial support of this work by the American Cancer Society (RSG-06-117-01), the Arnold and Mabel Beckman Foundation, Boehringer Ingelheim, Eli Lilly & Co. and the National Institutes of Health–NIGMS (GM80266).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For a discussion of strategy in complex molecule synthesis, along with detailed case studies, see: Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. John Wiley & Sons, Inc; New York: 1989. Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. VCH; New York: 1996.
- 2.For a recent review of advances in catalysis, see: Cornils B, Herrmann WA, Muhler M, Wong C-H. Catalysis from A to Z. Wiley-VCH; New York: 2007.
- 3.For a current review of allylation chemistry, see: Denmark SE, Almstead NG. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; New York: 2000. p. 299.For a current review on the application of the allylation reaction to the synthesis of natural products, see: Chemler SR, Roush WR. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; New York: 2000. p. 403.
- 4.Notable exceptions include bifunctionalized allylmetal reagents and complex crotylsilanes. For recent discussions, see: Flamme EM, Roush WR. J Am Chem Soc. 2002;124:13644. doi: 10.1021/ja028055j.and references therein; and Masse CE, Panek JS. Chem Rev. 1995;95:1293.
- 5.For a review of catalyt enantioselective addition of allylic organometallic reagents to aldehydes and ketones, see: Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.For a review of catalytically generated allylic metal reagents that serve as electrophiles, see: Lu Z, Ma S. Angew Chem Int Ed. 2008;47:258. doi: 10.1002/anie.200605113.
- 6.For the cross-coupling of two internal alkynes, see: Ryan J, Micalizio GC. J Am Chem Soc. 2006;128:2764. doi: 10.1021/ja057352w.For the cross-coupling of internal alkynes with substituted alkenes, see: Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440. doi: 10.1002/anie.200603515.For the cross-coupling of allenes with internal alkynes, see: Shimp HL, Micalizio GC. Chem Commun. 2007:4531. doi: 10.1039/b708256h.Shimp HL, Hare A, McLaughlin M, Micalizio GC. Tetrahedron. 2008;64:6831. doi: 10.1016/j.tet.2008.02.015.For cross-coupling of internal alkynes with aromatic imines, see: McLaughlin M, Takahashi M, Micalizio GC. Angew Chem Int Ed. 2007;46:3912. doi: 10.1002/anie.200605060.For cross-coupling of internal alkenes with aromatic imines, see: Takahashi M, Micalizio GC. J Am Chem Soc. 2007;129:7514. doi: 10.1021/ja071974v.
- 7.For a discussion of the burden of protecting groups in complex molecules synthesis, see: Baran PS, Maimone TJ, Richter JM. Nature. 2007;466:404. doi: 10.1038/nature05569.
- 8.For other examples of bimolecular C–C bond formation via formal metallo-[3,3] rearrangement, see: allylic alcohol–vinylsilane: Belardi JK, Micalizio GC. J Am Chem Soc. 2008;130:16870–16872. doi: 10.1021/ja8074242.allylic alcohol–internal alkyne: Kolundzic F, Micalizio GC. J Am Chem Soc. 2007;129:15112. doi: 10.1021/ja075678u.allenic alcohol–aromatic imine: McLaughlin M, Shimp HL, Navarro R. Synlett. 2008:735. doi: 10.1055/s-2008-1042808.and d) allenic alcohol–internal alkyne: see ref 6d.
- 9.For recent examples of imine allylation using preformed allylic silanes, see: Huber JD, Perl NR, Leighton JL. Angew Chem Int Ed. 2008;47:3037. doi: 10.1002/anie.200705621.Berger R, Rabbat PMA, Leighton JL. J Am Chem Soc. 2003;125:9596. doi: 10.1021/ja035001g.Schaus JV, Jain N, Panek JS. Tetrahedron. 2000;56:10263.For recent examples using preformed allylic stannanes, see: Gastner T, Ishitani H, Akiyama R, Kobayashi S. Angew Chem Int Ed. 2001;40:1896.Hallett DJ, Thomas EJ. Tetrahedron: Asym. 1995;6:2575.Keck GE, Enholm EJ. J Org Chem. 1985;50:146.For recent examples employing preformed allylic boron reagents, see: Wu TR, Chong JM. J Am Chem Soc. 2006;128:9646. doi: 10.1021/ja0636791.Li S, Batey RA. Chem Commun. 2004:1382. doi: 10.1039/b403759f.Lou S, Moquist PM, Schaus SE. J Am Chem Soc. 2007;129:15398. doi: 10.1021/ja075204v.Itsuno S, Watanabe K, Ito K, El-Shehawy AA, Sarhan AA. Angew Chem Int Ed Eng. 1997;36:109.For recent examples using in situ generated allylic boron reagents, see: Selander N, Kipke A, Sebelius S, Szabó KJ. J Am Chem Soc. 2007;129:13723. doi: 10.1021/ja074917a.Shimizu M, Kimura M, Watanabe T, Tamaru Y. Org Lett. 2005;7:637. doi: 10.1021/ol047609f.For examples using in situ generated allylic palladium reagents, see: Fernandes RA, Stimac A, Yamamoto Y. J Am Chem Soc. 2003;125:14133. doi: 10.1021/ja037272x.Hopkins C, Malinakova HC. Org Lett. 2006;8:5971. doi: 10.1021/ol0624528.For recent examples employing allylic zinc reagents, see: Wipf P, Kendall C. Org Lett. 2001;3:2773. doi: 10.1021/ol0163880.Wipf P, Pierce JG. Org Lett. 2005;7:3537. doi: 10.1021/ol051266j.For a recent example using in situ generated allylic titanium reagents, see: Gao Y, Sato F. J Org Chem. 1995;60:8136.For reviews on the synthesis of homoallylic amines via allylic metal reagents, see: Ramachandran PV, Burghardt TE. Pure Appl Chem. 2006;78:1397.Kouznetsov VV, Méndez LYV. Synthesis. 2008:491.Yamamoto T, Asao N. Chem Rev. 1993;93:2207.
- 10.Prenylation of imines has represented a significant challenge in organic chemistry. For a route to products like 5 based on the reactivity of allylic barium reagents, see: Yanagisawa A, Ogasawara K, Yasue K, Yamamoto H. J Chem Soc, Chem Commun. 1996:367.
- 11.The relative stereochemistry of 48 is proposed based on the model depicted in Figure 3, and is supported by observations made in related titanium-mediated reductive cross-coupling reactions of stereodefined allylic alcohols. For additional information, see: ref 7.
- 12.The empirical model described is based on the minimization of A-1,2 strain in a formal metallo-[3,3] rearrangement process, and does not account for the geometry at titanium. The proposed empirical model does not exclude a mechanism that follows from directed carbometalation and syn-elimination. For a review of allylic strain as a principle for stereochemical control, see: Hoffmann RW. Chem Rev. 1989;89:1841.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.