

Abstract
Purpose
Systemic chemotherapy fails to access much of the tumor burden in patients with advanced cancer, significantly limiting its efficacy. In preclinical studies, brief high doses of tyrosine kinase inhibitors (TKIs) targeting the Human Epidermal Growth Factor Receptor (HER) family can prime tumor vasculature for optimal chemotherapeutic delivery and efficacy. This study investigates the clinical relevance of this approach.
Experimental Design
A phase I clinical study of escalating doses of the HER TKI lapatinib given as a 2-day pulse prior to a weekly infusion of nab-paclitaxel (100 mg/m2) was conducted in patients with advanced solid tumors.
Results
Twenty-five patients were treated. Treatment was associated with grade 1-2 toxicities including diarrhea, nausea, rash, neutropenia, neuropathy, fatigue, alopecia, and anemia. The two dose-limiting toxicities were grade 3 vomiting and grade 4 neutropenia and the maximum tolerated dose (MTD) of lapatinib was defined as 5250 mg/day in divided doses. Lapatinib concentrations increased with increasing dose. Dynamic Contrast Enhanced MRI (DCE-MRI) studies in a subset of patients confirmed a decrease in tumor vascular permeability immediately following a lapatinib pulse. Sixty-five percent of evaluable patients experienced a partial or stable response on this therapy, 72% of whom were previously taxane-refractory.
Conclusion
A two day pulse of high dose lapatinib given prior to weekly nab-paclitaxel is a feasible and tolerable clinical regimen, suitable for testing this novel vascular-priming chemosensitization hypothesis developed in preclinical models.
INTRODUCTION
One of the significant barriers to the highly effective treatment of patients with metastatic cancer is the failure of chemotherapeutic agents to efficiently reach all the tumor cells and produce their cytotoxic effects on a significant fraction of the whole tumor burden. The lack of access to much of the disease is due to highly abnormal tumor vasculature, characterized by high permeability and very poor architecture resulting in tissue hypertension, hypoxia, acidosis, and absence of osmotic gradients resulting in poor exchange of nutrients and drugs (1). One of the principal challenges in clinical cancer research is to understand the mechanisms underlying this therapeutic barrier and develop adjunctive therapies to overcome it. Measurable progress has been made with therapies that target the VEGF signaling pathway. The effects of this class of agents on tumor vasculature have been mechanistically defined in preclinical models (2, 3) and their ability to enhance chemotherapeutic efficacy validated in a number of clinical studies (4-6). A role for other vascular targets remains to be defined.
The Human Epidermal Growth Factor Receptor (HER) family of receptor tyrosine kinases are important drivers of tumor endothelial signaling, in particular PI3K/Akt pathway activation (7-10). Upstream signals that activate HER family signaling in tumor endothelial cells include paracrine signaling (11, 12), autocrine signaling (13), and cross talk from G-protein coupled receptor activation (14). In preclinical models we found that the effects of HER-family tyrosine kinase inhibitor (TKI) therapy on tumor vasculature are at least as profound as those reported by VEGFR targeting therapies (15). This includes a reduction in transendothelial permeability, similar to VEGF-targeting agents, accompanied by an increase in fractional plasma volume, not seen with VEGF-targeting agents. Importantly, these effects are transient and reversible and seen within 48 hours of high dose TKI therapy, but not seen with continuous lower dosing. Consistent with the noted improved vascular function, the addition of a 2-day high dose pulse of TKI immediately prior to chemotherapy led to enhancement of chemotherapeutic efficacy while continuous TKI dosing at maximum tolerated dose had little effect. These data suggest that HER family TKIs may have chemosensitizing effects that are distinct from VEGF pathway targeting agents.
In phase III clinical studies, HER TKIs in continuous dosing have not enhanced the efficacy of cytotoxic chemotherapeutics (16, 17). A vascular or chemosensitizing effect with continuous dosing was not seen in preclinical models either, and it is unlikely that this activity can be harnessed at low dose and continuous schedule (15). The TKI itself is subject to delivery barriers and it cannot target the tumor vasculature if it cannot access it. Consistent with this, their vascular effects become apparent at high doses in preclinical models. In order to begin to test the clinical relevance of these findings, we conducted a phase I study of the HER TKI, lapatinib, given as a high dose pulse immediately prior to weekly intravenous albumin-bound paclitaxel (Abraxane; Abraxis BioScience Inc.; Santa Monica, CA) in patients with advanced or metastatic solid tumors. Lapatinib (Tykerb; GlaxoSmithKline; Research Triangle Park, NC) is a reversible HER TKI with a highly selective target profile and potentially wide therapeutic index permissive to significant dose escalation. Albumin-bound paclitaxel is a new formulation of paclitaxel that overcomes many of the delivery barriers created by the Cremophor® vehicle of the previous paclitaxel formulation, including complex non-linear pharmacokinetics and reduced volume of distribution, which may be due to plasma micelle entrapment and reduced tissue deposition (18-23). This formulation allows optimal testing of adjunctive strategies to further improve tissue delivery of paclitaxel.
Patients and Methods
Patient Eligibility
Patients were enrolled after this study protocol was approved by the Committee on Human Research. All participants provided written informed consent. Patients had advanced solid tumor malignancies. There were no restrictions on prior number of therapies, and prior paclitaxel and EGFR- or HER2-targeted therapies were allowed. Evaluable disease was required, but measureable disease was not required. Inclusion criteria included age >18 years, ECOG performance status of 0-2, life expectancy > 3 months, adequate organ function, and at least one month since prior therapy of any kind. Exclusions included pre-existing grade 2 neuropathy, significant cardiac disease, and progressing brain metastases.
Baseline Evaluation
Pretreatment evaluation included hematologic and biochemical blood work, chest/abd/pelvis CT imaging, and EKG and MUGA evaluations within 5 weeks of enrollment.
Treatment Plan
Patients self-administered lapatinib orally on days 1 and 2 followed by the intravenous administration of nab-paclitaxel at a fixed standard dose of 100 mg/m2 on day 3 of weeks 1, 2, and 3 of a 4-week cycle. Lapatinib was the experimental therapy and was administered using a dose escalation design guided by toxicity evaluation (see below). Lapatinib was administered in twice-daily dosing because of the higher AUC and reduced pill burden experienced with this schedule (24). Patients were instructed to take the lapatinib pills on an empty stomach to avoid any potential confounding effects caused by concomitant food intake. Patients continued on therapy as long as they remained free of progression and unacceptable toxicities. Treatment response was assessed both clinically and using Response Criteria to Treatment in Solid Tumors (RECIST) (25) every 2 months. Clinical activity was determined using results from history and physical examinations, serum biomarkers, chest/abd/pelvis CT scans, and occasional bone scans.
Dose Escalation Design
The lapatinib dose was escalated according to an accelerated titration design1 starting with 1000 mg/day in twice daily dosing (dose level 0) (26). Subsequent lapatinib dose levels were designated as the following: dose level 1 = 1500 mg/day, dose level 2 = 2000 mg/day, dose level 3 = 2750 mg/day, dose level 4 = 3750 mg/day, dose level 5 = 5250 mg/day, dose level 6 = 7500 mg/day. At least one month of treatment and toxicity data were required for all patients in order to determine tolerability. The accelerated phase continued in cohorts of one until a patient experienced a dose limiting toxicity (DLT), or two patients experienced ≥ grade 2 toxicity. When one of these criteria was met, the accelerated dose-escalation phase closed and the standard dose-escalation phase was initiated. The standard phase continued in cohorts of 3, expandable to 6, until the highest dose at which ≤1 of 6 patients experienced a DLT was determined. There was no intra-patient dose escalation. Toxicity was assessed according to the Common Toxicity Criteria (CTC) version 3.0. DLT was defined as toxicity that occurred during the first cycle of therapy and was attributable to treatment. Toxicities meeting criteria for a DLT include 1) grade 4 neutropenia (ANC <500) for ≥ 96 hours or grade 3 neutropenia with fever (ANC <1000 and T>38.5 °C); 2) grade 4 thrombocytopenia (<25,000) or a bleeding episode requiring platelet transfusion support; 3) any grade 3 or greater non-hematologic toxicity excluding fatigue and sensory neuropathy. Diarrhea and nausea/vomiting were graded in the setting of maximal anti-diarrheals and anti-emetics, respectively; 4) grade 3 or greater sensory neuropathy despite dose reduction of nab-paclitaxel; 5) grade 3 or greater fatigue lasting longer than 1 week; and 6) any lapatinib- or nab-paclitaxel-related toxicity causing treatment delay of greater than 2 weeks.
Toxicity Assessment, Toxicity Management and Dose Modifications
Toxicity assessments were made on days 3, 10, and 17 of the first two cycles and monthly thereafter. Complete blood counts were evaluated with each nab-paclitaxel infusion. Radiologic and cardiac imaging was done every 8 weeks. For patients with grade 3 diarrhea or rash despite maximal supportive care, or a second recurrence of intolerable grade 2 diarrhea or rash despite maximal supportive care, the lapatinib dose was decreased to one level lower and nab-paclitaxel was held until symptoms improved to a grade 1 toxicity level. GCSF support was added in the first instance of grade ≥2 neutropenia. Dose reduction of nab-paclitaxel was incurred in the second instance of grade ≥2 neutropenia despite GCSF, and in the first instance of grade ≥2 neuropathy after resolution to grade 1. For all other non-hematologic grade ≥3 toxicities, treatment was held until improvement to grade 1 and then resumed at the same dose of nab-paclitaxel and one dose level lower of lapatinib.
Plasma Lapatinib Levels and Paclitaxel Pharmacokinetics
Blood samples for assessment of lapatinib plasma concentration were obtained at baseline, 4 hours after the first dose of lapatinib (approximate Tmax), and 48 hours after the first lapatinib dose (highest trough concentration for this regimen) prior to the nab-paclitaxel infusion. Samples were analyzed for lapatinib plasma concentration using a previously described method (27) with a sensitivity of 1ng/mL and precision and accuracy within 15%.
After the maximum tolerated dose (MTD) was defined, 3 additional patients were enrolled at the MTD for paclitaxel pharmacokinetic studies. In these three patients, the administration of lapatinib was omitted from the first week of nab-paclitaxel, but was given prior to the second and all the subsequent weekly infusions. Blood samples were drawn before and at ½, 1, 2, 4, 7, and 24 hours after the beginning of the nab-paclitaxel infusion on day 3 and again on day 10 of the first cycle. An additional sample was also collected 24 hours after the 3rd infusion of nab-paclitaxel on day 18 of the first cycle.
Paclitaxel concentration was measured using a liquid chromatography/ tandem mass spectrometry system consisting of a 717 plus autosampler (Waters Corporation, Milford, MA) and Quattro Ultima (Micromass, Manchester, UK) detector with electospray positive ionization mode. The multiple reaction monitor was set at: 854.4 – 509.2 m/z for paclitaxel and 859.4 – 509.2 m/z for internal standard (paclitaxel-d5). The sample cone voltage and collision energy for paclitaxel and internal standard were set at 35 V and 15 eV, respectively. Chromatography was performed on BDS C18 column (50 × 4.6 mm, 5mm particle size, Thermo Electron Corporation, Bellefonte, PA). The mobile phase was CH3CN: H2O: CH3COOH, (50: 50: 0.15) (v/v) containing 4 mM NH4CH3COO. The flow rate was 1.0 ml/min with 25% of the flow liquid split into the mass system. The plasma sample (0.2 ml) was extracted with 1 ml of methyl tert-butyl ether. The organic layer was then transferred to another tube, evaporated by N2 gas, reconstituted with 0.15 ml of mobile phase, and 10 μl was injected onto the column. The standard curve was from 20-5000 ng/ml. The relative standard deviations for low, medium, and high concentration QC samples were less than 15%. Pharmacokinetic analyses were performed using WinNonlin software Version 4.1.
MR Perfusion Studies
Dynamic contrast enhanced MRI (DCE-MRI) scans of measureable non-pulmonary and non-CNS metastases were obtained within 2 weeks of starting lapatinib therapy and again on day 3 of cycle 1 immediately following the 2-day lapatinib pulse (defined as scans 1 and 2, respectively) in a subset of eligible patients. DCE-MRI scans were also obtained on days 15 (pre-lapatinib) and 17 (post-lapatinib) of the first cycle during the standard phase of the protocol (defined as scans 3 and 4).
All scans were obtained on a 1.5 T scanner (General Electric Healthcare, Milwaukee, WI). Initial unenhanced MR images were obtained to localize a non-pulmonary non-CNS metastasis at least 2 cm in craniocaudad diameter. A solid portion of the tumor was designated as the indicator lesion at the discretion of the study radiologist. For the dynamic contrast technique, a three-dimensional (3D) fast spoiled gradient-recalled (SPGR) echo sequence (256 × 128 matrix; eight 5-mm slices acquired every 11 seconds for 5.53 minutes; 22-36 cm axial field of view) was used to acquire T1-weighted images before, during, and following intravenous administration of 0.1 mmol/kg gadopentetate dimeglumine injected at 3 cc/sec and followed by a 10 cc saline flush at 3 cc/sec. For each scan, the dynamic contrast enhanced images were transferred to a desktop computer (Dell Dimension 4700, Round Rock, TX) for post-processing using a commercially available image analysis software program (MIStar, Apollo Medical Imaging, Melbourne, Australia). All MR perfusion studies were analyzed by a single radiologist. Signal-intensity time curves were generated by averaging all voxels in each region of interest at each time point. Because signal intensity varies nearly linearly with tracer concentration, using the pulse sequences and concentrations expected in this study, it was determined that conversion to tracer concentration via T1 mapping would increase rather than decrease measurement variability. These data were fit to the Tofts model to determine tumor vascular permeability (Ktrans) and the fractional plasma volume (Vp) for each tumor.
The Wilcoxon signed-rank test for matched paired data was used to compare the Ktrans and Vp values between pre- and post-lapatinib DCE-MRI studies.
RESULTS
Patient Characteristics
Twenty-eight patients were accrued. Three of the patients were withdrawn from study within 1 day due to rapidly progressing disease or withdrawal of consent and contributed little data to the study. Twenty-five patients with advanced or solid tumor malignancies were treated on study between February 2006 and February 2008. Patient characteristics are outlined in Table 1. The group encompasses a heavily pre-treated population with diverse cancer subtypes, typical of a phase I referral pattern. The majority of patients were taxane-refractory and the lung cancer patients deemed appropriate for EGFR TKI therapy had previously failed such therapy.
Table 1.
Patient Characteristics
| No. of patients | 25 |
|
| |
| Median age (years) | 57 |
|
| |
| Male | 16 |
|
| |
| Median # of prior chemotherapy regimens |
3 |
|
| |
| # with previous taxane therapy | 18 (72%) |
|
| |
| # previous EGFR TKI therapy | 5 (20%) |
|
| |
| Tumor type | |
| Lung | 9 |
| Esophageal | 3 |
| Prostate | 3 |
| Pancreatic | 2 |
| Melanoma | 2 |
| Breast | 2 |
| Mesothelioma | 1 |
| Sarcoma | 1 |
| Ovarian | 1 |
| Bladder | 1 |
Toxicity
All 25 treated patients received at least one cycle of therapy. The median number of cycles completed per patient was 4. Dose levels 0-3 were escalated under the accelerated design until conversion criteria were met (two grade 2 toxicities). Dose-escalation was converted to the standard design beginning with dose level 3 (2750 mg/day). At dose level 6 (7500 mg/day), two of six patients experienced DLTs consisting of grade 4 neutropenia with fever, and grade 3 nausea and vomiting. Subsequent expansion of dose level 5 (5250 mg/day) encountered no DLTs in a cohort of 6 patients establishing this dose to be the MTD of lapatinib for this combination.
Five patients had serious adverse events (SAE) which required hospitalization: hemorrhoidal bleeding (confirmed by colonoscopy), pre-syncope, grade 3 vomiting, and 2 patients with grade 4 febrile neutropenia. Only the vomiting and grade 4 febrile neutropenia were attributable to treatment, and were all seen at dose level 6. The patient with pre-syncope was diagnosed with pneumonia and dehydration. Table 2 lists all drug-related grade 1-4 toxicities found in ≥10% of patients. The most severe toxicity for each patient is listed for each category. Most toxicities were low grade and manageable. The diarrhea, nausea, and vomiting were transient and were experienced within 24 to 48 hours of lapatinib administration. Two patients required two dose reductions each, the first due to diarrhea in later cycles and the second due to diarrhea and febrile neutropenia. None of the grade 1-2 non-hematologic toxicities demonstrated obvious worsening, either in frequency or intensity, with the escalation of lapatinib from dose level 0-6. There was also no relationship found between dose level and nab-paclitaxel-associated toxicities such as neuropathy and arthralgias. MUGA scans repeated every 8 weeks while on study did not reveal any clinically significant change in cardiac ejection fraction at any dose level.
Table 2.
Grade 1-4 Toxicities (in ≥ 10% of patients)
| Total # of Patients (n = 25) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Toxicity | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total | |||||
| # | % | # | % | # | % | # | % | # | % | |
| Diarrhea | 14 | 56 | 9 | 36 | 2 | 8 | 25 | 100 | ||
| Anemia | 9 | 36 | 5 | 20 | 1 | 4 | 15 | 60 | ||
| Fatigue | 6 | 24 | 2 | 8 | 8 | 32 | ||||
| Nausea | 7 | 28 | 3 | 12 | 1 | 4 | 11 | 44 | ||
| Neutropenia | 1 | 4 | 7 | 28 | 5 | 20 | 3 | 12 | 16 | 64 |
| Vomiting | 3 | 12 | 7 | 28 | 1 | 4 | 11 | 44 | ||
| Acneiform Rash | 8 | 32 | 6 | 24 | 14 | 56 | ||||
| Alopecia | 11 | 44 | 4 | 16 | 15 | 60 | ||||
| Neuropathy | 3 | 12 | 2 | 8 | 1 | 4 | 6 | 24 | ||
| Anorexia | 3 | 12 | 1 | 4 | 4 | 16 | ||||
| Nail changes | 3 | 12 | 3 | 12 | ||||||
| Arthralgia | 2 | 8 | 2 | 8 | ||||||
| Non-acneiform rash | 2 | 8 | 2 | 8 | ||||||
| Fever and neutropenia | 1 | 4 | 1 | 4 | ||||||
Diarrhea is the most common toxicity of lapatinib, and has been shown to be dose-related (28). Incidence of diarrhea was higher in this study, consistent with the high doses used, but severity was mitigated by the short duration of pulse dosing, and was manageable with anti-diarrheal treatment (Figure 1).
Figure 1.

Incidence of diarrhea in relation to lapatinib dose
Therapy was discontinued in 15 patients due to disease progression. Four patients were removed or withdrew from study because of cumulative toxicities including grade 2 peripheral sensory neuropathy (2 patients), grade 2 vomiting and diarrhea (1 patient), and grade 2 fatigue (1 patient) after 6, 4, 7, and 2 cycles, respectively. Other reasons for study discontinuation included intercurrent illness requiring radiation (2 patients) or surgery (1 patient), patient withdrawal (1 patient), and symptoms related to underlying disease (1 patient).
Clinical Activity
Two patients were removed from study prior to initial treatment response assessment. One patient was removed for surgical management of orthopedic hardware failure, and the second patient was removed because of rapid disease progression at the time of study entry and preference for hospice care. Twenty-three of 25 patients were evaluable for initial response after 8 weeks of therapy. Five of 23 patients (22%) had a partial response (PR) after 8 weeks of treatment. Another 10 patients (43%) had stable disease (SD) (Table 3). As such, a total of 15 of 23 patients (65%) experienced either a PR or SD after 8 weeks of therapy. Eleven of these 15 patients (73%) had previously received taxane therapy, and 5 (33%) had previously received EGFR TKI therapy. Patients with either a PR or SD remained on study for a median 162 days (range 51-282 days).
Table 3.
Patients with Partial Responses and Stable Disease
| Tumor Type | Lapatinib dose (mg/day) |
Duration on study |
Previous Taxane therapy |
Previous EGFR TKI |
# Prior Regimens |
|---|---|---|---|---|---|
| Partial Response | |||||
| NSCLC | 1000 | 6 months | N | N | 3 |
| Breast | 5250 | 7 months | Y | N | 6 |
| NSCLC | 7500 | 6 months | Y | Y | 3 |
| Breast | 7500 | 4 months | Y | N | 1 |
| Esophageal | 7500 | 8 months | Y | N | 2 |
| Stable Disease | |||||
| Prostate1 | 2000 | 4 months | Y | N | 5 |
| gastric-esophageal | 2750 | 10 months | Y | N | 2 |
| NSCLC | 2750 | 4 months | N | Y | 3 |
| NSCLC | 3750 | 4 months | N | Y | 4 |
| NSCLC | 5250 | 2 months | Y | N | 5 |
| Mesothelioma | 5250 | 3 months | N | N | 2 |
| NSCLC | 5250 | 4 months | Y | Y | 3 |
| NSCLC | 5250 | 8 months | Y | Y | 3 |
| Bladder | 7500 | 4 months | Y | N | 1 |
| Prostate1 | 7500 | 7 months | Y | N | 6 |
All patients received nab-paclitaxel 100 mg/m2
non-measureable bone-only disease
Pharmacokinetics
Lapatinib plasma concentrations at 48 hours after administration, representing the highest trough concentration, increased with increasing dose in a less than proportional manner, as shown by the best-fit curve (r=0.702) in Figure 2a. Variability in dose-normalized concentrations (CV 60%) was consistent with previous reports (24,29). Despite the less than proportional increase, plasma concentrations achieved by the high-dose pulse regimen were on average 3-fold higher than those achieved by the maximum tolerated dose administered twice daily in early Phase I trials (Figure 2a). Mean paclitaxel plasma concentrations (AUC) appeared to be higher after a lapatinib pulse (Figure 2b), but the statistical significance of this apparent difference could not be assessed. Mean lapatinib concentrations at increasing dose levels and mean paclitaxel pharmacokinetic parameters are listed in Table 4a-b.
Figure 2.
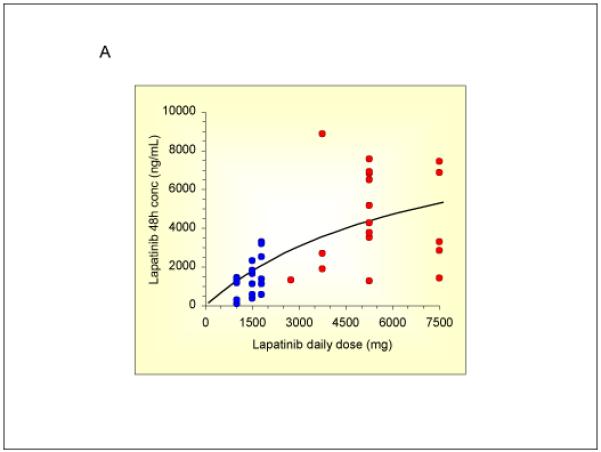

Lapatinib and paclitaxel pharmacokinetic studies
A) 48 hour lapatinib plasma concentrations in individual patients (red dots). Historical data from previous lapatinib studies employing twice daily, continuous dosing (blue dots).
B) Plasma paclitaxel concentrations with or without a preceding two day lapatinib 5250 mg/day pulse (n = 3 patients).
Table 4.
| a: Paclitaxel Pharmacokinetics | ||
|---|---|---|
| Parameter | Paclitaxel alone | Paclitaxel + Lapatinib |
| mean AUC (ng·h/mL) | 3242 ± 214 | 4751 ± 1965 |
| CL (L/h/m2) | 30.9 ± 2.0 | 23.4 ± 8.6 |
| Vss (L/m2) | 111 ± 104 | 69.5 ± 47.4 |
| Half-life (h) | 4.53 ± 3.12 | 3.52 ± 0.68 |
| b: Mean Lapatinib Plasma Concentrations | ||
|---|---|---|
| Lapatinib dose | 4 hour conc (ng/mL) | 48 hour conc (ng/mL) |
| 2750 mg/day | 315 | 1330 |
| 3750 mg/day | 2060 ± 824 | 4488 ± 3820 |
| 5250 mg/day | 2238 ± 814 | 4916 ± 2020 |
| 7500 mg/day | 2287 ± 1894 | 4649 ± 2965 |
Dynamic Contrast Enhance MRI
In order to pilot the utility of DCE-MRI in capturing the vascular effect of a high dose pulse of lapatinib, 8 of 25 patients underwent DCE-MRI studies prior to and 48 hours after lapatinib administration (before nab-paclitaxel infusion). DCE-MRI studies were performed at baseline (pre-lapatinib, scan 1) and on day 3 (post-lapatinib, scan 2) as well as on day 15 (pre-lapatinib, scan 3) and day 17 (post-lapatinib, scan 4) during the first cycle of therapy. Of the 8 patients, 1 patient had two sets of evaluable MRIs (scans 1 and 2 as well as scans 3 and 4); 2 patients had evaluable MRIs at baseline and day 3 only; and 5 patients had evaluable MRIs on day 15 and day 17 only. There were 9 paired MRI studies in total (3 pairs of scans 1 and 2; 6 pairs of scans 3 and 4). Of the 9 paired MRI studies, 8 showed a decrease in tumor vascular permeability (Ktrans) after a 48 hour pulse of lapatinib. The one patient who underwent 2 paired MRI scans showed a decrease in Ktrans on both sets. Of the 8 patients whose tumors demonstrated a decrease in Ktrans after a 2-day pulse of lapatinib, 3 patients demonstrated a simultaneous increase in tumor vascular perfusion volume (Vp) (Figure 3 a-b).
Figure 3.


DCE-MRI studies
A) Tumor vascular permeability (Ktrans) values in individual patients. Scans 1 and 2 were pre- and post- first lapatinib pulse (baseline and day 3 of first cycle), respectively. Scans 3 and 4 were pre- and post- third lapatinib pulse (days 15 and 17 of first cycle), respectively.
B) Tumor vascular perfusion volume (Vp) values in individual patients.
When data from the pre-lapatinib scans (scans 1 and 3) were pooled and compared to the data from the post-lapatinib scans (scans 2 and 4) using the Wilcoxon signed-rank test, a statistically significant decrease in Ktrans was seen (mean 954 versus 728 per min per 1000, respectively, p=0.0202). No significant change in Vp was found (mean 15.9 versus 22.9 per 1000, respectively, p=0.767).
DISCUSSION
This study demonstrates that a 2-day pulse of lapatinib prior to weekly intravenous nab-paclitaxel chemotherapy (100 mg/m2) is safe and tolerable in patients with advanced or metastatic solid tumor malignancies. The MTD of lapatinib is 5250 mg/day given through twice daily dosing. This is considerably higher than the MTD of lapatinib when given continuously. The higher MTD in pulse dosing is associated with higher incidences of grade 1 and 2 toxicities, but generally manageable. Prior lapatinib studies using continuous dosing schedules have described rates of common toxicities such as diarrhea, nausea, and rash on the order of 36-42%, 10-13%, and 27-31%, respectively (28, 29). A phase 1 study of daily lapatinib 1250 mg/day and every three week docetaxel reported the incidence of diarrhea, nausea, and rash as 56%, 25%, and 52% respectively (30). The higher incidence of low grade diarrhea in our study is consistent with the high lapatinib doses used, and also with previous studies showing that diarrhea is dose-related (28).
Lapatinib dose-escalation produced increasing plasma concentrations despite a less than proportional relationship and intrinsically high variability in bioavailability. Mean paclitaxel concentrations appeared to be higher after a 2-day high-dose lapatinib pulse, however additional data are required to assess the significance of such an effect. The slightly higher rates of grade 3-4 neutropenia in our study compared to studies of weekly nab-paclitaxel 100 mg/m2 given alone (32% vs 24%) (31-33) may, in part, be due to lapatinib inhibition of paclitaxel efflux by P-glycoprotein.
The anti-tumor efficacy seen in this cohort of largely taxane-refractory patients is a promising signal of biological activity that requires further study. The mechanistic evidence from DCE-MRI data in preclinical models suggests that this is due to improved tumor vascular function (15). This is difficult to confirm in patients since clinically approved macromolecular contrast media are not yet available. The results in this pilot exploratory analysis show that DCE-MRI does pick up some change in signal and may be worthwhile to pursue. Seven of the 8 patients demonstrated a transient decrease in tumor vascular permeability 48 hours after lapatinib administration and 3 of them had a concomitant increase in tumor vascular perfusion. A statistically significant decrease in Ktrans is found when all pre-lapatinib DCE-MRI scans are compared to all post-lapatinib DCE-MRI scans, however the small sample size makes further statistical considerations difficult. An elevated endothelial transfer constant is a pathologic feature of hyperpermeable malignant lesions (34). Hyperpermeability leads to interstitial hypertension, hypoxia, and acidosis which prevent the efficient delivery of chemotherapy into tumor cells (1). A reduction in tumor vascular permeability reduces interstitial hypertension, re-establishes physiologic gradients, and creates a more favorable microenvironment for chemodelivery.
Fury et al performed a similar phase I study with intermittent escalating doses of gefitinib on days 1 and 2 followed by fixed-dose docetaxel (75 mg/m2) in patients with advanced solid tumors (35). They escalated the gefitinib dose significantly above the standard dose, and found toxicity profiles similar to those in this study. Only a weak signal of biological activity was seen in the gefitinib study, although the differences in study population as well as the TKI and chemotherapy agents used make direct comparison to the present lapatinib study difficult.
In summary, a 2 day pulse of lapatinib at 5250mg/day in divided doses prior to nab-paclitaxel (100 mg/m2) is a safe and feasible treatment regimen in patients with advanced solid tumor malignancies with a promising signal of biological efficacy in treatment-refractory patients. Phase II studies are being planned to assess the anti-tumor efficacy of this regimen, and additional preclinical studies are ongoing to develop even more effective approaches to enhance chemodelivery into tumor tissues.
Translational Relevance.
The ability of biologic agents to induce temporary improvements in tumor vascular function and enhance the effects of chemotherapy has been demonstrated with agents that target the VEGF signaling pathway. Recent data suggest that HER family tyrosine kinase inhibitors (TKIs) also have significant effects on tumor vascular function and that the way in which they are administered may play an important physiologic role. HER TKIs inhibit a number of signaling pathways only transiently which suggests that some of their biologic activity is short-lived. In preclinical models, tumor vascular permeability and perfusion characteristics are transiently optimized following a brief pulse of high dose HER TKI therapy, and chemotherapy effects are enhanced when delivered after such a pulse. Our study is the initial translation of this chemosensitization hypothesis, and shows that targeting HER family receptors through alternative scheduling strategies is clinically feasible and may be a novel approach to improving chemodelivery.
Acknowledgements
AJC is supported by an ASCO Young Investigator Award and an American Cancer Society post-doctoral fellowship. MMM is supported by the National Institute of Health.
Research Support:
UCSF Hellen Diller Comprehensive Cancer Center, Atwater Foundation
Footnotes
REFERENCES
- 1.Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- 2.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 3.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–6. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 4.Hurwitz HI, Fehrenbacher L, Hainsworth JD, et al. Bevacizumab in combination with fluorouracil and leucovorin: an active regimen for first-line metastatic colorectal cancer. J Clin Oncol. 2005;23:3502–8. doi: 10.1200/JCO.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 5.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–76. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 6.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 7.Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006;66:2173–80. doi: 10.1158/0008-5472.CAN-05-3387. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki T, Kitadai Y, Nakamura T, et al. Inhibition of epidermal growth factor receptor and vascular endothelial growth factor receptor phosphorylation on tumor-associated endothelial cells leads to treatment of orthotopic human colon cancer in nude mice. Neoplasia. 2007;9:1066–77. doi: 10.1593/neo.07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta VB, Besner GE. HB-EGF promotes angiogenesis in endothelial cells via PI3-kinase and MAPK signaling pathways. Growth Factors. 2007;25:253–63. doi: 10.1080/08977190701773070. [DOI] [PubMed] [Google Scholar]
- 10.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–50. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 11.Kuwai T, Nakamura T, Sasaki T, et al. Phosphorylated epidermal growth factor receptor on tumor-associated endothelial cells is a primary target for therapy with tyrosine kinase inhibitors. Neoplasia. 2008;10:489–500. doi: 10.1593/neo.08200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker CH, Kedar D, McCarty MF, et al. Blockade of epidermal growth factor receptor signaling on tumor cells and tumor-associated endothelial cells for therapy of human carcinomas. Am J Pathol. 2002;161:929–38. doi: 10.1016/S0002-9440(10)64253-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semino CE, Kamm RD, Lauffenburger DA. Autocrine EGF receptor activation mediates endothelial cell migration and vascular morphogenesis induced by VEGF under interstitial flow. Exp Cell Res. 2006;312:289–98. doi: 10.1016/j.yexcr.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 14.Lin CI, Chen CN, Huang MT, et al. Lysophosphatidic acid upregulates vascular endothelial growth factor-C and tube formation in human endothelial cells through LPA(1/3), COX-2, and NF-kappaB activation- and EGFR transactivation-dependent mechanisms. Cell Signal. 2008 doi: 10.1016/j.cellsig.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Moasser MM, Wilmes LJ, Wong CH, et al. Improved tumor vascular function following high-dose epidermal growth factor receptor tyrosine kinase inhibitor therapy. J Magn Reson Imaging. 2007;26:1618–25. doi: 10.1002/jmri.21196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giaccone G, Herbst RS, Manegold C, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol. 2004;22:777–84. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Herbst RS, Giaccone G, Schiller JH, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 2. J Clin Oncol. 2004;22:785–94. doi: 10.1200/JCO.2004.07.215. [DOI] [PubMed] [Google Scholar]
- 18.Yeh TK, Lu Z, Wientjes MG, Au JL. Formulating paclitaxel in nanoparticles alters its disposition. PharmRes. 2005;22:867–74. doi: 10.1007/s11095-005-4581-4. [DOI] [PubMed] [Google Scholar]
- 19.van ZL, Karlsson MO, Verweij J, et al. Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer Chemotherapy and Pharmacology. 2001;47:309–18. doi: 10.1007/s002800000215. [DOI] [PubMed] [Google Scholar]
- 20.van TO, Huizing MT, Panday VR, Schellens JH, Nooijen WJ, Beijnen JH. Cremophor EL causes (pseudo-) non-linear pharmacokinetics of paclitaxel in patients. BrJ Cancer. 1999;81:330–5. doi: 10.1038/sj.bjc.6690696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knemeyer I, Wientjes MG, Au JL. Cremophor reduces paclitaxel penetration into bladder wall during intravesical treatment. Cancer Chemotherapy and Pharmacology. 1999;44:241–8. doi: 10.1007/s002800050973. [DOI] [PubMed] [Google Scholar]
- 22.Sparreboom A, van ZL, Brouwer E, et al. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer Res. 1999;59:1454–7. [PubMed] [Google Scholar]
- 23.Sparreboom A, van TO, Nooijen WJ, Beijnen JH. Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer Res. 1996;56:2112–5. [PubMed] [Google Scholar]
- 24.Koch KMLD, Mangum S. Pharmacokinetics of GW572016 in an ascending dose tolerability study of phase I cancer patients. Eur J Cancer. 2003;1:S169. poster 559. [Google Scholar]
- 25.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 26.Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC. Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst. 1997;89:1138–47. doi: 10.1093/jnci/89.15.1138. [DOI] [PubMed] [Google Scholar]
- 27.Hsieh STT, Koch K, Dunn J. Increasing throughput of parallel on-line extraction liquid chromatography/electrospray ionization tandem mass spectrometry system for GLP quantitative bioanalysis in drug development. Rapid Commun Mass Spectrom. 2004;18:285–292. doi: 10.1002/rcm.1327. [DOI] [PubMed] [Google Scholar]
- 28.Burris HA, 3rd, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib ( GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23:5305–13. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- 29.Gomez HL, Doval DC, Chavez MA, et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J Clin Oncol. 2008;26:2999–3005. doi: 10.1200/JCO.2007.14.0590. [DOI] [PubMed] [Google Scholar]
- 30.LoRusso PM, Jones SF, Koch KM, et al. Phase I and pharmacokinetic study of lapatinib and docetaxel in patients with advanced cancer. J Clin Oncol. 2008;26:3051–6. doi: 10.1200/JCO.2007.14.9633. [DOI] [PubMed] [Google Scholar]
- 31.Crown JPBH, Jones S, Koch KM, Fittipaldo A, Parikh R, Koehler M. Safety and tolerability of lapatinib in combination with taxanes in patients with breast cancer. J Clin Oncol. 2007;25(18S):1027. [Google Scholar]
- 32.Jones SF, Burris HA, DA Y. Lapatinib (an oral dual kinase inhibitor) plus weekly or every 3 week paclitaxel. Breast Cancer Res Treat. 2004;88(suppl 1):1069. [Google Scholar]
- 33.Nyman DW, Campbell KJ, Hersh E, et al. Phase I and pharmacokinetics trial of ABI-007, a novel nanoparticle formulation of paclitaxel in patients with advanced nonhematologic malignancies. J Clin Oncol. 2005;23:7785–93. doi: 10.1200/JCO.2004.00.6148. [DOI] [PubMed] [Google Scholar]
- 34.Daldrup-Link HE, Rydland J, Helbich TH, et al. Quantification of breast tumor microvascular permeability with feruglose-enhanced MR imaging: initial phase II multicenter trial. Radiology. 2003;229:885–92. doi: 10.1148/radiol.2293021045. [DOI] [PubMed] [Google Scholar]
- 35.Fury MG, Solit DB, Su YB, et al. A phase I trial of intermittent high-dose gefitinib and fixed-dose docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2007;59:467–75. doi: 10.1007/s00280-006-0286-6. [DOI] [PubMed] [Google Scholar]