

Abstract
Dyskeratosis congenita (DC) is a rare genodermatosis characterised by a classic triad of dystrophic nails, reticular skin pigmentation and mucous membrane leukoplakic patches, which have a high rate of malignant transformation. The case report presented here deals with a sporadic case of DC without similar clinical presentation in the first-degree and second-degree relatives. Of note in this case, there was rapid malignant transformation in the non-homogeneous nodulo-speckled leukoplakic patch on the dorsum of the tongue.
Background
Dyskeratosis congenita (DC), though having a classic triad of clinical features in majority of cases, often escapes the experienced eyes of a doctor. Even the patient ignores the early clinical features and correlates them with certain other coincidences, such as snake bite in this case, thus seeking a late medical consultation. There have been X-linked recessive, autosomal recessive and autosomal dominant modes of inheritance reported in the literature previously.1 However, a sufficient number of sporadic cases due to unidentified mutations have also been reported.2 The significance of the condition lies in premature mortality arising from either bone marrow failure or malignant changes in the mucocutaeneous leukoplakia. Thus, careful early diagnosis and an appropriate treatment plan are essential to reduce the morbidity and mortality in such cases.
Here, a sporadic case of DC is reported with no related disorder in the siblings of the patient, or the first-degree and second-degree relatives. The patient exhibited the classic triad with mild bone marrow suppression. Significant in the present case was the malignant transformation in the leukoplakic patch over a short duration of 7 months.
Case presentation
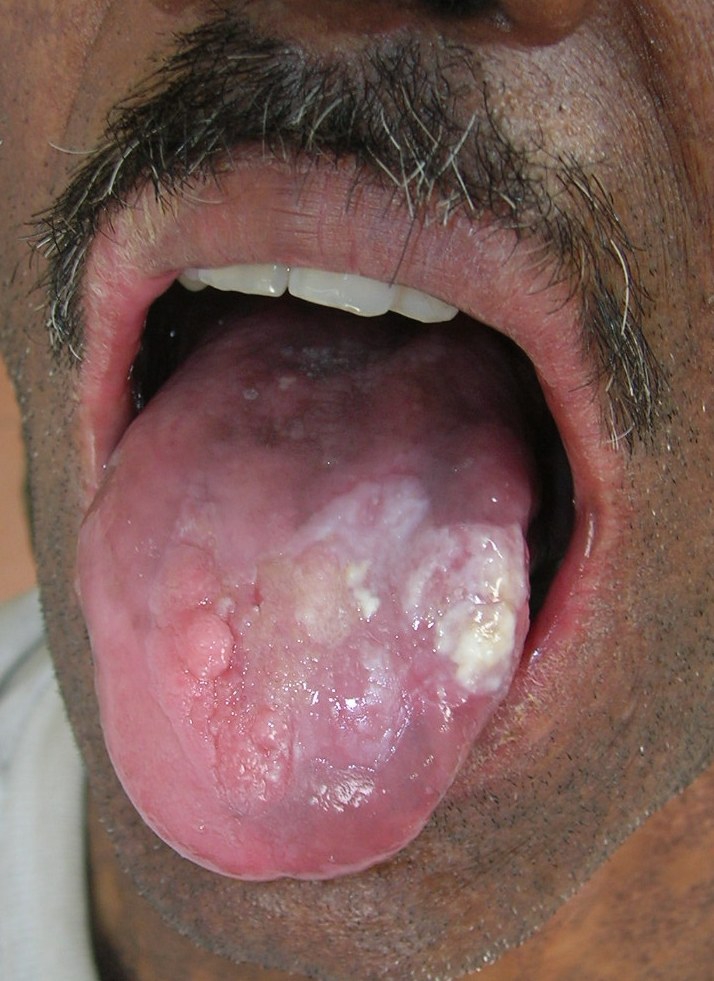
A 38-year-old man was referred to the Department of Oral Pathology, Dr R Ahmed Dental College & Hospital, Kolkata, by a local dental practitioner who was treating the patient, considering his issue of prolonged pain and a burning sensation on the tongue while consuming hot and spicy food as chronic oral candidiasis. The problem was not relieved by topical and systemic antifungal treatment with Fluconazole, administered intermittently for the last year. On intraoral examination, atrophy and depapillation was observed with a non-homogenous nodulo-speckled irregular leukoplakic patch on the dorsum and left lateral border of tongue without any positive history of tobacco habit (figure 1). Also, the patient had multiple missing teeth with a history of spontaneous exfoliation. Brownish reticular skin hyperpigmentation was present over the face, neck, chest, back and limbs since childhood when he was around 10 years old. This hyperpigmentation was interpreted as a consequence of snake bite by a local medical practitioner at that time. Further examination showed diffuse reticular hyperpigmented macules with islands of normal skin present all over the body including the genitalia (figures 2 and 3). The skin over the palms and soles was thickened with scaling and absence of dermatoglypics (figures 4–6). Anonychia was present over the fingernails and toenails and some nails were dystrophic (figures 4 and 6). Skin biopsy taken from the right upper lip region and left arm region was suggestive of hyperkeratotic lesion with mild lichenoid inflammation. An ophthalmological examination revealed pteryguim in the right eye with epiphora and ectropion over both the eyes (figure 7).
Figure 1.

Dorsum of tongue with non-homogenous nodulo-speckled leukoplakic patch.
Figure 2.

Frontal view of the patient revealing reticular skin pigmentation and eye abnormalities.
Figure 3.
Dorsal view showing reticular pattern of pigmentation.
Figure 4.

Plantar surface of hands.
Figure 6.

Feet showing the areas of hyperpigmentation and dystrophic nails.
Figure 7.
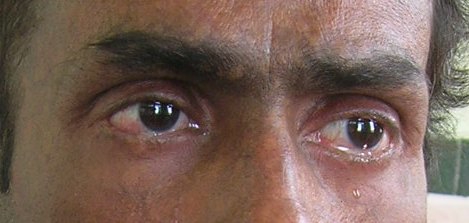
Eyes showing epiphora and ectropion
Figure 5.

Palmar surface of hands.
His medical history revealed hospitalisation for nephritis and blood transfusion for anaemia (4.6 g%) 8 years previously. At the same time bone marrow aspiration showed hypocellularity. A chronologically ordered sequence of the appearance of clinical manifestations is given in table 1.
Table 1.
Sequelae of events in the development of disease
| Age | Signs and symptoms |
|---|---|
| 10 years | Skin hyperpigmentation |
| 12 years | Dystrophic nail changes |
| 20 years | Faciaphotosensitivity, epiphora and redness of eyes |
| 25 years | Spontaneous exfoliation of teeth |
| 30 years | Nephritis, anaemia and hypocellular bone marrow |
| 37 years | Mucosal leukoplakic patch |
| 38 years | Malignant transformation of the mucosal lesion |
The patient gave a personal history of being a non-smoker and non-drinker, and was unmarried. On exploring the family history we could not trace any Mendelian pattern of inheritance and the patient was clinically diagnosed as having a sporadic case of DC.
Investigations
Routine haematological investigations revealed only a mild degree of anaemia (10.2 g%). An oral incisional biopsy from the leukoplakic patch on the dorsum of tongue revealed the presence of hyperparakeratotic stratified squamous epithelium with acanthosis and mild dysplasia. The underlying connective tissue was richly vascular with mild chronic inflammatory cell infiltration under 10× magnification (figure 8). Overall the histolopathological picture was suggestive of a mild degree of epithelial dysplasia. The patient was referred to the Department of Oral and Maxillofacial Surgery for the excision of the lesion. However, the patient failed to report back.
Figure 8.

A 10× view of the section stained with H&E.
Then, 7 months later the patient re-presented with an exophytic growth on the tongue for the last month. On examination, a 5×4.5 cm ulceroproliferative exophytic lesion with surface keratin deposition was observed on the left lateral border of the tongue crossing the midline (figure 9). Evaluation for lymph nodes revealed level II (2×2 cm) and level IB (1×1 cm) palpable cervical lymph nodes on left and right side, respectively. An abdominal examination revealed no other organopathy. Complete blood count was within the normal range except for haemoglobin, which was 10.2 g%. An incisional biopsy from the representative site was performed and was corroborative with the histological diagnosis of moderately differentiated squamous cell carcinoma (figure 10). The patient was then referred to the Department of Oncosurgery.
Figure 9.
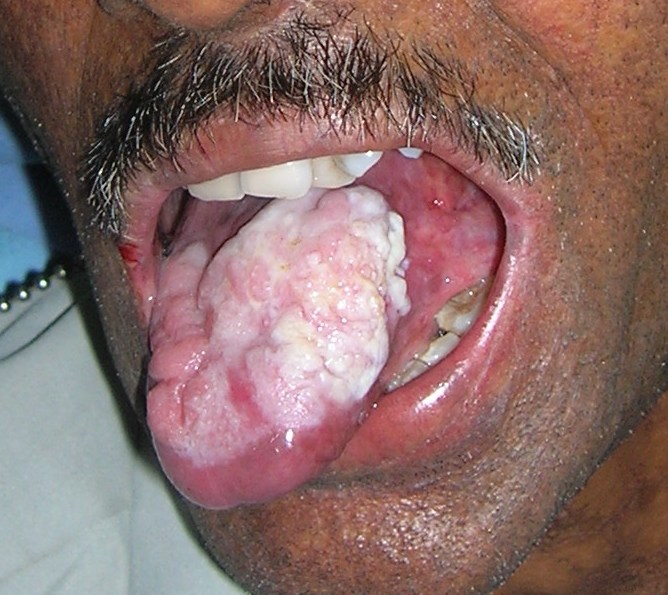
Dorsum of tongue showing ulceroproliferative growth 7 months later.
Figure 10.

A 10× view of the section stained with H&E.
Differential diagnosis
The differential diagnoses include Fanconi's anaemia, pachyonychia congenita and graft versus host disease (GVHD). Oral lesions need to be differentially diagnosed from oral candidiasis, tobacco-associated leukoplakia, white sponge nevus and hypertrophic lichen planus.3
Treatment
Excision of the tongue lesion with 1.5 cm safe margins along with dissection of cervical neck nodes, which showed evidence of metastasis on fine-needle aspiration cytology, was performed under general anaesthesia. Peroperative frozen sections were taken to evaluate marginal clearance, which revealed the tumour mass infiltrating beyond the margins. Thus the peroperative decision was taken to excise the tongue followed by subsequent radiotherapy. Postoperative histopathological examination confirmed the initial diagnosis of moderately differentiated squamous cell carcinoma and metastatic foci in the dissected lymph nodes (figure 11).
Figure 11.

A 40× view of the dissected lymph node section stained with H&E.
Outcome and follow-up
The patient is on regular follow-up and further progress will be reported subsequently.
Discussion
DC was first described by Zinsser in 1906. Later on it was subsequently described by Engman in 1926 and Cole in 1930. DC, which is also designated as Zinsser–Engman–Cole syndrome, is a triad consisting of nail dystrophy, mucosal leukoplakia and abnormal skin pigmentation.4 The precise incidence of DC is as yet unknown, however the prevalence has been estimated to be approximately 1 in 1 000 000.5
As per the English literature review, DC is an established multisystem disorder in which affected patients often present with haematological, ophthalmological, dermatological and neurological features with a marked tendency for spontaneous malignant transformation.6 Clinical features usually become evident by the age of 10 years. A reticular pattern of skin pigmentation develops first, followed by nail dystrophy. Intraorally, leukoplakic patches and progressive periodontal diseases are frequently observed.1 Patients with DC have a recognised increased risk of malignancy from pre-existing mucosal leukoplakia (hyperkeratosis), and the incidence of this transformation is in the order of approximately 35%, as compared to 1% in non-syndromic patients with tobacco-associated leukoplakia.7 8 The main causes of death include bone marrow failure/immunodeficiency (60% to 70% of cases), pulmonary complications (10% to 15%) and malignancies (5% to 10%).9 A brief compilation of the major clinical features is given in table 2.
Table 2.
Clinical findings in dyskeratosis congenita
| Systemic findings | Oral findings |
|---|---|
| Skin hyperpigmentation | Soft tissues: |
| Nail dystrophy | Leukoplakia |
| Bone marrow failure | Lichenoid lesions |
| Epiphora | Hyperpigmentation |
| Mental retardation | Atrophic glossitis |
| Cerebral hypoplasia | Oral ulcerations |
| Microcephaly | Malignancies |
| Pulmonary diseases | Hard tissues: |
| Premature hair loss | Hypodontia |
| Hyperhidrosis | Delayed eruption |
| Hypogonadism | Periodontitis |
| Oesophageal stricture | Dental caries |
| Osteoporosis | Thin enamel |
| Deafness | Taurodontism |
| Photophobia | Root: crown higher |
| Malignancy |
Clinically there appears to be three patterns of inheritance for DC: autosomal dominant (AD), autosomal recessive (AR) and X-linked recessive (XLR).10 Apart from these causes, sporadic cases of DC have also been reported.2 Among the inheritance causes, XLR is the most common mode of inheritance and thus results in a striking male predeliction.1The presumed underlying defect in DC is an inability to preserve telomere length secondary to mutations in the genes for RNA or proteins composing this ribonucleoprotein complex. Chromosome ends are capped by telomeres, which are protective DNA–protein complexes. Telomeres shorten progressively with successive cell divisions in most cells, and cell division ceases when telomere length becomes critically short.6
Mutations in genes encoding the RNA template (telomerase RNA component; TERC), telomerase catalytic reverse transcriptase (TERT) and dyskerin, a component of the telomerase complex, have been identified in various DC families.9 11 The DKC1 gene is located on chromosome Xq28, which encodes the nucleolar protein dyskerin and has been reported to be associated with X-linked recessive DC. Mutations in TERC and TERT have been identified in some autosomal dominant DC, but in the majority of cases no mutations have been identified.10
An atypical form of DC, also known as Hoyeraal–Hreidarsson (HH) syndrome is a severe multisystem disorder that can present in neonatal period and infancy. It is characterised by severe growth retardation, immune deficiency, bone marrow failure and neurological abnormalities.9
Treatment of this syndrome revolves around the main causes of premature mortality such as malignancy and bone marrow failure. Early diagnosis of patients with DC will enable harvesting and storage of bone marrow before the onset of marrow failure in susceptible individuals. The current treatment for bone marrow failure is allogenic haemopoietic stem cell transplantation. It is extremely important to regularly screen for malignancies, especially of the gastrointestinal system. The management of oral malignancies is centred on regular review and biopsy of the premalignant hyperkeratotic lesions and surgical laser excision of any suspicious areas. Confirmed malignancies require prompt surgical resection and ipsilateral node clearance, with or without adjuvant radiotherapy. Regular medical and dental follow-up is of utmost importance.6
Learning points.
-
▶
Dyskeratosis congenita (DC) is a rare genodermatosis with fatal outcomes and a wide range of clinical manifestations that require appropriate attention and prompt treatment.
-
▶
Although onset may occur in early childhood, diagnosis is often delayed until clinical signs are apparent.
-
▶
Oral leukoplakic patches in DC have a higher malignant transformation rate, so any leukoplakic patch with or without history of tobacco consumption concomitant with the triad of features of DC should be dealt carefully.
-
▶
Early diagnosis can enable the patient to use the available treatment modalities to his or her best and thus reduce the associated morbidity and mortality.
Footnotes
Competing interests None.
Patient consent Obtained.
References
- 1.Neville BW, Damm DD, Allen CM, et al. Oral and maxillofacial pathology. 2nd edn Philadelphia: Saunders, 2002:648–649 [Google Scholar]
- 2.Vulliamy TJ, Marrone A, Knight SW, et al. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood 2006;107:2680–5 [DOI] [PubMed] [Google Scholar]
- 3.Auluck A. Dyskeratosis congenita. Report of a case with literature review. Med Oral Patol Oral Cir Bucal 2007;12:E369–73 [PubMed] [Google Scholar]
- 4.Tanaka A, Kumagai S, Nakagawa K, et al. Cole-Engman syndrome associated with leukoplakia of the tongue: a case report. J Oral Maxillofac Surg 1999;57:1138–41 [DOI] [PubMed] [Google Scholar]
- 5.Handley TP, Ogden GR. Dyskeratosis congenita: oral hyperkeratosis in association with lichenoid reaction. J Oral Pathol Med 2006;35:508–12 [DOI] [PubMed] [Google Scholar]
- 6.Vulliamy T, Dokal I. Dyskeratosis congenita. Semin Hematol 2006;43:157–66 [DOI] [PubMed] [Google Scholar]
- 7.Cannell H. Dyskeratosis congenita. Br J Oral Surg 1971;9:8–20 [DOI] [PubMed] [Google Scholar]
- 8.Holmstrup P, Vedtofte P, Reibel J, et al. Long-term treatment outcome of oral premalignant lesions. Oral Oncol 2006;42:461–74 [DOI] [PubMed] [Google Scholar]
- 9.Dokal I, Vulliamy T. Dyskeratosis congenita. Orphanet Encyclopedia 2004;1–11 [Google Scholar]
- 10.Atkinson JC, Harvey KE, Domingo DL, et al. Oral and dental phenotype of dyskeratosis congenita. Oral Dis 2008;14:419–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vulliamy TJ, Walne A, Baskaradas A, et al. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis 2005;34:257–63 [DOI] [PubMed] [Google Scholar]