

Abstract
The potential neuroprotective role of sex hormones in chronic neurodegenerative disorders and acute brain ischemia following cardiac arrest and stroke is of a great therapeutic interest. Long-term pretreatment with estradiol and other estrogens affords robust neuroprotection in male and female rodents subjected to focal and global ischemia. However, the receptors (e.g., cell surface or nuclear), intracellular signaling pathways and networks of estrogen-regulated genes that intervene in neuronal apoptosis are as yet unclear. We have shown that estradiol administered at physiological levels for two weeks before ischemia rescues neurons destined to die in the hippocampal CA1 and ameliorates ischemia-induced cognitive deficits in ovariectomized female rats. This regimen of estradiol treatment involves classical intracellular estrogen receptors, transactivation of IGF-1 receptors and stimulation of the ERK/MAPK signaling pathway, which in turn maintains CREB activity in the ischemic CA1. We also find that a single, acute injection of estradiol administrated into the brain ventricle immediately after an ischemic event reduces both neuronal death and cognitive deficits. Because these findings suggest that hormones could be used to treat patients when given after brain ischemia, it is critical to determine whether the same or different pathways mediate this form of neuroprotection. We find that an agonist of the membrane estrogen receptor GPR30 mimics short latency estradiol facilitation of synaptic transmission in the hippocampus. Therefore, we are testing the hypothesis that GPR30 may act together with intracellular estrogen receptors to activate cell signaling pathways to promote neuron survival after global ischemia.
Keywords: estrogens, hippocampus, global ischemia, apoptosis, GPR30
Introduction
It is now well established that estrogens exert profound protective effects in animal models of focal and global ischemia (for review, see 1-4). Global ischemia in humans or induced experimentally in animals causes selective and delayed neuronal death in pyramidal neurons of the hippocampal CA15. Histological evidence of degeneration is not observed until 2-3 days after ischemia in rats or 3-4 days in gerbils6, 7. Although the mechanisms underlying ischemia-induced death are as yet unclear, the substantial delay between insult and onset of cell death suggests a critical role for transcriptional changes activating specific apoptotic pathways. Thus, classical estrogen receptors (ERs), which are ligand-activated transcription factors, might mediate neuroprotection by directly regulating the transcription of pro- and anti-apoptotic molecules. Genomic screens for estrogen responses element (ERE) motifs reveal genes such as IGF-1 (insulin like growth factor-1), nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and anti-apoptotic members of the Bcl-2 family known to be involved in neural survival8, 9. However, the likely existence of classical ER-mediated mechanisms does not rule out the possibility that cell signaling initiated by estrogen-binding molecules located at the plasma membrane could also block apoptotic cascades via ER-independent regulation of gene transcription.
Indeed, estrogens produce rapid actions in neurons, on the order of seconds to minutes, consistent with a role for membrane receptors10, 11. Estradiol rapidly activates cellular pathways associated with growth factor signaling, such as the ERK/MAPK and PI3K/Akt cascades, and promotes accumulation of second messengers in a G protein-dependent manner. One scenario is that classical ER-α and ER-β are expressed at the plasma membrane12; however, the mechanisms by which they might act to regulate second messenger production is unclear. Moreover, findings that estradiol can elicit rapid actions on neurons even in the presence of ER-α and ER-β blockade or in neurons lacking ER-α and ER-β (for review see13) suggest that estradiol signaling depends on receptors in addition to ER-α and ER-β. Whether classical ERs (ER-α and ER-β) located in the nucleus or on the plasma membrane, or other membrane estrogen receptors are critical mediators of the neuroprotective actions of estradiol in an animal model of global ischemia is the topic of this review.
Estradiol and Global Ischemia
1) Long-term exposure to estradiol: histological and behavioral neuroprotection
We and others have shown that estradiol, chronically administrated at physiological levels, affords robust protection against ischemia-induced cell death in hippocampal CA1 of intact male gerbils14 and ovariectomized female rats15, 16 and gerbils16. The histological outcomes after ischemic insult were quite similar in estradiol-treated animals irrespective of sex or species. Dramatic cell loss (close to 90%) compared to sham-operated animals is observed in the pyramidal layer of the hippocampal CA1 at 7 days after a 5-10 min episode of global ischemia. Cell loss is significantly reduced by about 60% when animals subjected to ischemia are implanted with pellets providing physiological levels of 17β-estradiol (E2) beginning 2 weeks before and continuing for 1 week after ischemic insult. We verified that this chronic treatment with physiological E2 also confers behavioral neuroprotection17. Performance on memory tasks that depend on the hippocampus and that show deficits after ischemia was improved by chronic exposure to E2 in ovariectomized rats. Long-term E2 prevented the ischemia-induced deficit in visual working memory, maintaining normal performance in tests with retention intervals of up to 1 hr. Long-term E2 treatment also prevented ischemia-induced deficits in spatial memory tests with short (1 and 7 min), but not longer (15 min) retention intervals.
2) Long-term exposure to estradiol: Cellular pathways of neuroprotection
Both ER-α and ER-β are involved
Determining whether ER-α, ER-β, and/or other membrane estrogen receptors mediate estradiol neuroprotection following global ischemia is of great interest both for the development of therapeutic strategies and for elucidating underlying molecular mechanisms of delayed neuronal death. Our published work using chronic administration of selective agonists for either ER-α or ER-β in female rats indicated that the activation of either receptor was able to rescue hippocampal CA1 pyramidal neurons following transient global ischemia15. However, only ERα was upregulated in the CA1 by E2 and ischemia. We also showed that ICI 182,780, a competitive antagonist for both ER-α or ER-β, completely blocks long-term E2 neuroprotection when administered in the early post-ischemic period (Figure 1). These findings confirm that neuroprotection afforded by chronic treatment with E2 likely involves activation of the classical receptors ER-α and ER-β. However, this does not rule out the possibility that other estrogen receptors, which could also be sensitive to ICI 182,780, may play a role in long term E2 neuroprotection following ischemic insult.
Figure 1. Long-term estradiol protection requires both estrogen receptors and IGF-1 receptors.

Ovariectomized female gerbils were implanted with placebo or estradiol pellets 2 weeks before global ischemia or sham operation. They received icv infusions of the broad spectrum estrogen receptor antagonist ICI 182,780 (100 μg), the IGF-1 receptor antagonist JB-1 (10 μg), or vehicle at 0 and 12 h after ischemia. Plasma estradiol levels at the time of death were 15.4 ± 1.4 pg/ml in the placebo group and 136.35 ± 12.9 pg/ml in the estradiol group. Neither ICI 182,780 nor JB-1 affected neuronal survival in ischemic or sham animals implanted with placebo pellets, but they abolished the neuroprotective actions of estradiol. Data are from Jover-Mengual et al., 2007.
ERK/MAPK signaling is critical for estradiol neuroprotection
Neuronal apoptosis can proceed via two parallel pathways, the intrinsic pathway (mitochondrial) and the extrinsic pathway (involving death receptors), to activate caspase-3 and lead to DNA fragmentation and other hallmarks of cell suicide18. Our early study on E2 neuroprotection in male gerbils suggested that chronic hormone treatment rescued neurons destined to die by interfering with apoptotic death cascades that activate caspase-314. Under conditions of transient brain ischemia, the mitochondrial pathway predominates, with activation of pro-apoptotic members of the Bcl-2 family triggering the release of killer proteins (smac/DIABLO, apoptosis inducing factor AIF, cytochrome C) from the mitochondrial intermembranous space by outer membrane permeabilization (for review see18). Promoting the transcription of anti-apoptotic Bcl-2 family members and thereby shifting the balance in favor of anti-apoptotic versus pro-apoptotic can elicit neuroprotection by stabilizing the mitochondrial membrane and attenuating the release of killer proteins. Transcription of the anti-apoptotic factor Bcl-2 is positively regulated by activated cAMP response element binding protein (CREB) 19, 20, which is thought to play a major role in neuronal survival and neurogenesis following ischemic insult21. For example, CREB activation is required for the acquisition of ischemic tolerance in gerbil CA1 neurons after global ischemia22. Moreover, Hu et al.23 showed that after transient global ischemia, CREB phosphorylation was induced in the ischemia-resistant dentate granule cells of the adult rat hippocampus.
Activation of the ERK/MAPK pathway by extracellular stimuli, including growth factors, neurotransmitters, and notably ischemic stress, can lead to activation of CREB by phosphorylation at residues Ser133 and Ser14420, 24. As E2 can activate the ERK/MAPK pathway in vitro and in vivo25, we tested the hypothesis that E2 blocks ischemia-induced apoptosis in hippocampal CA1 pyramidal cells through the activation of ERK/MAPK, promoting the sustained phosphorylation of CREB and subsequent up-regulation of the anti-apoptotic protein Bcl-2. We showed that intracerebroventricular (icv) injection of the MEK inhibitor PD98059 abrogated the neuroprotective action of chronically administered E2 (Figure 2). Ischemia induced dephosphorylation of ERK1 and CREB in animals treated with placebo, and E2 treatment maintained p-ERK1 and p-CREB levels in post-ischemic CA1 (Figure 3). Ischemia also promoted down-regulation of the anti-apoptotic protein Bcl2, and this was prevented by long-term E2 treatment. Surprisingly, whereas ERK/MAPK signaling was required for CREB activation and neuronal survival, the ability of E2 to maintain high levels of Bcl-2 was ERK-independent. That is, the MEK inhibitor PD98059 administered immediately after ischemia did not block the ability of E2 to maintain normal levels of Bcl-2 in CA1 even though neuroprotection was abolished. Thus, although Bcl-2 may be necessary for cell survival after global ischemia, it is clearly not sufficient. The two major CREB targets that are known to promote neuron survival following global ischemia are Bcl-2 and BDNF19, 26. Thus, it would be interesting to determine whether E2 activation of CREB phosphorylation in the post-ischemic hippocampus enhances BDNF transcription.
Figure 2. Neuroprotection conferred by long-term estradiol pretreatment is blocked by the MEK inhibitor PD98059.
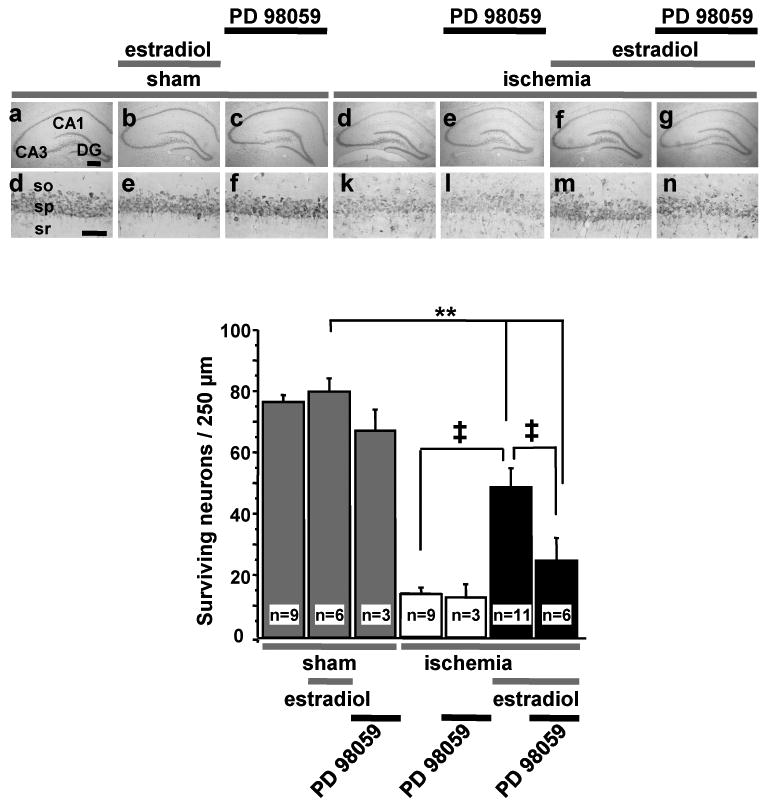
Ovariectomized female rats were treated as described in Figure 1, except that the duration of global ischemia was 10 min. When animals were killed for histological evaluation, they exhibited physiological serum estradiol levels (33 ± 2.9 pg/ml). The upper panel shows representative low (4×) and high (20×) photomigrographs of CA1 pyramidal cells 7 days after ischemia or sham operation. The lower panel illustrates quantitation of surviving pyramidal neurons in CA1 at 7 days after surgery. Estradiol significantly increased the number of surviving neurons. Icv administration of the MEK inhibitor PD98059 (3 μg) at 0 and 12 h after reperfusion prevented the ability of estradiol to increase neuronal survival. Data are from Jover-Mengual et al., 2007. so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum.
Scale bars, Lower magnification, 400 μm; higher magnification, 60 μm.
**, P <0.01 vs. all sham groups; ‡, P <0.01 ischemia + estradiol vs. ischemia or ischemia+estradiol+PD98059.
Figure 3. Long-term estradiol treatment blunts ischemia-induced ERK1 dephosphorylation and acts via MAPK to prevent ischemia-induced CREB dephosphorylation.

Animals were treated as described in Figure 2. They were infused icv with vehicle or the MEK inhibitor PD98059 immediately after reperfusion, killed 1 or 3 h later, and the hippocampal CA1 microdissected. Total cell lysates or nuclear fractions were immunoblotted for total and phospho- (p) ERK-1 and CREB. (A) Ischemia induced dephosphorylation of ERK1 at 1 h after global ischemia. Estradiol significantly increased basal p-ERK1 in shams and maintained levels of p-ERK1 in ischemic rats at 1h. PD98059 reversed the ability of estradiol to maintain p-ERK1 in postischemic CA1. (B) Ischemia promoted dephosphorylation of CREB in the nuclei of CA1 neurons. Estradiol maintained the phosphorylated, activated state of CREB in the CA1 of ischemic rats. PD98059 blocked the ability of estradiol to maintain p-CREB levels in postischemic CA1. Data are from Jover-Mengual et al., 2007.
*, P<0.05; **, P<0.01.
Cross talk with IGF-1
There is considerable evidence that crosstalk between ERα and IGF-1 receptors is important for the cellular actions of both E2 and IGF-1. E2 and IGF-1 act synergistically to regulate synaptic remodeling, neuronal differentiation and survival27. IGF-1-dependent neuronal differentiation and survival require ERs, and E2 neuroprotection requires ongoing IGF-1 signaling28. For example, IGF-1 receptors are required for E2 protection of hilar neurons from seizure-induced injury29. Upon stimulation with E2, ERα and IGF-1 receptors form a macromolecular signaling complex, which recruits and activates downstream kinases such as PI3K and MAPK30, 31. We have shown that the neuroprotective effect of long-term E2 pretreatment in global ischemia is abolished when animals receive icv injections of a specific IGF-1 receptor antagonist (JB1; see Figure 1) in the early post-ischemic period. These results strongly suggest that transactivation of IGF-1 receptors by membrane-associated estrogen receptors activate the ERK/MAPK signaling pathway, leading to CREB phosphorylation and subsequent neuroprotection.
3) Acute versus long-term E2 neuroprotection: are the same receptors and cellular pathways involved?
The importance of postmenopausal estrogen therapy (mimicked experimentally by long-term hormone treatment of ovariectomized animals) for protection against the neuronal death associated with cardiac arrest or stroke remains controversial. A critical issue is whether long-term estrogen exposure and/or prior availability affect the frequency and/or severity of ischemic insults. A finding that a single injection of estradiol administered after an ischemic event has neuroprotective effects is of great clinical relevance, even if a supra-physiological dose is required. We therefore examined the ability of estradiol administered immediately after global ischemia and reperfusion to protect hippocampal CA1 neurons17 (Figure 4A). Ovariectomized female rats were subjected to global ischemia or sham surgery and estradiol (50 μg) or vehicle was injected icv immediately after reperfusion. Ischemia induced extensive death of pyramidal cells in the hippocampal CA1 at 7 days post-ischemia. E2 did not detectably alter the number or morphology of CA1 neurons in sham-operated rats, but significantly reduced the ischemia-induced neuronal loss (P < 0.01). Moreover, acute estradiol significantly improved visual memory assessed at short retention intervals, although it did not prevent deficits in spatial memory (Figure 4B). These promising results raise the possibility that estradiol and other related compounds could be potential therapeutic agents following stroke and cardiac arrest. Whether neuroprotection afforded by E2 administered acutely after ischemia is mediated by the same cellular pathways as those involved in long-term E2 neuroprotection is currently under examination in our lab.
Figure 4. Estradiol administered immediately after global ischemia promotes cellular and behavioral neuroprotection.

Ovariectomized female rats were subjected to global ischemia (10 min) or sham surgery and received a single icv infusion of 50 μg of estradiol or the vehicle immediately after reperfusion. (A) Estradiol afforded significant protection against ischemia-induced cell death measured 7 or 9 days after ischemia (animals killed at 9 days underwent behavior testing). Horizontal bars indicate the mean cell count. Significant differences between ischemia and ischemia + estradiol are indicated by * (P < 0.05) and between sham and ischemia + estradiol by ** (P < 0.05). (B) During days 6–9 after ischemia, visual memory was assessed in the object recognition task (see Gulinello et al., 2006 for details). Data are reported as the exploratory preference score (new object exploration/total object exploration × 100) for 3-min trials. At retention times greater than 1 min, ischemic rats perform at chance levels (i.e., 50%). A single icv infusion of estradiol markedly improved visual preference scores in ischemic rats, assessed at the 10- and 20- but not at the 60-min retention intervals. Sample sizes are denoted on individual bars. *P < 0.05. Data are from Gulinello et al., 2006.
Could the PI3K/Akt pathway play a role in acute E2 neuroprotection?
The survival promoting effects of hormones and growth factors are executed, at least in part, through the PI3K-Akt pathway32, and E2 promotes activation of this cellular pathway in various cell types, including neurons11. Hence, it is reasonable to propose that E2 given acutely after an ischemic insult might mediate its neuroprotective effect via activation of the PI3K/Akt pathway33. PI3K acts by phosphorylation and activation of the serine-threonine kinase Akt. It is well known that Akt promotes cell survival by suppressing genes implicated in apoptotic cell death (for review see34). In each case, Akt phosphorylates and thereby inactivates its target. Targets of Akt known to play a role in neuronal apoptosis following brain ischemia include pro-apoptotic proteins of the Bcl-2 family, pro-caspase-9, which is cleaved to yield caspase-9, an initiator of the caspase death cascade, members of the Forkhead family of transcription factors, which promote transcription of pro-death genes, and GSK-3β (for review see35, 36). The ability of neurotrophins such as BDNF to promote neuronal survival requires functional PI3K/Akt signaling37. Therefore, one can propose a possible mechanism for acute E2 protection involving a synergistic activation of both the ERK/MAPK and PI3K-Akt pathway by estrogens. The activation of the ERK/MAPK signaling pathway could lead to up-regulation of BDNF, which in turn would stimulate the PI3K/Akt pathway through the activation of its target receptor trkB. Moreover, because trkB receptors can promote neuron survival through the activation of both the MAPK and PI3K/Akt pathways38-40, such a mechanism could engage a powerful positive feedback loop for suppressing pro-apoptotic protein transcription by maintaining sustained activation of Akt.
Is there a potential role for GPR30?
GPR30, an orphan G-protein coupled receptor that binds E2 with an affinity similar to ER-α and ER-β41, is widely expressed in the brain, including the hippocampus 42-45. Many of the short latency E2 effects on hippocampal neurons involve second messenger systems that are difficult to reconcile with the intracellular events traditionally associated with nuclear ERs, but would be consistent with the actions of a G protein-coupled receptor. For example, E2 rapidly potentiates kainate currents in hippocampal CA1 pyramidal neurons. This effect is mediated by cAMP46, is evident in hippocampal neurons from mice lacking both ER-α and ER-β and is not blocked by the ER-α/ ER-β antagonist ICI 182,78047. Direct application of E2 can also increase excitatory postsynaptic potentials (EPSPs) elicited by Schaffer collateral stimulation and recorded in the CA1 layer of hippocampal slices from male rats48 and gonadectomized male and female mice lacking a functional ER-α49. Moreover, ICI 182,780, an antagonist of both ER-α and ER-β, does not block E2 potentiation of synaptic responses in slices from ER-α knockout mice. Indeed, in 5/12 slices from ER-α knockout mice, ICI 182,780 alone increased EPSPs. Because new evidence indicates that ICI 182,780 may be a partial agonist at GPR30, which can induce cAMP production50, these findings strongly suggest a role of GPR30 in mediating the short latency increase in hippocampal synaptic transmission observed in response to E2.
In support of this speculation, whole cell current recordings in our laboratory suggest that GPR30 could mediate rapid effects of E2 on synaptic transmission (Figure 5). Acute application of the selective GPR30 agonist G1, which does not bind ER-α and ER-β51, onto hippocampal slices of ovariectomized female rats significantly increased the amplitude of excitatory postsynaptic currents similar to that observed in response to E2 (Fig 5A). Moreover, both G1 and E2 occluded the effects of the other compound on synaptic currents (Fig 5B and 5C), and cells that did not respond to E2 also failed to respond to G1 (data not shown). These observations strongly suggest that E2-dependent increases in synaptic transmission in CA1 pyramidal neurons could be mediated by the activation of GPR30. There is so far no evidence that E2 dependent increases in synaptic transmission in the CA1 hippocampal cells are directly linked to E2 neuroprotection following ischemic insult. However, the observations that GPR30 is expressed and functionally active in the hippocampal CA1 provide a rationale for speculating that this receptor could be involved in the acute neuroprotective actions of E2, most likely through the activation of cellular pathways known to promote cell survival.
Figure 5. The GPR30 agonist G1 mimics short latency estradiol potentiation of hippocampal CA1 neuronal excitability.

Young adult female rats were killed one week after ovariectomy and hippocampal slices were prepared. Schaffer collaterals were activated by monopolar stimulation. Excitatory transmission was monitored by whole cell recordings of excitatory postsynaptic currents (EPSC) of CA1 pyramidal cells in voltage clamp configuration. EPSCs were recorded in the presence of picrotoxin (100 μM) to block inhibitory transmission. After establishing stable baseline EPSCs, 100 nM estradiol (E2) or G1 were bath applied to the slice. (A) shows enhanced responses of CA1 pyramidal neurons to Schaffer collateral stimulation in the presence of E2 or G1. In total, 4/7 cells responded to G1, and 3/5 cells responded to E2. This rate of response is comparable to what has previously been reported for E2 (ref). The latency to onset of the enhanced response and the magnitude of potentiation of the EPSC were identical for both compounds. (B) shows recordings from 2 cells that were potentiated by G1; additional application of E2 has no further effect on the EPSC. (C) shows recordings from 2 cells that were potentiated by E2; additional application of G1 has no further effect on the EPSC.
Thus, if acute E2 neuroprotection following global ischemia involves the rapid activation of the MAPK and/or PI3K/Akt cellular pathways, GPR30 could potentially play a crucial role. GPR30 is a Gαs-coupled receptor50 that can promote calcium release in neurons42 and mediate the phosphorylation of ERK1/ERK2 in breast cancer cell lines52. Thus, GPR30 could promote the activation of the MAPK signaling pathway in neurons through activation of either PKA or PKC53. GPR30 was also shown to directly activate PI3K in cell lines expressing the recombinant receptor54. Transactivation of tyrosine kinase receptors by G-protein coupled receptors in the absence of ligand is well documented55, 56. Therefore, GPR30 could also activate growth factor and neurotrophin receptors even in absence of ligand (e.g., BDNF)57 to act as an early effector of the elaborate anti-apoptotic machinery thought to underlie E2 neuroprotection (see Figure 6). Although GPR30 might be a good candidate to mediate long-term as well as acute E2 neuroprotection, two observations suggest that this receptor might not be involved in the neuroprotective effects of long-term E2 treatment. First, we have shown that chronic E2 neuroprotection is totally abrogated by the ER-α/ER-β antagonist ICI 182,780, a drug that was reported to be an agonist of GPR30 stimulation of cAMP production in vitro50. Second, chronic administration of either an ER-α or ER-β selective agonist provided the same level of neuroprotection as E2 in some treated animals, suggesting that activation of the classical ERs may be sufficient for neuroprotection15. However, it is notable that selective agonists for ER-α and ER-β were not neuroprotective in about half of the animals to which they were administered. Thus, we cannot rule out the possibility that ER-α and ER-β need an additional mediator, such as GPR30, to promote reliable E2 neuroprotection when hormone is administered in the long-term treatment paradigm.
Figure 6. Long-term and acute estradiol treatment may act through multiple cellular pathways to afford neuroprotection following global ischemia.

Our data suggest that long-term estradiol treatment promotes CA1 neuron survival by actions at classical ERs and by transactivation of IGF-1 receptors, resulting in sustained activation of the ERK/MAPK cascade and subsequent increases in p-CREB (pathway in red). Blockade of the ERK/MAPK pathway does not affect estradiol maintenance of Bcl-2 expression, indicating that this action may involve direct interactions of ERs with the Bcl-2 promoter. BDNF, a target of activated CREB, is thought to bind the receptor tyrosine kinase trk-B to activate both the MAPK and PI3K pathway to promote neuroprotection. Stimulation of the PI3K/Akt pathway by BDNF would inactivate the pro-apoptotic proteins (Bad, Bid, pro-caspase-9) to halt the apoptosis cascade. When administered acutely (immediately after reperfusion), estradiol may bind to either classical membrane ERs (ER-α, ER-β) and/or GPR30. GPR30 could block apoptotic cascades through the direct activation of ERK/MAPK and/or PI3K/Akt. This could also occur through the transactivation of receptor tyrosine kinases (IGF-1 receptors and/or Trk-B) by ERs or GPR30. Which of these multiple cellular pathways mediate acute estradiol neuroprotection is currently under examination in our laboratory.
Apaf, Apoptotic protease activating factors; Bax, Bcl-2 associated × protein; BIM, Bcl-2 interacting mediator of cell death; Bad, Bcl-2 associated death protein; Bid, BH3 interacting domain death agonist; cyt c, cytochrome c. BDNF: Brain derived neurotrophic factor.
Conclusion and Perspectives
There is still considerable controversy concerning the risks and benefits of long-term postmenopausal hormone treatment on the incidence and severity of cardiac arrest and stroke in humans. Therefore, in-depth investigation of the molecular mechanisms underlying the neuroprotective effects of E2 when administered acutely after an ischemic event is clinically relevant. Determining whether this form of neuroprotection involves activation of classical ERs or other membrane-associated estrogen receptors, such as GPR30, might also point the way to new therapeutic targets. Our present interests focus on delineating the molecular mechanisms underlying acute neuroprotection afforded by E2 and other pharmacological agents that activate membrane estrogen receptors such as GPR30. If such compounds promote neuroprotection through the activation of the PI3K/Akt pathway, it will be essential to identify which downstream Akt targets (e.g., Forkhead transcription factors, GSK3β) intervene in the apoptotic death cascade to promote neuron survival. Such studies are expected to accelerate development of therapeutic strategies to ameliorate neuronal death and cognitive deficits not only in global ischemia, but in other neurodegenerative disorders.
Acknowledgments
This work was supported by NIH grants R37 MH41414, R01 NS045693 and R01 AG027702 and a grant from the F.M. Kirby Foundation. RSZ is the F.M. Kirby Professor of Neural Repair and Protection.
Footnotes
Disclosure Statement: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilson ME, Dubal DB, Wise PM. Estradiol protects against injury-induced cell death in cortical explant cultures: a role for estrogen receptors. Brain Research. 2000;873:235–42. doi: 10.1016/s0006-8993(00)02479-3. [DOI] [PubMed] [Google Scholar]
- 2.McCullough LD, Hurn PD. Estrogen and ischemic neuroprotection: an integrated view. Trends Endocrinol Metab. 2003;14:228–35. doi: 10.1016/s1043-2760(03)00076-6. [DOI] [PubMed] [Google Scholar]
- 3.Gibson CL, Gray LJ, Murphy SP, Bath PM. Estrogens and experimental ischemic stroke: a systematic review. J Cereb Blood Flow Metab. 2006;26:1103–13. doi: 10.1038/sj.jcbfm.9600270. [DOI] [PubMed] [Google Scholar]
- 4.Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- 5.Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–6. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- 6.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–8. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 7.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 8.McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–74. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 9.Nilsson S, et al. Mechanisms of Estrogen Action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 10.Ronnekleiv OK, Malyala A, Kelly MJ. Membrane-initiated signaling of estrogen in the brain. Semin Reprod Med. 2007;25:165–77. doi: 10.1055/s-2007-973429. [DOI] [PubMed] [Google Scholar]
- 11.Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16:140–53. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- 12.Evinger AJ, 3rd, Levin ER. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005;70:361–3. doi: 10.1016/j.steroids.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Kelly MJ, Ronnekleiv OK. Membrane-initiated estrogen signaling in hypothalamic neurons. Mol Cell Endocrinol. 2008;290:14–23. doi: 10.1016/j.mce.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jover T, et al. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci. 2002;22:2115–24. doi: 10.1523/JNEUROSCI.22-06-02115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen Can Act via Estrogen Receptor {alpha} and {beta} to Protect Hippocampal Neurons against Global Ischemia-Induced Cell Death. Endocrinology. 2005;146:3070–3079. doi: 10.1210/en.2004-1515. [DOI] [PubMed] [Google Scholar]
- 16.Jover-Mengual T, Zukin RS, Etgen AM. MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia. Endocrinology. 2007;148:1131–43. doi: 10.1210/en.2006-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gulinello M, Lebesgue D, Jover-Mengual T, Zukin RS, Etgen AM. Acute and chronic estradiol treatments reduce memory deficits induced by transient global ischemia in female rats. Horm Behav. 2006;49:246–60. doi: 10.1016/j.yhbeh.2005.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rami A, Bechmann I, Stehle JH. Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog Neurobiol. 2008;85:273–96. doi: 10.1016/j.pneurobio.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Wilson BE, Mochon E, Boxer LM. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546–56. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meller R, et al. CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:234–46. doi: 10.1038/sj.jcbfm.9600024. [DOI] [PubMed] [Google Scholar]
- 21.Kitagawa K. CREB and cAMP response element-mediated gene expression in the ischemic brain. Febs J. 2007;274:3210–7. doi: 10.1111/j.1742-4658.2007.05890.x. [DOI] [PubMed] [Google Scholar]
- 22.Hara T, et al. CREB is required for acquisition of ischemic tolerance in gerbil hippocampal CA1 region. J Neurochem. 2003;86:805–14. doi: 10.1046/j.1471-4159.2003.01847.x. [DOI] [PubMed] [Google Scholar]
- 23.Hu BR, Fux CM, Martone ME, Zivin JA, Ellisman MH. Persistent phosphorylation of cyclic AMP responsive element-binding protein and activating transcription factor-2 transcription factors following transient cerebral ischemia in rat brain. Neuroscience. 1999;89:437–52. doi: 10.1016/s0306-4522(98)00352-2. [DOI] [PubMed] [Google Scholar]
- 24.Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–45. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Segars JH, Driggers PH. Estrogen action and cytoplasmic signaling cascades. Part I: membrane-associated signaling complexes. Trends in Endocrinology and Metabolism. 2002;13:349–354. doi: 10.1016/s1043-2760(02)00633-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–40. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 27.Cardona-Gomez GP, Mendez P, DonCarlos LL, Azcoitia I, Garcia-Segura LM. Interactions of estrogen and insulin-like growth factor-I in the brain: molecular mechanisms and functional implications. J Steroid Biochem Mol Biol. 2002;83:211–7. doi: 10.1016/s0960-0760(02)00261-3. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Segura LM, Sanz A, Mendez P. Cross-talk between IGF-I and estradiol in the brain: focus on neuroprotection. Neuroendocrinology. 2006;84:275–9. doi: 10.1159/000097485. [DOI] [PubMed] [Google Scholar]
- 29.Azcoitia I, Sierra A, Garcia-Segura LM. Neuroprotective effects of estradiol in the adult rat hippocampus: interaction with insulin-like growth factor-I signalling. J Neurosci Res. 1999b;58:815–22. doi: 10.1002/(sici)1097-4547(19991215)58:6<815::aid-jnr8>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 30.Song RX, et al. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor {alpha} to the plasma membrane. PNAS. 2004;101:2076–2081. doi: 10.1073/pnas.0308334100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendez P, Wandosell F, Garcia-Segura LM. Cross-talk between estrogen receptors and insulin-like growth factor-I receptor in the brain: Cellular and molecular mechanisms. Frontiers in Neuroendocrinology Estrogen, Growth Factors and Brain Function, N/A. 2006;27:391–403. doi: 10.1016/j.yfrne.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–44. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 33.Amantea D, Russo R, Bagetta G, Corasaniti MT. From clinical evidence to molecular mechanisms underlying neuroprotection afforded by estrogens. Pharmacological Research. 2005;52:119–132. doi: 10.1016/j.phrs.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Parcellier A, Tintignac LA, Zhuravleva E, Hemmings BA. PKB and the mitochondria: AKTing on apoptosis. Cell Signal. 2008;20:21–30. doi: 10.1016/j.cellsig.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 35.Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol. 2006;34:249–70. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]
- 36.Mullonkal CJ, Toledo-Pereyra LH. Akt in ischemia and reperfusion. J Invest Surg. 2007;20:195–203. doi: 10.1080/08941930701366471. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–91. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen N, Lee SB, Lee YS, Lee KH, Ahn JY. Neuroprotection by NGF and BDNF Against Neurotoxin-Exerted Apoptotic Death in Neural Stem Cells Are Mediated Through Trk Receptors, Activating PI3-Kinase and MAPK Pathways. Neurochem Res. 2008 doi: 10.1007/s11064-008-9848-9. [DOI] [PubMed] [Google Scholar]
- 39.Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J Neurochem. 1999;73:1409–21. doi: 10.1046/j.1471-4159.1999.0731409.x. [DOI] [PubMed] [Google Scholar]
- 40.Almeida RD, et al. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005;12:1329–43. doi: 10.1038/sj.cdd.4401662. [DOI] [PubMed] [Google Scholar]
- 41.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 42.Brailoiu E, et al. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193:311–21. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]
- 43.Sakamoto H, et al. Expression of G protein-coupled receptor-30, a G protein-coupled membrane estrogen receptor, in oxytocin neurons of the rat paraventricular and supraoptic nuclei. Endocrinology. 2007;148:5842–50. doi: 10.1210/en.2007-0436. [DOI] [PubMed] [Google Scholar]
- 44.Matsuda K, et al. Expression and intracellular distribution of the G protein-coupled receptor 30 in rat hippocampal formation. Neurosci Lett. 2008;441:94–9. doi: 10.1016/j.neulet.2008.05.108. [DOI] [PubMed] [Google Scholar]
- 45.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346:904–10. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 46.Gu Q, Moss RL. 17 beta-Estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J Neurosci. 1996;16:3620–9. doi: 10.1523/JNEUROSCI.16-11-03620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gu Q, Korach KS, Moss RL. Rapid action of 17beta-estradiol on kainate-induced currents in hippocampal neurons lacking intracellular estrogen receptors. Endocrinology. 1999;140:660–6. doi: 10.1210/endo.140.2.6500. [DOI] [PubMed] [Google Scholar]
- 48.Bi R, Broutman G, Foy MR, Thompson RF, Baudry M. The tyrosine kinase and mitogen-activated protein kinase pathways mediate multiple effects of estrogen in hippocampus. Proc Natl Acad Sci U S A. 2000;97:3602–7. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fugger HN, Kumar A, Lubahn DB, Korach KS, Foster TC. Examination of estradiol effects on the rapid estradiol mediated increase in hippocampal synaptic transmission in estrogen receptor alpha knockout mice. Neurosci Lett. 2001;309:207–9. doi: 10.1016/s0304-3940(01)02083-3. [DOI] [PubMed] [Google Scholar]
- 50.Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen Action Via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and cAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Mol Endocrinol. 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 51.Bologa CG, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 52.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 53.Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- 54.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 55.Delcourt N, Bockaert J, Marin P. GPCR-jacking: from a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol Sci. 2007;28:602–7. doi: 10.1016/j.tips.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Natarajan K, Berk BC. Crosstalk coregulation mechanisms of G protein-coupled receptors and receptor tyrosine kinases. Methods Mol Biol. 2006;332:51–77. doi: 10.1385/1-59745-048-0:51. [DOI] [PubMed] [Google Scholar]
- 57.Lee FS, Rajagopal R, Chao MV. Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev. 2002;13:11–7. doi: 10.1016/s1359-6101(01)00024-7. [DOI] [PubMed] [Google Scholar]