

Abstract
The Molecular Mechanics/Poisson Boltzmann Surface Area (MM/PBSA) and the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) methods calculate binding free energies for macromolecules by combining molecular mechanics calculations and continuum solvation models. To systematically evaluate the performance of these methods, we report here an extensive study of 59 ligands interacting with six different proteins. First, we explored the effects of the length of the molecular dynamics (MD) simulation, ranging from 400 to 4800 ps, and the solute dielectric constant (1, 2 or 4) to the binding free energies predicted by MM/PBSA. The following three important conclusions could be observed: (1). MD simulation lengths have obvious impact on the predictions, and longer MD simulations are not always necessary to achieve better predictions; (2). The predictions are quite sensitive to solute dielectric constant, and this parameter should be carefully determined according to the characteristics of the protein/ligand binding interface; (3). Conformational entropy showed large fluctuations in MD trajectories and a large number of snapshots are necessary to achieve stable predictions. Next, we evaluated the accuracy of the binding free energies calculated by three Generalized Born (GB) models. We found that the GB model developed by Onufriev and Case was the most successful model in ranking the binding affinities of the studied inhibitors. Finally, we evaluated the performance of MM/GBSA and MM/PBSA in predicting binding free energies. Our results showed that MM/PBSA performed better in calculating absolute, but not necessarily relative, binding free energies than MM/GBSA. Considering its computational efficiency, MM/GBSA can serve as a powerful tool in drug design, where correct ranking of inhibitors is often emphasized.
Keywords: MM/GBSA, MM/PBSA, Generalized Born, Poisson Boltzmann, Binding free energy
Introduction
Computational methods that combine molecular mechanics energy and implicit solvation models, such as Molecular Mechanics/Poisson Boltzmann Surface Area (MM/PBSA) and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA), have been widely exploited in free energy calculations.1–3 Compared with the rigorous methods such as free energy perturbation (FEP) and thermodynamic integration (TI) methods,4 MM/PBSA and MM/GBSA are more computationally efficient. A related approach is the linear interaction energy (LIE) method,5 which averages interaction energy from the MD simulations to estimate the absolute binding free energy. Similar to MM/PBSA and MM/GBSA, LIE restricts the simulations to the two end points of ligand binding. Different from most empirical scoring functions used in molecular docking MM/PBSA and MM/GBSA do not need a large training set to fit different parameter for each energy term.6–11 Moreover, both of MM/PBSA and MM/GBSA allow for rigorous free energy decomposition into contributions originating from different groups of atoms or types of interaction.12–14
In MM/PBSA or MM/GBSA, binding free energy (ΔGbind) between a ligand (L) and a receptor (R) to form a complex RL is calculated as:
| (1) |
| (2) |
| (3) |
where ΔEMM, ΔGsol and −TΔS are the changes of the gas phase MM energy, the solvation free energy and the conformational entropy upon binding, respectively. ΔEMM includes ΔEinternal (bond, angle and dihedral energies), electrostatic ΔEelectrostatic and van der Waals energies ΔEvdw. ΔGsolv is the sum of electrostatic solvation energy (polar contribution), ΔGPB/GB, and non-electrostatic solvation component (non-polar contribution), ΔGSA. The polar contribution is calculated using either GB or PB model, while the non-polar energy is estimated by solvent accessible surface area (SASA). The conformational entropy change −TΔS is usually computed by normal-mode analysis on a set of conformational snapshots taken from MD simulations.
A common strategy to reduce noise and cancel errors in simulations is to run molecular dynamics (MD) simulations on the complex only. Snapshots taken from this single trajectory of MD simulation are used to calculate each free energy component in the above equations. In such a single trajectory approach, ΔEinternal is canceled between ligand, receptor and complex, which can significantly reduce the noise in most cases. One can also use a separate trajectory approach to calculate the energy terms by taking snapshots from three individual MD simulations of complex, protein and ligand separately.1 In principle, this approach is more accurate than the single trajectory approach. Meanwhile, it is also more expensive in terms of computational cost.
MM/GBSA or MM/PBSA has been successfully applied to various protein-ligand15–23 or protein-protein/peptide complexes24–26 but their performance is system dependent27, 28 In addition, MM/PBSA or MM/GBSA is sensitive to simulation protocols, such as sampling strategy of generating snapshots and entropy calculation methods as well as other parameters, e.g. charge models, force fields, solute dielectric constant and radius parameters in continuum solvent models.1 For example, Weis and coworkers studied how the force fields and the methods to sample conformational space affected the calculated binding free energies of seven biotin analogues. They found that simulation results are not sensitive to force fields but explicit water molecules are indispensible in MD simulations.29
Here we systematically investigated the following issues in MM/PBSA and MM/GBSA methods: (1). The effect of the length of MD simulations; (2). The suitable solute dielectric constant to calculate the polar solvation energies; (3). The best way to perform the entropy calculations; (4). Comparison of the performances of different PB and GB models to evaluate the absolute binding free energy and rank affinities of ligands bound to the same protein.
For such an investigation, it is necessary to choose a set of reliable test systems. We performed MM/PBSA and MM/GBSA calculations with various protocols and parameters for 59 ligands bound to six different proteins. These systems were selected because they have been well characterized by X-ray crystallography, and reliable experimental binding free energies have been obtained for a number of ligands. Moreover, systems like avidin and P450cam have been studied by several theoretical techniques, such as FEP, LIE, and MM/PBSA,28–32 and we can compare our results with the previous studies.
Materials and Methods
1. Preparation of complexes
The MM/GBSA or MM/PBSA calculations were applied to six different protein systems, including α-thrombin (7 ligands), avidin (7 ligands), cytochrome C peroxidase (18 ligands), neuraminidase (8 ligands), P450cam (12 ligands) and penicillopepsin (7 ligands). The experimental binding data and the PDB entries for the six proteins are listed in Table S1 in the supporting materials. The chemical structures of the ligands are shown in Figure S1 in the supporting materials. The protonated states for all ligands are shown in Figure 1 in the Supporting Materials.
For ligands bound to α-thrombin, cytochrome C peroxidase, neuraminidase and penicillopepsin, MD simulations were performed based on the crystal structures of the complexes. The starting structures of the six avidin analogues (b2–b7) were generated based on the avidin-biotin complex (PDB entry: 1avd33). The biotin molecule in the crystal structure was manually mutated to the other ligands. It has been shown that the neutral form of the guanidinium group in b2 and b5 biotin analogues is dominant when it is bound to the protein.34 Therefore, the neutral form of the guanidinium group was used in our simulations. The crystal structures of the nine P450cam ligands were used for MD simulations. Starting structures of the other three P450 ligands (e3, e5 and e6) were obtained by manually modifying the ligand (e1) in the crystal structure of 2cpp35 with the conformation of the protein unaltered. The preparation of the models was accomplished in the SYBYL molecular simulation package.36
In the cytochrome C peroxidase complexes, the lone-pair electrons of the epsilon nitrogen in His175 form resonant bonds with the iron ion and the hydrogen atom is located at the delta nitrogen of His175. In the P450cam complexes, lone-pair electrons of the sulfur atom in Cys357 form resonant bonds with the iron ion and this cysteine residue is thus deprotonated. All the crystal water molecules were kept in the simulations.
The atomic partial charges of all ligands were derived by semiempirical AM1 geometry optimization and subsequent single-point Hartree-Fock (HF)/6-31G* calculations of the electrostatic potential, to which the charges were fitted using the RESP technique.37 The reason why we chose AM1 for optimization, not usually used HF/6-31G(d), is to reduce computational cost.38 The optimization and the electrostatic potential calculations were conducted by Gaussian03.39 Partial charges and force field parameters of the inhibitors were generated automatically using the antechamber program in AMBER9.0.40
In molecular mechanics (MM) minimizations and MD simulations, the AMBER03 force field was used for proteins41 and the general AMBER force field (gaff) was used for ligands.42 The force field parameters developed by Giammona were used for the heme groups in the cytochrome C peroxidase and the P450cam systems.43 To neutralize the systems, counter ions of Cl− or Na+ were placed in grids that had the largest positive or negative Coulombic potential around the protein. The whole system was immersed in a rectangular box of TIP3P water molecules. The water box was extended 9 Å from solute atoms in all three dimensions.
2. Molecular Dynamics (MD) Simulations
In MM minimization and MD simulations, particle mesh Ewald (PME) was employed to treat the long-range electrostatic interactions.44 The dielectric constant was set to 1.0 and the cutoff for nonbonded interactions was set to 8Å. Before MD simulations, the complexes were relaxed by 5,000 cycles of minimization procedure (500 cycles of steepest descent and 4500 cycles of conjugate gradient minimization). After minimization, the system was gradually heated in the NVT ensemble from 10 K to 300 K over 20 ps. Initial velocities were assigned from a Maxwellian distribution at the starting temperature. We then performed 5 ns NPT MD simulations with a target temperature of 300 K and a target pressure of 1 atm. Temperature was controlled by the Andersen temperature coupling scheme45 and the pressure was controlled by the isotropic position scaling protocol applied in AMBER.46 The SHAKE procedure was employed to constrain all hydrogen atoms, and the time step was set to 2.0 fs.47 After equilibration, the coordinates of the complexes were saved every 2 ps. The MM optimization and MD simulations were accomplished by using the sander program in AMBER9.0.46
3. MM/PBSA calculations
The gas-phase interaction energy between the protein and the ligand, ΔEMM, is the sum of electrostatic and van der Waals interaction energies. The solvation free energy ΔGsolvation is the sum of polar (ΔGPB) and non-polar (ΔGSA) parts. The ΔGPB term was calculated by solving the finite-difference Possion-Boltzmann equation using Delphi II.48 In Delphi calculations, the grid spacing was set to 0.5 Å, and the longest linear dimension of the grid was extended at least 20% beyond the protein. The Parse radii were employed for all atoms.49 The Pauling van der Waals radii 1.35 and 1.95 Å50 were used for F and Br, respectively. For Fe in the heme group, the van der Waals radius in the force field was used. The value of the exterior dielectric constant was set to 80, and the solute dielectric constant was set to one of the three values: 1, 2 or 4.24 The non-polar contribution was determined based on solvent-accessible surface area (SASA) using the LCPO method:51 ΔGSA=0.0072 × SASA. For the calculations of ΔEMM, ΔGGB, and ΔGSA, 625 snapshots evenly extracted from the single MD trajectory of complex from 0 to 5 ns were used.
3. MM/GBSA Calculations
In the MM/GBSA calculations, the gas-phase interaction energy (ΔEMM) and the non-polar (ΔGSA) part of the solvation energy were calculated in the same way as in the MM/PBSA calculations. The electrostatic solvation energy (ΔGGB) was calculated by using GB models. A value of 80 was used for the exterior dielectric constant, and 1, 2 or 4 was used for the solute dielectric constant for comparison. Three GB models implemented in AMBER9.0 were used, including the pairwise GB model of Hawkins and coworkers (referred as GBHCT)52, 53 with parameters developed by Tsui and Case,54 and two modified GB models developed by Onufriev and colleagues (referred as GBOBC1 and GBOBC2).55
5. The entropy calculations
The normal-mode analysis was performed to evaluate the conformational entropy change upon ligand binding (−TΔS) using the nmode program in AMBER9.0.46 Because the normal mode analysis is computationally expensive, we only considered the residues within a 12 Å sphere centered at the ligand and these residues were retrieved from an MD snapshot for each ligand-protein complex. The open valences were saturated by adding hydrogen atoms using the tleap program of AMBER9.0.46 The corresponding ligand and receptor were extracted from the reduced complex structure. Then each structure was fully minimized for 100,000 steps using a distance-dependent dielectric of 4rij (rij is the distance between two atoms) to mimic the solvent dielectric change from the solute to solvent until the root-mean-square of the elements of the gradient vector was less than 5 × 10−4 kcal·mol−1·Å−1. To reduce the computational demand, 125 snapshots were taken from 0 to 5 ns to estimate the contribution of the entropy to binding. The final conformational entropy was obtained from the average over the snapshots. It should be noted that different from the other energy terms the entropy contribution is computed in a way independent of the internal dielectric constant.
Results and Discussion
1. Binding free energies calculated by MM/PBSA
We first calculated the binding free energies of the six protein systems using the MM/PBSA method. As the first try, the binding entropy was averaged over 45 snapshots from 0.2 to 2 ns, and the other energy terms were averaged over 225 snapshots from 0.2 to 2 ns. We investigated how the interior dielectric constant affected the performance of MM/PBSA. Three different solute dielectric constants (1, 2 and 4) in the PB calculations were used and the best value was determined by comparing the correlation coefficients between the calculated and the experimental binding free energies.
For the six systems studied here, the best correlations are shown in Figure 1, and the calculated binding free energies and five energy components are listed in Table 1. In terms of the correlation coefficients, the accuracy of MM/PBSA varied for different systems. The binding free energies between avidin and its ligands from simulation correlate very well with experimental values (correlation coefficient r=0.92). For three other systems, α-thrombin, neuraminidase and P450cam, MM/PBSA achieved relatively satisfactory performance (r = 0.68~0.81). MM/PBSA did not perform well on cytochrome C peroxidase (r = 0.27) and penicillopepsin (r = 0.41).
Figure 1.

The correlations between the binding free energies calculated by MM/PBSA and the experimental values for (a) α-thrombin inhibitors, (b) avidin, (c) cytochrome C peroxidase, (d) neuraminidase, (e) P450cam and (f) penicillopepsin.
Table 1.
The experimental and the calculated binding free energies (kcal/mol) using MM/PBSA for the six protein-ligand systems.
| No. | ΔEvdw | ΔEele | ΔGPB | ΔGSA | ΔEele+ΔGPB | ΔEvdw+ΔGSA | TΔS | ΔGcal. | ΔGexp. |
|---|---|---|---|---|---|---|---|---|---|
| α-thrombin (εin = 4) | |||||||||
| a1 | −58.86±1.09a | −4.37±0.24 | 13.66±0.29 | −7.61±0.17 | 9.28±0.04 | −66.47±1.27 | −22.81±1.57 | −34.38±0.35 | −12.39 |
| a2 | −62.42±1.69 | −3.64±2.36 | 14.29±1.72 | −7.65±0.03 | 10.65±0.64 | −70.06±1.72 | −20.20±3.01 | −39.22±4.09 | −10.08 |
| a3 | −52.69±1.18 | −2.15±0.27 | 11.32±0.43 | −6.68±0.24 | 9.16±0.16 | −59.37±1.42 | −20.01±0.65 | −30.19±1.92 | −8.92 |
| a4 | −50.58±2.58 | −0.46±0.07 | 8.58±0.19 | −6.42±0.13 | 8.12±0.12 | 57.00±2.71 | −17.97±1.06 | −30.92±1.53 | −7.68 |
| a5 | −18.73±0.24 | −33.35±0.28 | 44.56±0.10 | −2.79±0.00 | 11.21±0.38 | −21.51±0.24 | −15.50±0.82 | 5.20±0.20 | −3.98 |
| a6 | −57.79±0.46 | −18.08±0.11 | 28.14±0.09 | −7.11±0.01 | 10.06±0.02 | −64.89±0.45 | −24.99±1.73 | −29.84±2.20 | −10.6 |
| a7 | −58.27±0.17 | −40.62±0.06 | 57.20±0.21 | −7.02±0.04 | 16.58±0.28 | −65.29±0.13 | −22.53±0.33 | −26.18±0.08 | −11.57 |
| avidin (εin = 1) | |||||||||
| b1 | −30.31±0.30 | −172.54±4.72 | 177.89±4.58 | −4.21±0.01 | 5.35±0.14 | −34.52±0.31 | −18.69±0.22 | −10.48±0.39 | −20.4 |
| b2 | −30.10±0.83 | −156.76±4.74 | 167.42±4.55 | −4.27±0.04 | 10.66±0.19 | −34.57±0.79 | −20.44±0.65 | −3.27±0.33 | −14.3 |
| b3 | −28.80±0.19 | −165.47±5.49 | 168.64±2.62 | −4.21±0.01 | 3.17±2.86 | −33.01±0.17 | −21.33±0.43 | −8.50±2.61 | −14.0 |
| b4 | −40.31±0.14 | −28.99±0.50 | 55.52±1.49 | −5.55±0.06 | 26.53±1.99 | −45.86±0.19 | −17.64±1.61 | −1.87±3.41 | −8.8 |
| b5 | −28.52±1.39 | −21.67±7.19 | 39.47±4.80 | −4.09±0.04 | 17.80±2.39 | −32.61±1.43 | −15.19±1.03 | 0.37±1.98 | −8.2 |
| b6 | −25.53±1.00 | −21.97±0.20 | 36.26±1.36 | −3.69±0.28 | 14.29±1.16 | −29.22±1.28 | −14.30±1.61 | 0.63±1.48 | −5.0 |
| b7 | −11.99±0.16 | −25.11±0.30 | 28.23±0.06 | −2.12±0.01 | 3.12±0.09 | −14.10±0.16 | −13.57±1.08 | 2.59±0.83 | −4.5 |
| cytochrome C peroxidase (εin = 1) | |||||||||
| c1 | −25.60±0.02 | −352.29±0.21 | 365.41±0.32 | −2.81±0.01 | 13.12±0.11 | −28.41±0.03 | −15.50±0.09 | −15.29±0.14 | −3.85 |
| c2 | −25.84±0.06 | −354.14±0.11 | 363.18±0.80 | −2.79±0.02 | 9.03±0.69 | −28.63±0.04 | −16.87±1.10 | −19.60±0.73 | −4.78 |
| c3 | −17.90±0.91 | −361.70±3.03 | 369.11±10.1 | −1.93±0.43 | 7.41±7.09 | −19.82±1.34 | −14.53±0.06 | −12.41±8.43 | −4.81 |
| c4 | −22.28±0.15 | −353.88±1.88 | 364.08±3.20 | −2.62±0.00 | 10.20±1.32 | −24.90±0.16 | −15.05±1.02 | −14.70±1.16 | −5.86 |
| c5 | −18.07±0.07 | −365.62±6.40 | 377.83±2.66 | −2.47±0.02 | 12.21±3.74 | −20.53±0.05 | −13.19±1.28 | −8.32±3.79 | −3.96 |
| c6 | −17.74±0.48 | −377.44±0.95 | 381.86±1.05 | −2.86±0.44 | 4.42±0.09 | −20.60±0.05 | −15.63±0.03 | −16.18±0.05 | −6.00 |
| c7 | −17.94±0.12 | −372.64±3.54 | 376.95±3.28 | −2.40±0.01 | 4.31±0.26 | −20.34±0.12 | −15.73±0.04 | −16.02±0.38 | −5.99 |
| c8 | −21.33±0.03 | −360.68±2.15 | 374.32±2.77 | −2.49±0.00 | 13.64±0.62 | −23.83±0.03 | −15.50±0.40 | −10.19±0.59 | −4.96 |
| c9 | −17.09±0.41 | −372.32±1.53 | 371.75±0.19 | −2.44±0.00 | −0.57±1.73 | −19.52±0.41 | −15.44±0.14 | −20.09±1.31 | −5.21 |
| c10 | −20.72±0.31 | −372.36±0.79 | 376.73±2.16 | −3.29±0.43 | 4.37±1.37 | −24.01±0.74 | −15.45±2.08 | −19.64±2.11 | −4.92 |
| c11 | −21.89±0.08 | −363.77±3.15 | 370.83±3.33 | −2.64±0.01 | 7.06±0.19 | −24.53±0.06 | −14.50±0.33 | −17.47±0.25 | −4.92 |
| c12 | −18.40±0.08 | −379.84±2.79 | 380.67±2.49 | −2.48±0.00 | 0.83±0.30 | −20.88±0.08 | −14.39±0.48 | −20.05±0.38 | −7.07 |
| c13 | −16.64±0.04 | −381.35±2.81 | 382.24±3.07 | −2.78±0.42 | 0.89±0.26 | −19.42±0.46 | −13.81±0.31 | −18.54±0.72 | −5.02 |
| c14 | −19.34±0.12 | −366.62±1.23 | 367.85±0.56 | −2.62±0.00 | 1.22±0.66 | −21.96±0.12 | −15.22±0.93 | −20.74±0.55 | −4.73 |
| c15 | −11.42±0.03 | −372.89±0.16 | 365.15±0.18 | −2.42±0.44 | −7.73±0.01 | −13.84±0.42 | −12.09±0.29 | −21.57±0.43 | −4.33 |
| c16 | −15.83±0.06 | −375.74±2.87 | 375.48±1.02 | −2.26±0.00 | −0.26±1.86 | −18.09±0.06 | −15.19±0.04 | −18.35±1.91 | −5.82 |
| c17 | −16.35±0.00 | −370.52±1.42 | 367.86±0.32 | −2.34±0.00 | −2.66±1.10 | −18.69±0.01 | −14.95±0.68 | −21.34±1.11 | −5.94 |
| c18 | −15.64±0.13 | −369.60±2.73 | 369.33±0.75 | −2.35±0.01 | −0.27±3.47 | −17.980.14 | −16.93±0.07 | −18.25±1.32 | −6.06 |
| neuraminidase (εin = 4) | |||||||||
| d1 | −22.63±0.30 | −29.30±0.41 | 29.57±0.89 | −3.99±0.38 | 0.27±0.49 | −26.61±0.08 | −21.97±1.02 | −4.37±1.59 | −4.09 |
| d2 | −22.95±0.57 | −33.69±1.21 | 32.14±0.84 | −4.65±0.03 | −1.55±0.36 | −27.60±0.59 | −15.78±2.65 | −13.36±2.88 | −7.23 |
| d3 | −21.19±0.17 | −26.07±0.54 | 27.55±0.43 | −4.45±0.00 | 1.48±0.97 | −25.64±0.17 | −21.54±0.07 | −2.62±0.72 | −3.74 |
| d4 | −21.84±0.52 | −22.36±0.17 | 25.43±0.18 | −4.64±0.01 | 3.06±0.01 | −26.49±0.53 | −20.75±2.30 | −2.68±2.84 | −4.84 |
| d5 | −20.12±0.64 | −23.62±0.04 | 25.55±0.64 | −4.58±0.04 | 1.92±0.60 | −24.71±0.68 | −19.03±0.08 | −3.75±1.20 | −6.61 |
| d6 | −28.40±0.03 | −22.53±3.39 | 26.83±1.90 | −4.99±0.03 | 4.30±1.49 | −33.39±0.05 | −19.29±1.83 | −9.80±3.26 | −10.2 |
| d7 | −27.83±0.38 | −3.53±0.35 | 9.44±0.80 | −4.98±1.16 | 6.11±0.45 | −32.81±1.55 | −22.86±2.01 | −3.84±0.05 | −7.73 |
| d8 | −29.43±0.01 | −13.36±0.42 | 19.34±0.24 | −4.49±0.47 | 5.98±0.18 | −33.92±0.46 | −17.72±2.16 | −10.22±1.89 | −11.45 |
| P450cam (εin = 1) | |||||||||
| e1 | −27.57±0.61 | −9.10±0.27 | 21.78±0.57 | −2.78±0.58 | 12.68±0.83 | −30.35±0.03 | −15.25±0.85 | −2.43±0.01 | −7.90 |
| e2 | −26.94±0.28 | −0.14±0.04 | 13.22±0.56 | −3.33±0.03 | 13.09±0.52 | −30.27±0.25 | −16.63±0.62 | −0.56±0.15 | −5.91 |
| e3 | −29.03±0.24 | −8.59±0.12 | 25.51±0.79 | −3.38±0.00 | 16.92±0.90 | −32.41±0.24 | −14.45±1.76 | −1.04±2.42 | −6.54 |
| e4 | −20.35±0.05 | −1.85±0.10 | 12.69±0.50 | −2.79±0.02 | 10.84±0.40 | −23.14±0.03 | −15.04±0.27 | 2.74±0.63 | −5.57 |
| e5 | −21.13±0.24 | −0.23±0.08 | 13.92±0.50 | −2.37±0.55 | 13.69±0.57 | −23.50±0.78 | −14.40±0.32 | 4.60±0.11 | −5.52 |
| e6 | −27.68±0.36 | −9.02±2.34 | 22.91±0.38 | −3.56±0.01 | 13.89±1.96 | −31.25±0.35 | −15.31±0.54 | −2.04±1.06 | −5.93 |
| e7 | −29.42±0.10 | −4.32±0.16 | 20.62±0.68 | −3.48±0.01 | 16.29±0.51 | −32.89±0.10 | −13.24±0.42 | −3.36±0.83 | −7.53 |
| e8 | −26.36±0.04 | −0.03±0.01 | 12.65±0.03 | −2.60±0.55 | 12.62±0.04 | −28.96±0.51 | −13.99±0.56 | −2.34±0.01 | −5.90 |
| e9 | −28.09±0.11 | −2.87±0.07 | 16.98±0.08 | −3.15±0.01 | 14.11±0.01 | −31.23±0.10 | −15.10±0.60 | −2.03±0.69 | −7.40 |
| penicillopepsin (εin = 2) | |||||||||
| f1 | −50.93±1.01 | −32.01±2.12 | 59.90±1.36 | −8.59±0.00 | 27.89±3.48 | −59.52±1.02 | −22.43±1.30 | −9.20±3.20 | −12.83 |
| f2 | −41.34±1.58 | −17.23±3.30 | 35.94±0.53 | −6.78±0.18 | 18.70±3.83 | −48.12±1.76 | −22.01±0.44 | −7.41±1.62 | −10.51 |
| f3 | −48.82±1.24 | −33.58±0.82 | 48.29±1.66 | −7.34±0.13 | 14.71±0.84 | −55.16±1.37 | −24.89±0.64 | −15.57±1.16 | −12.27 |
| f4 | −40.62±2.30 | −25.06±3.58 | 51.78±1.11 | −6.79±0.29 | 26.72±2.47 | −47.41±2.59 | −16.09±1.52 | −14.60±2.45 | −10.91 |
| f5 | −38.63±4.78 | −7.32±0.62 | 23.49±4.96 | −5.48±0.85 | 16.17±4.34 | −44.10±5.63 | −20.06±0.16 | −7.87±1.45 | −8.37 |
| f6 | −42.23±4.33 | −8.76±0.99 | 26.45±3.94 | −6.60±0.05 | 17.69±2.96 | −48.83±4.28 | −21.31±0.23 | −9.83±1.09 | −7.03 |
| f7 | −45.28±2.23 | −15.07±2.75 | 34.59±5.31 | −7.51±0.48 | 19.52±2.56 | −52.79±2.72 | −23.67±2.29 | −9.60±2.13 | −6.80 |
The statistical error was estimated based on the deviation between block averages.
The success of MM/PBSA on the avidin system is not surprising. The seven ligands have homologous chemical structures and their experimental binding free energies span a wide range, from −4.5 to −20.4 kcal/mol. The system has been widely studied by several methods, including FEP,30 LIE,32 and MM/PBSA.18, 20, 28, 29 All the computational results showed good correlation with the experimental values. In this system, it was observed that the large change of the electrostatic interaction upon binding was compensated by the desolvation energy (Table 1), which is common in many systems.
The correlation coefficient (r = 0.72 at εin= 1 and r = 0.69 at εin= 2) of the P450cam system is acceptable. Almlöf et al. studied the P450cam using the LIE method and they achieved a correlation coefficient of 0.96.31 It should be noted in the LIE analysis, two scaling parameters (α and β) for van der Waals and electrostatic interactions, and a constant term (γ) were adjusted by linear correlation. That is to say, in LIE, different energy has different weight. Therefore, it is not surprising to see that the correlation given by MM/PBSA is worse than that given by LIE.
The correlation coefficient for neuraminidase (r = 0.68 at εin= 4) is not as good as that for avidin. One possible reason is that, compared with the avidin test set, the binding free energies of the neuraminidase ligands fall into a much narrower range, from −3.7 to −11.5 kcal/mol, than those of the avidin ligands.
There are 18 ligands in the cytochrome C peroxidase system, and their binding free energies range from −3.9~−7.0 kcal/mol. As shown in Figure 1, the correlation coefficient is only 0.27, which means that MM/PBSA is not efficient to rank the binding affinities. The predictions on cytochrome C peroxidase are poor, since the ligands are dissimilar, while those of, for example, avidin are similar, and the binding free energy ranges relatively narrowly (−3.85 to −7.07 kcal/mol). Moreover, the force field parameters used for the heme group and the radii used in the PB calculations for the Fe ion are not as accurate as those for other molecules. Similar as cytochrome C peroxidase, P450cam has a heme group in the binding pocket. The prediction on P450cam is better due to the following reasons. First, the binding free energies of the P450cam inhibitors fall into a wider range, from −5.5 to −11.8 kcal/mol. Second, the P450cam inhibitors are hydrophobic. The relatively small contribution of the electrostatic and polar solvation energy to binding effectively reduces the magnitude of fluctuations.
Compared with avidin and neuraminidase, the other two systems, α-thrombin and penicillopepsin, are more challenging because their ligands have quite diverse chemical structures (Figure S1 in the supporting materials). With respect to the correlation coefficient, the prediction for α-thrombin is satisfactory. It should be noted that in Figure 1a the data are not evenly distributed and one sample (a5, benzamidine) deviates from the others significantly. In order to estimate the capability of MM/PBSA to rank the binding affinities of the candidate molecules, the Spearman correlation coefficient (rs), which calculates the correlation between two sets of rankings, has been calculated for the α-thrombin inhibitors. However, the low Spearman correlation coefficient of 0.29 means that the predictions from MM/PBSA cannot give effective ranking for the studied molecules. The calculated binding free energy for a5 is unfavorable (5.20 kcal/mol) compared with other ligands. If we exclude benzamidine in correlation, the correlation coefficient will decrease from 0.81 to 0.01. In summary, for this case, MM/PBSA did not make reliable predictions for ligands with diverse structures. Ligands a5 and a7 are positively charged in neutral aqueous solution, while a1 to a4 do not have charged centers. Ligand a6 has two charged centers, but its net charge is zero. The charged states of a5 and a7 might be the essential reason for the poor predictions. The polarization of the charged molecules to the binding interface is quite different from the neutral ones. Therefore, it is necessary to use different solute dielectric constants to characterize the local environment of the binding interfaces. With the same parameters for all ligands, the systematic errors associated with different types of ligands cannot be canceled efficiently.
The correlation coefficient (r=0.41 at εin= 2) indicates that the prediction for the penicillpepsin system is not satisfactory either. Although the charged state of all penicillpepsin ligands was the same, diverse chemical structures still led to unsatisfactory predictions.
In summary, we found that MM/PBSA achieved satisfactory performance on ligands with similar chemical structures, such as in the cases of avidin and P450. For ligands with diverse structures such as those of α-thrombin and penicillpepsin, the accuracy of MM/PBSA calculations varied upon systems.
2. Impact of the value of the solute dielectric constant
In MM/GBSA or MM/PBSA, the dielectric constant (εin) of unity (1.0) is normally used for solute. But for macromolecules, especially for the highly-charged binding interface, higher εin values are used for considering electronic polarization effect. We have compared the binding free energies computed using three different solute dielectric constants, 1.0, 2.0 and 4.0. The correlation coefficients between the calculated binding free energies and the experimental data are listed in Table 2. It is obvious that the predictions are sensitive to the value of solute dielectric constant. For example, for the avidin system, εin=1 made the best correlation (r = 0.92), which was much better than those using εin=2 or 4 (r = 0.67 or 0.51). For the six systems studied here, if the correlation between the experimental binding free energies and the predicted value was used as the criterion, εin= 1 performed best for avidin, cytochrome C peroxidase, and P450cam; εin= 2 performed best for penicillopepsin; while εin= 4 performed best for the other two systems, neuraminidase and α-thrombin. One thing we need to point out is that MM/PBSA or MM/GBSA is usually used to rank the binding affinities for some systems rather than give the accurate predictions of the absolute binding free energies. Here, we used the single trajectory protocol to estimate the total binding free energies, and the changes of the conformational energy for ligand and protein upon ligand binding were not included. So it is reasonable that the prediction will be quite different from the true values.
Table 2.
The correlation coefficients and unsigned mean errors (UMEs) using different solute dielectric constants in the MM/PBSA calculations.
| εin=1 | εin=2 | εin=4 | ||||
|---|---|---|---|---|---|---|
| r | UME | r | UME | r | UME | |
| α-thrombin | 0.45 | 23.26 | 0.69 | 13.83 | 0.81 | 19.81 |
| avidin | 0.92 | 7.63 | 0.67 | 3.85 | 0.51 | 4.99 |
| cytochrome C peroxidase | 0.27 | 3.62 | 0.00 | 2.88 | −0.33 | 4.51 |
| neuraminidase | −0.20 | 13.83 | 0.28 | 6.15 | 0.68 | 2.26 |
| P450cam | 0.72 | 5.86 | 0.69 | 2.76 | 0.68 | 5.13 |
| penicillopepsin | 0.29 | 19.87 | 0.41 | 2.84 | 0.32 | 10.09 |
The unsigned mean errors (UMEs) of the predictions are listed in Table 2. The lowest UME was coincident with the highest correlation coefficient in the cases of cytochrome C peroxidase, neuraminidase and penicillopepsin. Therefore, an optimal solute dielectric constant is not only important for ranking ligands but also for calculating the absolute binding free energies. For the six systems we studied, three had a UME value with the best correlation lower than 4.0 kcal/mol, one between 5.0 and 6.0 kcal/mol, one between 7.0 and 8.0 kcal/mol and one larger than 19 kcal/mol. These relatively large UMEs compared with the binding free energies suggested that absolute binding free energy calculated by MM/PBSA usually have large deviation from the experimental values, although it made reasonable predictions for some systems.
Unfortunately, there is no universal dielectric constant suitable for all the six protein systems. We thus investigated the inhibitor/protein binding interfaces in order to find a possible way to choose an optimal solute dielectric constant.
For neuraminidase, εin= 4 made the best predictions. Neuraminidase has a highly-charged binding pocket (Figure 2), characterized by nine charged residues, including five Arg’s (Arg119, Arg153, Arg226, Arg294, Arg372, net charge is +1), three Glu’s (Glu120, Glu278 and Glu279, net charge is −1), and one Asp (Asp152, net charge is −1). More importantly, three charged residues (Arg119, Arg294, and Arg372) can form strong ion-ion interactions with the negatively charged center of the ligand.56 Therefore, a relatively larger value of solute dielectric constant is justified to consider the strong electronic polarization of the binding interface. This observation is consistent with the previous studies for the predictions of the ligand/DNA system, where εin= 4 was shown to be a better choice than lower values.57
Figure 2.
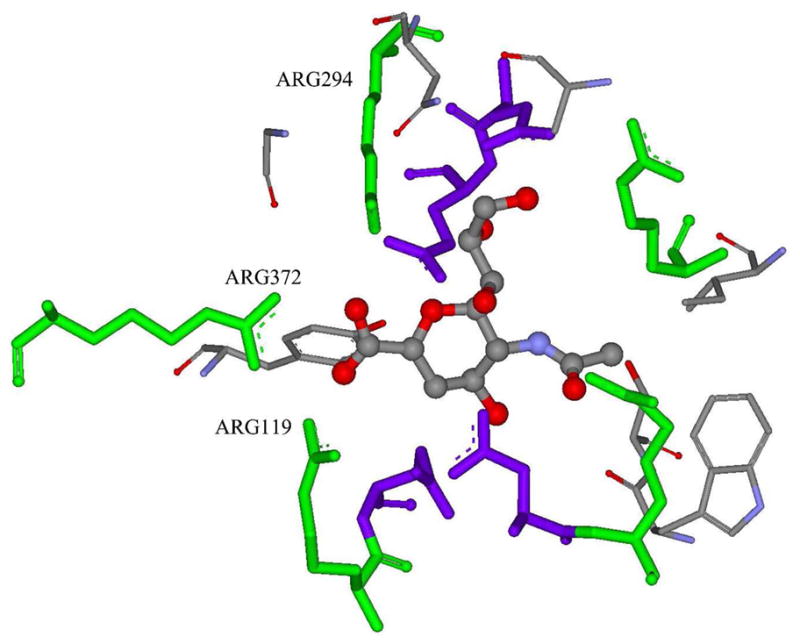
The 3-D structure of the neuraminidase binding site. The ligand d1 is shown in ball-and-stick representation, the Arg residues are shown in green stick representation, and the Glu and Asp residues are shown in purple stick representation. The three Arg residues that can form strong ion-ion interactions with ligands are labeled. The figure was generated using the Discovery Studio molecular simulation package.61
For α-thrombin, εin= 4 also achieved the best correlation coefficient (r = 0.81). But as noted above, the high correlation was due to the unbalanced distribution of the ligand samples. It is not conclusive whether εin= 4 is the best choice for all α-thrombin ligands. The binding pocket of α-thrombin includes three charged residues, Asp189, Asp194 and Glu217. Strong ion-ion interactions were found between Asp and the positively charged center of a5/a7 in the crystal structures. In contrast, no ion-ion interactions were found between the protein and the ligands a1~a4. It is questionable to use a universal solute dielectric constant to characterize the complicated binding interfaces of the α-thrombin complexes. The calculated binding free energies for the α-thrombin ligands based on εin = 1 and εin = 2 are listed in Table S2 in the supporting materials, and those based on εin= 4 are listed in Table 1. As shown in Table S2, εin = 1 or 2 is a good choice for ligands a1 to a4, but not for ligands a5 and a7. It is clear that ligands a5 and a7 prefer a larger dielectric constant. When εin = 1, the predicted binding free energies for a5 and a7 are very positive, and they are only marginally positive when εin = 4. In a5/α-thrombin complex, strong ion-ion interactions are observed between a5 and Asp189, and a crystal water molecule is observed to mediate the interactions between a5 and Phe227. The strongly polarized binding interface of protein/a5 needs to be characterized by a high εin value. Therefore, the solute dielectric constant is not only dependent on the target protein but also on the ligand. Even for the same target protein, different εin values may be necessary to characterize different target-ligand binding interfaces. According to the UMEs, maybe εin = 2 is a better choice than εin = 4.
For penicillopepsin, εin = 2 is a good choice. In the binding site of penicillopepsin, one negatively charged residue, Asp77, is observed to form strong ion-ion interactions with the positively charged center of the inhibitors.
For avidin, P450cam and cytochrome C peroxidase, εin=1 showed the best performance. For avidin, no charged residues are founded in the binding pocket. For cytochrome C peroxidase, one charged residue (Asp235) is found in the binding pocket, but no ion-ion interactions were found between the protein and the ligands. Similarly, for P450cam, the binding pocket has one charged residue (Asp297), but no ion-ion interactions were found between P450cam and ligands.
Based on our analysis, a high solute dielectric constant, such as 4, is usually recommended when the binding interface is highly charged, for example, several charged residues exist in the binding pocket and especially strong ion-ion interactions are formed between the protein and the ligand. When the binding interface is moderately charged, for example, two or three charged residues exist in the binding pocket or effective ion-ion interactions are formed between the protein and the ligand, a moderate solute dielectric constant, such as 2, is preferred. When the binding pocket is hydrophobic and no ion-ion interactions are formed between the protein and the ligand, the solute dielectric constant of 1 is the best choice.
Obviously, the solute dielectric constant is directly determined by the binding interface. Therefore we characterized the interface properties, and studied the quantitative relationships between the interface properties and the values of the solute dielectric constant. The interface properties were characterized by the following steps: (1). Define the strong polar/polar interaction pairs between the protein and the ligand. In the calculations, O, N and S were defined to be the polar atoms. If the distance between a polar protein atom and a polar ligand atom is less than a cutoff (5 or 6 Å was used here), the interactions for this polar/polar atom pair are assumed to be strong. (2). Calculate the solvent accessible surface area (SASA) for each complex and the corresponding uncomplexed protein using the MSMS program with a probe radius of 1.4 Å.58 The SASA difference (SASAD) of the protein is the interface area. (3). Calculate the total SASAD for the protein atoms which were found in the strong polar/polar interaction pairs (defined as PSASAD). Figure 3 shows the correlations between the PSASAD and the dielectric constants for the six protein/ligand systems. Good correlation can be observed. It suggests that PSASAD may be a useful measurement to help choose the solute dielectric constant in the MM/PBSA calculations. Here we only analyzed six protein/ligand systems. There is no doubt that further studies are necessary to correlate the relation between the interface properties and the values of the solute dielectric constant.
Figure 3.

The correlation between the best performed solute dielectric constants and the PSASAD values for six complexes, α-thrombin/a1, avidin/b1, cytochrome C peroxidase/c1, neuraminidase/d1, P450cam/e1 and penicillopepsin/f1. PSASAD was calculated based on a cutoff of (a) 5 Å or (b) 6 Å to define polar/polar interactions between the protein and the ligand. In this figure, the solute dielectric constant εin is 1 for avidin, cytochrome C peroxidase and P450cam, 2 for penicillopepsin and α-thrombin, and 4 for neuraminidase. For α-thrombin, εin= 4 is chosen based on the correlation coefficient; but as noted in the text, εin= 1 or 2 may be a better choice for ligands a1 to a4.
3. Impact of the length of MD simulations
In order to investigate the influence of simulation length on predictions, we conducted a comparative study of free energy calculations by using ten different lengths of MD simulations: 0.2~0.6 ns (50 snapshots), 0.2~1.0 ns (100 snapshots), 0.2~2.0 ns (225 snapshots), 0.2~3.0 ns (350 snapshots), 0.2~4.0 ns (475 snapshots), 0.2~5.0 ns (600 snapshots), 1.0~3.0 ns (250 snapshots), 1.0~5.0 ns (500 snapshots), 2.0~5.0 ns (375 snapshots), and 3.0~5.0 ns (250 snapshots). The correlations between the experimental binding free energies and the predicted values and the standard deviations of the ten correlation coefficients are summarized in Table 3. The standard deviations of four systems, including α-thrombin, cytochrome C peroxidase, neuraminidase and P450cam, are larger than 0.05; that is to say, for these four systems, the length of MD simulations shows obvious impacts on the predictions. Moreover, it is interesting to observe that extending the simulation time does not always improve the correlations between the predicted binding free energies and the experimental values. For some systems, such as neuraminidase, the predictions based on relatively short MD simulations are even slightly better than those based on longer MD simulations. After comparing the r0.2~0.6ns and the r0.2~5ns values, we found that for four systems, including α-thrombin, avidin, neuraminidase and penicillopepsin, the predictions based on the much longer MD simulations (4.8 ns) are obviously worse than those based on the shorter MD simulations (400 ps). It should be noted that in the comparison the predictions for different lengths of MD simulations, the snapshots were even extracted and then the numbers of snapshots for different MD simulations are different. In order to check the effect of different number of snapshot for the same MD simulation length, we compared the predictions based on 600 snapshots from 0.2~6.0 ns and those only based on 100 snapshots from 0.2~6.0 ns. According to our calculations, the two predictions yield the same correlation coefficients. That is to say, instead of using 600 snapshots 100 snapshots can also generate stable predictions.
Table 3.
The correlation between the experimental and the calculated binding free energies using three different lengths of MD simulationsa
| r0.2~0.6ns | r0.2~1ns | r0.2~2ns | r0.2~3ns | r0.2~4ns | r0.2~5ns | r1~3ns | r1~5ns | r2~5ns | r3~5ns | sdb | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| α-thrombin | εin=1 | 0.60 | 0.58 | 0.54 | 0.52 | 0.50 | 0.49 | 0.49 | 0.47 | 0.46 | 0.45 | 0.050 |
| εin =2 | 0.81 | 0.80 | 0.77 | 0.73 | 0.72 | 0.71 | 0.70 | 0.69 | 0.68 | 0.68 | 0.048 | |
| εin =4 | 0.91 | 0.90 | 0.87 | 0.85 | 0.84 | 0.83 | 0.82 | 0.81 | 0.80 | 0.80 | 0.039 | |
| avidin | εin =1 | 0.93 | 0.95 | 0.92 | 0.86 | 0.87 | 0.89 | 0.80 | 0.87 | 0.87 | 0.92 | 0.044 |
| εin =2 | 0.78 | 0.80 | 0.77 | 0.74 | 0.73 | 0.74 | 0.71 | 0.73 | 0.73 | 0.75 | 0.027 | |
| εin =4 | 0.64 | 0.66 | 0.66 | 0.63 | 0.62 | 0.63 | 0.62 | 0.62 | 0.61 | 0.62 | 0.017 | |
| cytochrome C peroxidase | εin =1 | 0.07 | 0.20 | 0.30 | 0.27 | 0.25 | 0.19 | 0.26 | 0.17 | 0.12 | 0.08 | 0.081 |
| εin =2 | −0.10 | −0.01 | 0.08 | 0.06 | 0.05 | 0.03 | 0.08 | 0.04 | 0.01 | 0.01 | 0.053 | |
| εin =4 | −0.19 | −0.16 | −0.13 | −0.14 | −0.14 | −0.13 | −0.14 | −0.13 | −0.13 | −0.11 | 0.022 | |
| neuraminidase | εin =1 | −0.26 | −0.35 | −0.42 | −0.39 | −0.38 | −0.42 | −0.38 | −0.41 | −0.39 | −0.42 | 0.048 |
| εin =2 | 0.36 | 0.11 | 0.06 | 0.08 | 0.10 | 0.04 | 0.07 | 0.03 | 0.04 | 0.00 | 0.101 | |
| εin =4 | 0.78 | 0.65 | 0.65 | 0.62 | 0.60 | 0.54 | 0.58 | 0.50 | 0.45 | 0.42 | 0.107 | |
| P450cam | εin =1 | 0.49 | 0.43 | 0.59 | 0.56 | 0.53 | 0.57 | 0.59 | 0.58 | 0.54 | 0.57 | 0.051 |
| εin =2 | 0.63 | 0.59 | 0.65 | 0.61 | 0.60 | 0.62 | 0.61 | 0.62 | 0.60 | 0.63 | 0.018 | |
| εin =4 | 0.67 | 0.63 | 0.65 | 0.61 | 0.61 | 0.62 | 0.61 | 0.61 | 0.59 | 0.62 | 0.023 | |
| penicillopepsin | εin =1 | 0.29 | 0.26 | 0.25 | 0.23 | 0.21 | 0.21 | 0.22 | 0.21 | 0.19 | 0.20 | 0.031 |
| εin =2 | 0.37 | 0.35 | 0.35 | 0.34 | 0.33 | 0.33 | 0.33 | 0.33 | 0.32 | 0.32 | 0.016 | |
| εin =4 | 0.35 | 0.34 | 0.33 | 0.32 | 0.32 | 0.31 | 0.32 | 0.31 | 0.30 | 0.30 | 0.016 | |
The entropy term was not included in the total binding free energy for correlation analysis
sd represents the standard deviation of the ten correlation coefficients.
The time evolutions of the RMSD values for the six avidin/ligand systems are shown in Figure 4. The initial structures for these six complexes were manually modeled, which justified the longer MD equilibration. The RMSD plots indicate that among all these six avidin/ligand systems avidin/b5 achieved equilibrium after about 1.5 ns and avidin/b6 achieved equilibrium after about 200 ps. Some of them, such as b3, b4 and b7, seemly did not reach equilibrium throughout the entire MD simulations. The fluctuations of enthalpies for these six avidin/ligand complexes are shown in Figure 5. In order to assess the convergence of the enthalpy, the cumulated mean values are also shown in Figure 5. The enthalpies are quite variable, but the averaged values became stable quickly after a short length of MD simulations for most cases (b4 to b7). Comparison between Figures 4 and 5 shows that the conformations of the avidin complexes are continuously tuned to be more stable but the interaction between the ligand and the protein is not so sensitive to this fine conformational adjustment. It is well-known that effective sampling is coupled with good force field. When the force field is not precise enough, long-time conformational collection is also not meaningful. Therefore, long MD simulations do not necessarily lead to accurate binding free energy calculations when the single-trajectory protocol is used.
Figure 4.

The RMSD values in the MD trajectories of the six avidin/ligand complexes.
Figure 5.

The fluctuations and the accumulated mean values of enthalpies for the six avidin/ligand complexes.
4. Impact of the conformational entropy
In order to investigate the impact of the conformational entropy on calculating relative binding free energies, we compared the correlations between the calculated and the experimental binding free energies with and without including the conformational entropy term (Tables 2 and 4). We found that the inclusion of the entropy term did not always improve the prediction accuracy, as shown in the cases of α-thrombin, avidin and cytochrome C peroxidase. For the other two systems of neuraminidase and P450cam, considering conformational entropy obviously improved the correlations. For penicillopepsin, the inclusion of the entropy term only improved the correlations slightly. This observation is consistent with many previous studies that, without considering conformational entropy, MM/PBSA could still achieve satisfactory accuracy of ranking ligand affinities.
Table 4.
The correlations between the enthalpies and the experimental binding free energies
| εin=1 | εin=2 | εin=4 | |
|---|---|---|---|
| α-thrombin | 0.54 | 0.77 | 0.87 |
| avidin | 0.92 | 0.77 | 0.66 |
| cytochrome C peroxidase | 0.30 | 0.08 | −0.13 |
| neuraminidase | −0.42 | 0.06 | 0.65 |
| P450cam | 0.59 | 0.65 | 0.65 |
| penicillopepsin | 0.25 | 0.35 | 0.32 |
In this study, normal mode analysis (NMA) was used to estimate conformational entropy. Although widely used, NMA is not perfect due to not considering anharmonic effects. One observation from our calculations is that the calculated entropies fluctuate significantly between MD snapshots (Figure 6). Such a large fluctuation is usual for the vibrational entropies computed by NMA.21, 59 Large fluctuations in entropy predicted by NMA may arise from the mismatch between the minimized geometries of the complex and of the receptor or ligand.59 Therefore, a large number of snapshots is needed to obtain stable predictions.
Figure 6.

The fluctuations and the accumulated mean values of entropies for the six protein-ligand systems.
As noticed in many previous studies as well as in the current study, the success of MM/PBSA relies on the cancellation of errors in individual energy components. Limited by the computational cost of normal mode analysis, we took 45 snapshots for conformational entropy estimation, far less than the 200 snapshots used in enthalpy calculation when we calculated the binding free energies using the single trajectory from 0.2 to 2.0 ns. It is possible that the fluctuation of entropy is synchronized with other terms and errors cannot be canceled when using different set of snapshots. Therefore, we did another test by calculating the enthalpic and entropic contributions by using the same 40 snapshots. The correlations between the predicted and the experimental binding free energies are similar with or without entropies when compared with the corresponding values shown in Tables 2 and 4. This observation of no improvement of correlation implied that the errors of normal mode analysis probably did not get canceled with other terms. However, we need to emphasize that the single trajectory approach may not be a good choice for the estimates of the conformational entropy. If the difference between the ligand free in solution and when it is bound to the protein is significant, the prediction error may be significant. Moreover, as shown in Figure 6, the entropy shows larger fluctuation, we must emphasize that the estimation of conformational entropy based on 45 snapshots may not enough to get converged predictions for some systems.
5. Comparison between MM/PBSA and MM/GBSA
The PB model is theoretically more rigorous than the GB models and MM/PBSA is often considered to be naturally superior to MM/GBSA for predicting binding free energies. Here, we compared the performance of these two methods on ranking binding affinities of the six protein-ligand systems.
The three GB models examined in this study were the pairwise GB model developed by Hawkins et al. (GBHCT)52, 53 and two modified GB models developed by Onufriev et al. (GBOBC1 and GBOBC2),55 which were implemented in the AMBER software package. Based on the correlation coefficients between the calculated and the experimental binding free energies, GBOBC1 performed better than the other two GB models (Table 5).
Table 5.
The correlations between the experimental and calculated binding free energies using three different GB models and three different solute dielectric constants
| GBHCT | GBOBC1 | GBOBC2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| εin=1 | εin=2 | εin=4 | εin=1 | εin=2 | εin=4 | εin=1 | εin=2 | εin=4 | |
| α-thrombin | 0.58 | 0.88 | 0.90 | 0.71 | 0.91 | 0.90 | 0.73 | 0.78 | 0.83 |
| avidin | 0.78 | 0.49 | 0.37 | 0.93 | 0.73 | 0.49 | 0.92 | 0.91 | 0.77 |
| cytochrome C peroxidase | 0.17 | −0.11 | −0.27 | 0.21 | −0.04 | −0.48 | 0.35 | 0.22 | −0.41 |
| neuraminidase | 0.21 | 0.55 | 0.78 | −0.15 | 0.20 | 0.60 | −0.16 | −0.02 | 0.24 |
| P450cam | 0.61 | 0.65 | 0.66 | 0.58 | 0.63 | 0.65 | −0.42 | −0.37 | −0.21 |
| penicillopepsin | 0.51 | 0.62 | 0.65 | 0.68 | 0.73 | 0.74 | 0.23 | 0.40 | 0.55 |
Another interesting observation is that the performances of GBOBC1 and GBOBC2 were quite different. The theoretical frameworks of GBOBC1 and GBOBC2 are the same, and the only difference between the two models is the values of three parameters for calculating the inverse of the effective Born radii. The performance of these two GB models on MD simulations tested on several proteins were comparative in the previous studies,55 although further tests on an extensive set of protein structures revealed that GBOBC2 agreed better with PB in calculating the electrostatic solvation free energy.60 For the six protein-ligand systems studied here, GBOBC1 performed better than GBOBC2 obviously. GBOBC2 and GBOBC1 gave comparative predictions for avidin, and GBOBC2 performs better than GBOBC1 on cytochrome C peroxidase. But for the other four systems, GBOBC1 gave much better predictions than GBOBC2. Overall, our calculations showed that the GBOBC1 model was the most accurate one among the three GB models to calculate binding free energies.
The predicted binding free energies and the corresponding energy terms calculated by MM/GBSA based on GBOBC1 are listed in Table S3 in the supporting materials. Same as the results of Gohlke and Case,26 the electrostatic solvation energy computed by GB was consistently larger than that computed by PB, leading to lower binding free energy (Table S3 and Table 1). The possible reason is that the PARSE radii are on average smaller than the modified Bondi radii, which in turn leads to the dielectric boundary being closer to the charge centre.27 The unsigned mean errors of the predicted binding free energies given by MM/GBSA were 21.31 kcal/mol for α-thrombin, 9.30 kcal/mol for avidin, 5.62 kcal/mol for cytochrome C peroxidase, 4.42 kcal/mol for neuraminidase, 6.07 kcal/mol P450cam, and 10.58 kcal/mol penicillopepsin. These values were larger than those given by MM/PBSA (Table 2). Namely, MM/PBSA performed better than MM/GBSA on calculating the absolute binding free energies for the six protein-ligand systems.
We also examined the performance of MM/PBSA and MM/GBSA to rank binding affinities of ligands, which is often more important in many applications such as drug design than the absolute binding free energies. As shown in Table 5, MM/GBSA based GBOBC1 gave better correlations than MM/PBSA for two systems, α-thrombin and penicillopepsin; MM/GBSA based on GBHCT gave better correlations than MM/PBSA for neuraminidase. For avidin, the correlations given by MM/GBSA and MM/PBSA are similar. For cytochrome C peroxidase and P450cam, the correlations given by MM/PBSA were better. The poor predictions of MM/GBSA for cytochrome C peroxidase and P450cam might be due to the lack of accurate GB parameters for the Fe ions. For the systems without metal ions in the binding sites, the MM/GBSA calculations based on GBOBC1 performed better than MM/PBSA to rank the binding affinities of the ligands.
In this work, we observed that MM/GBSA gives better correlations than MM/PBSA in most systems. In principle, PB is more theoretically rigorous than GB, but it does not mean that MM/PBSA can give better predictions than MM/GBSA. Maybe two reasons can be used to explain the observations. First, here, the Parse parameter set was used to define the dielectric boundary. However, previous studies have shown that Parse parameter set cannot give good predictions of solvation free energies for amino acid side chain analogs and some relatively complicated functional groups.1, 61 It is well known that the Parse radii are based on the Pauling van der Waals radii.49 In Parse radii set, each element is defined as an atom type. It is possible that a single radius for an element is insufficient. For example, the single radius for carbon works well for simple aliphatics, while usually less well on heterocyclic and aromatic systems.49 It is necessary to develop more reliable radii for PB based on a larger set of experimental data for a set of more elaborately defined atom types. Second, the GB parameters were usually fitted from the experimental values. The prediction errors of GB can be well reduced by defining more atom types.
Conclusions
In the present study, we have evaluated the performance of MM/PBSA to calculate and rank the binding free energies of 59 ligands for six different protein systems. Our results showed that MM/PBSA gives good predictions for homologous ligands, and has a variable performance for ligands with diverse structures. We found that the MM/PBSA predictions are very sensitive to solute dielectric constant, which is directly related to the characteristics of the binding interface. In general, for highly-charged binding interface, a higher solute dielectric constant (εin = 4) is preferred; for moderately-charged binding interface, a moderate solute dielectric constant (εin = 2) is preferred; and for hydrophobic binding interface, a low solute dielectric constant (εin = 1) is preferred. The optimal solute dielectric constant is critical for different protein/inhibitor systems. We proposed a simple measurement, called PSASAD, to guide the choice of this important parameter. There is no doubt that further tests on a larger data set are necessary to choose the most optimal solute dielectric constant. Compared with the solute dielectric constant, the predictions of MM/PBSA are less sensitive to the length of MD simulations. However, the MD simulation lengths still have obvious impact on the predictions; moreover, longer MD simulations are not always necessary to achieve better predictions.
Our calculations showed that inclusion of the conformational entropy was crucial for predicting absolute binding free energies but not for ranking the binding affinities of similar ligands. Since the conformational entropy calculations showed large fluctuations, a large number of snapshots are required for reliable calculations, and it is necessary to develop more efficient methods to evaluate conformational entropy.
Finally, we compared the MM/PBSA and the MM/GBSA approaches. Three GB models were used to calculate the electrostatic solvation energy, and they showed variable performances. The GB model developed by Onufriev (GBOBC1) gave better accuracy than the other two GB models (GBOBC2 and GBHCT). Although MM/GBSA achieved worse predictions for the absolute binding free energies than MM/PBSA, it showed better performance to rank the binding affinities for systems without metals.
Supplementary Material
Table S1. The PDB entries, the description of the complexes and the experimental binding free energies;
Table S2. The binding free energies for the α-thrombin ligands based on two different solute dielectric constants (εin= 1 and εin= 2) calculated by MM/PBSA;
Table S3. The experimental and the calculated binding free energies as well as the corresponding energy components calculated by MM/GBSA;
Figure S1. Chemical structures of the 59 ligands of α-thrombin (a1~a7), avidin (b1~b7), cytochrome C peroxidase (c1~c18), neuraminidase(d1~d8), P450cam (e1~e12) and penicillopepsin (f1~f7). The name of each ligand can be found in Table S1 in the supporting materials. This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The project was supported by the Natural Science Foundation of China (No. 20973121) and the National Institutes of Health Grant (No. R01GM085188).
References
- 1.Wang JM, Hou TJ, Xu XJ. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Current Computer-Aided Drug Design. 2006;2(3):287–306. [Google Scholar]
- 2.Wang W, Donini O, Reyes CM, Kollman PA. Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu Rev Bioph Biom. 2001;30:211–243. doi: 10.1146/annurev.biophys.30.1.211. [DOI] [PubMed] [Google Scholar]
- 3.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Accounts Chem Res. 2000;33(12):889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 4.Beveridge DL, Dicapua FM. Free-Energy Via Molecular Simulation - Applications to Chemical and Biomolecular Systems. Annu Rev Biophys Bio. 1989;18:431–492. doi: 10.1146/annurev.bb.18.060189.002243. [DOI] [PubMed] [Google Scholar]
- 5.Aqvist J, Medina C, Samuelsson JE. New Method for Predicting Binding-Affinity in Computer-Aided Drug Design. Protein Eng. 1994;7(3):385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 6.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions.1. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aid Mol Des. 1997;11(5):425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 7.Bohm HJ. The Development of a Simple Empirical Scoring Function to Estimate the Binding Constant for a Protein Ligand Complex of Known 3-Dimensional Structure. J Comput Aid Mol Des. 1994;8(3):243–256. doi: 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- 8.Wang RX, Liu L, Lai LH, Tang YQ. SCORE: A new empirical method for estimating the binding affinity of a protein-ligand complex. J Mol Model. 1998;4(12):379–394. [Google Scholar]
- 9.Muegge I. Effect of ligand volume correction on PMF scoring. J Comput Chem. 2001;22(4):418–425. [Google Scholar]
- 10.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J Med Chem. 1999;42(5):791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 11.Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol. 2000;295(2):337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 12.Gohlke H, Kiel C, Case DA. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RaIGDS complexes. J Mol Biol. 2003;330(4):891–913. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- 13.Hou TJ, Zhang W, Case DA, Wang W. Characterization of domain-peptide interaction interface: A case study on the amphiphysin-1 SH3 domain. J Mol Biol. 2008;376(4):1201–1214. doi: 10.1016/j.jmb.2007.12.054. [DOI] [PubMed] [Google Scholar]
- 14.Hou TJ, Xu Z, Zhang W, McLaughlin WA, Case DA, Xu Y, Wang W. Characterization of Domain-Peptide Interaction Interface: a generic structure-based model to decipher the binding specificity of SH3 domains. Mol Cell Proteomics. 2009;8(4):639–649. doi: 10.1074/mcp.M800450-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou TJ, Guo SL, Xu XJ. Predictions of binding of a diverse set of ligands to gelatinase-A by a combination of molecular dynamics and continuum solvent models. J Phys Chem B. 2002;106(21):5527–5535. [Google Scholar]
- 16.Wang W, Kollman PA. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. P Natl Acad Sci USA. 2001;98(26):14937–14942. doi: 10.1073/pnas.251265598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lepsik M, Kriz Z, Havlas Z. Efficiency of a second-generation HIV-1 protease inhibitor studied by molecular dynamics and absolute binding free energy calculations. Proteins-Structure Function and Bioinformatics. 2004;57(2):279–293. doi: 10.1002/prot.20192. [DOI] [PubMed] [Google Scholar]
- 18.Kuhn B, Kollman PA. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J Med Chem. 2000;43(20):3786–3791. doi: 10.1021/jm000241h. [DOI] [PubMed] [Google Scholar]
- 19.Huo SH, Wang JM, Cieplak P, Kollman PA, Kuntz ID. Molecular dynamics and free energy analyses of cathepsin D-inhibitor interactions: Insight into structure-based ligand design. J Med Chem. 2002;45(7):1412–1419. doi: 10.1021/jm010338j. [DOI] [PubMed] [Google Scholar]
- 20.Brown SP, Muchmore SW. High-throughput calculation of protein-ligand binding affinities: Modification and adaptation of the MM-PBSA protocol to enterprise grid computing. J Chem Inf Model. 2006;46(3):999–1005. doi: 10.1021/ci050488t. [DOI] [PubMed] [Google Scholar]
- 21.Stoica I, Sadiq SK, Coveney PV. Rapid and accurate prediction of binding free energies for saquinavir-bound HIV-1 proteases. J Am Chem Soc. 2008;130(8):2639–2648. doi: 10.1021/ja0779250. [DOI] [PubMed] [Google Scholar]
- 22.Hou TJ, Zhu LL, Chen LR, Xu XJ. Mapping the binding site of a large set of quinazoline type EGF-R inhibitors using molecular field analyses and molecular docking studies. J Chem Inf Comp Sci. 2003;43(1):273–287. doi: 10.1021/ci025552a. [DOI] [PubMed] [Google Scholar]
- 23.Hou TJ, Yu R. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: Mechanism for binding and drug resistance. J Med Chem. 2007;50(6):1177–1188. doi: 10.1021/jm0609162. [DOI] [PubMed] [Google Scholar]
- 24.Wang W, Kollman PA. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J Mol Biol. 2000;303(4):567–582. doi: 10.1006/jmbi.2000.4057. [DOI] [PubMed] [Google Scholar]
- 25.Hou TJ, Chen K, McLaughlin WA, Lu BZ, Wang W. Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain. Plos Comput Biol. 2006;2(1):46–55. doi: 10.1371/journal.pcbi.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gohlke H, Case DA. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J Comput Chem. 2004;25(2):238–250. doi: 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- 27.Pearlman DA. Evaluating the molecular mechanics Poisson-Boltzmann surface area free energy method using a congeneric series of ligands to p38 MAP kinase. J Med Chem. 2005;48(24):7796–7807. doi: 10.1021/jm050306m. [DOI] [PubMed] [Google Scholar]
- 28.Kuhn B, Gerber P, Schulz-Gasch T, Stahl M. Validation and use of the MM-PBSA approach for drug discovery. J Med Chem. 2005;48(12):4040–4048. doi: 10.1021/jm049081q. [DOI] [PubMed] [Google Scholar]
- 29.Weis A, Katebzadeh K, Soderhjelm P, Nilsson I, Ryde U. Ligand affinities predicted with the MM/PBSA method: Dependence on the simulation method and the force field. J Med Chem. 2006;49(22):6596–6606. doi: 10.1021/jm0608210. [DOI] [PubMed] [Google Scholar]
- 30.Miyamoto S, Kollman PA. Absolute and Relative Binding Free-Energy Calculations of the Interaction of Biotin and Its Analogs with Streptavidin Using Molecular-Dynamics Free-Energy Perturbation Approaches. Proteins-Structure Function and Bioinformatics. 1993;16(3):226–245. doi: 10.1002/prot.340160303. [DOI] [PubMed] [Google Scholar]
- 31.Almlof M, Brandsdal BO, Aqvist J. Binding affinity prediction with different force fields: Examination of the linear interaction energy method. J Comput Chem. 2004;25(10):1242–1254. doi: 10.1002/jcc.20047. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Dixon R, Kollman PA. Ranking ligand binding affinities with avidin: A molecular dynamics-based interaction energy study. Proteins. 1999;34(1):69–81. [PubMed] [Google Scholar]
- 33.Pugliese L, Coda A, Malcovati M, Bolognesi M. 3-Dimensional Structure of the Tetragonal Crystal Form of Egg-White Avidin in Its Functional Complex with Biotin at 2.7-Angstrom Resolution. J Mol Biol. 1993;231(3):698–710. doi: 10.1006/jmbi.1993.1321. [DOI] [PubMed] [Google Scholar]
- 34.Green NM. Thermodynamics of Binding of Biotin and Some Analogues by Avidin. Biochem J. 1966;101(3):774. doi: 10.1042/bj1010774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poulos TL, Finzel BC, Howard AJ. High-Resolution Crystal-Structure of Cytochrome-P450cam. J Mol Biol. 1987;195(3):687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 36.SYBYL molecular simulation package. 2004 http://www.sybyl.com.
- 37.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges - the Resp Model. J Phys Chem-Us. 1993;97(40):10269–10280. [Google Scholar]
- 38.Bren U, Hodoscek M, Koller J. Development and validation of empirical force field parameters for netropsin. Journal of Chemical Information and Modeling. 2005;45(6):1546–1552. doi: 10.1021/ci050151r. [DOI] [PubMed] [Google Scholar]
- 39.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JAJ, Vreven T, Kudin KN, Burant JC, Millam JM, Lyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken B, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liashenko A, Liashenko A, Piskorz P, Komaromi I, Martin RL, FOx DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Gaussian Inc; Wallingford CT: 2004. [Google Scholar]
- 40.Wang JM, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25(2):247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 41.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong GM, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang JM, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24(16):1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 42.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 43.Giammona DA. Ph D. University of California; Davis: 1984. An examination of conformational flexibility in porphyrin and bulky-ligand binding in myoglobin. [Google Scholar]
- 44.Darden T, York D, Pedersen L. Particle Mesh Ewald - an N. Log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
- 45.Andersen HC. Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys. 1980;72:2384–2393. [Google Scholar]
- 46.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23(3):327–341. [Google Scholar]
- 48.Rocchia W, Alexov E, Honig B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions. J Phys Chem B. 2001;105(28):6507–6514. [Google Scholar]
- 49.Sitkoff D, Sharp KA, Honig B. Accurate Calculation of Hydration Free-Energies Using Macroscopic Solvent Models. J Phys Chem-Us. 1994;98(7):1978–1988. [Google Scholar]
- 50.Batsanov SS. Van der Waals radii of elements. Inorg Mater+ 2001;37(9):871–885. [Google Scholar]
- 51.Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO) J Comput Chem. 1999;20(2):217–230. [Google Scholar]
- 52.Hawkins GD, Cramer CJ, Truhlar DG. Pairwise Solute Descreening of Solute Charges from a Dielectric Medium. Chem Phys Lett. 1995;246(1–2):122–129. [Google Scholar]
- 53.Hawkins GD, Cramer CJ, Truhlar DG. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J Phys Chem-Us. 1996;100(51):19824–19839. [Google Scholar]
- 54.Tsui V, Case DA. Theory and applications of the generalized Born solvation model in macromolecular Simulations. Biopolymers. 2000;56(4):275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 55.Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins-Structure Function and Bioinformatics. 2004;55(2):383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 56.Udommaneethanakit T, Rungrotmongkol T, Bren U, Frecer V, Stanislav M. Dynamic Behavior of Avian Influenza A Virus Neuraminidase Subtype H5N1 in Complex with Oseltamivir, Zanamivir, Peramivir, and Their Phosphonate Analogues. Journal of Chemical Information and Modeling. 2009;49(10):2323–2332. doi: 10.1021/ci900277r. [DOI] [PubMed] [Google Scholar]
- 57.Misra VK, Honig B. On the Magnitude of the Electrostatic Contribution to Ligand-DNA Interactions. P Natl Acad Sci USA. 1995;92(10):4691–4695. doi: 10.1073/pnas.92.10.4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanner MF, Olson AJ, Spehner JC. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers. 1996;38(3):305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 59.Page CS, Bates PA. Can MM-PBSA calculations predict the specificities of protein kinase inhibitors? J Comput Chem. 2006;27(16):1990–2007. doi: 10.1002/jcc.20534. [DOI] [PubMed] [Google Scholar]
- 60.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem. 2004;25(2):265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 61.Zhang W, Hou TJ, Qiao XB, Xu XJ. Parameters for the generalized born model consistent with RESP: atomic partial charge assignment protocol. J Phys Chem B. 2003;107(34):9071–9078. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The PDB entries, the description of the complexes and the experimental binding free energies;
Table S2. The binding free energies for the α-thrombin ligands based on two different solute dielectric constants (εin= 1 and εin= 2) calculated by MM/PBSA;
Table S3. The experimental and the calculated binding free energies as well as the corresponding energy components calculated by MM/GBSA;
Figure S1. Chemical structures of the 59 ligands of α-thrombin (a1~a7), avidin (b1~b7), cytochrome C peroxidase (c1~c18), neuraminidase(d1~d8), P450cam (e1~e12) and penicillopepsin (f1~f7). The name of each ligand can be found in Table S1 in the supporting materials. This information is available free of charge via the Internet at http://pubs.acs.org.