

Abstract
Nicotinamidase/Pyrazinamidase (PncA) is involved in the NAD+ salvage pathway of Mycobacterium tuberculosis and other bacteria. In addition to hydrolyzing nicotinamide into nicotinic acid, PncA also hydrolyzes the pro-drug pyrazinamide to generate the active form of the drug, pyrazinoic acid, which is an essential component of the multidrug treatment of TB. A coupled enzymatic activity assay has been developed for PncA that allows for the spectroscopic observation of enzyme activity. The enzyme activity was essentially pH independent under the conditions tested, however, the measurement of the pH dependence of iodoacetamide alkylation revealed a pK value of 6.6 for the active site cysteine. Solvent deuterium kinetic isotope effects revealed an inverse value on kcat of 0.64, reconfirming the involvement of a thiol group in the mechanism. A mechanism is proposed for PncA catalysis that is similar to the mechanisms proposed for members of the Nitrilase superfamily, in which nucleophilic attack by the active site cysteine generates a tetrahedral intermediate that collapses with the loss of ammonia and subsequent hydrolysis of the thioester bond by water completes the cycle. An inhibitor screen identified the competitive inhibitor 3-pyridine carboxaldehyde with a Ki of 290 nM. Additionally, pyrazinecarbonitrile was found to be an irreversible inactivator of PncA, with a kinact/KI of 975 M−1sec−1.
Keywords: Pyrazinamide, Nicotinamidase, Pyrazinamidase, PncA, tuberculosis
Nicotinamidase (PncA) is responsible for the hydrolysis of nicotinamide to nicotinic acid and ammonia in the NAD+ salvage pathway of Mycobacterium tuberculosis and other bacteria (Scheme 1) (1). This enzyme is of particular interest because it also hydrolyzes the prodrug pyrazinamide (PZA) to the active bactericidal compound pyrazinoic acid (2, 3). Mutations in the pncA gene of M. tuberculosis have been shown to generate clinical resistance to PZA (4–7). PZA, in combination with rifampicin and isoniazid, is the current short course treatment for tuberculosis recommended by the World Health Organization (3, 8). The addition of PZA to this regimen leads to a significant reduction in the length of chemotherapy, from 9 or more months to 6 months (3). PZA has been shown to inhibit M. tuberculosis fatty acid synthetase type 1 and disrupt both membrane function and acidification of the cytoplasm, although its exact mechanism of bactericidal activity still remains unclear (3, 9).
Scheme 1.

The NAD+ salvage (bottom) and de novo (top) pathway in Mycobacterium tuberculosis.
Recent studies have shed light on the biochemical activity (8) as well as the structural details of PncA from Pyrococcus horkoshii (10), and Acinetobacter baumanii (11), however, no detailed kinetic analysis has been reported. Due to emerging antibiotic resistant strains of M. tuberculosis, a detailed understanding of the enzymatic activation of PZA is important. Moreover, a detailed mechanistic and structural understanding of all the enzymes involved in both the synthetic and salvage pathways of NAD+ biosynthesis has the potential to produce novel targets for the development of therapeutics for the treatment of tuberculosis. (1)
Previously, reported enzymatic activity assays for PncA involved end point assays, in which the enzymatic reaction was quenched at various time points and the mixture was separated by HPLC; the peaks for substrate and product are then integrated and compared to standards. We found this method both tedious and time consuming and sought to develop a simple, spectroscopic enzymatic assay. Toward this end, we have developed a coupled enzymatic assay that allowed us to directly monitor the steady-state enzymatic activity of Mycobacterium tuberculosis PncA (MtPncA). Using this assay system, we have characterized aspects of the kinetics, mechanism and inhibition by analogues of nicotinamide and pyrazinamide.
MATERIALS AND METHODS
Materials
All chemicals, buffers, salts, and L-glutamic dehydrogenase (GDH), were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). All enzymes used for molecular biology were supplied by New England Biolabs (Ipswich, MA). PCR primers and E. coli strain BL21 (DE3) cells were obtained from Invitrogen (Carlsbad, CA). Deuterated water was obtained from Cambridge Isotope Laboratories (Andover, MA). DNA sequencing was performed by Genewiz (South Plainfield, NJ).
Cloning, Overexpression, and Purification of PncA
The open reading frame of the Rv2043C (pncA gene) was amplified from M. tuberculosis H37Rv genomic DNA by standard PCR techniques using the primers 5’-ATCCCGCTCATATGCGGCGTT-GATCATCGTCGAC-3’ and 5’-ATCCCGCTCTCGAGTCAGGAGCTGCA-AACCAACTCGAC-3’ containing the underlined NdeI and XhoI restriction sites, respectively. The PCR product was cloned into pET-28a(+), and the recombinant PncA was expressed in E. coli strain BL21 (DE3). From a 100 mL overnight culture, 5 mL was used to inoculate 1L cultures of LB medium supplemented with kanamycin (50 μg/mL). Cultures were grown to mid-log phase (A600 ~ 0.8), cooled to 18°C, induced with 0.5 mM IPTG, and further incubated overnight at 18°C. Soluble expression of the protein was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
All protein purification steps were carried out at 4°C. The cells were harvested by centrifugation at 6000g, resuspended in 50 mM HEPES, pH 7, containing 250 mM NaCl, 5 mM imidazole, protease inhibitors and DNase I (0.1 μg/ml) and stirred for 30 min. The cells were lysed by sonication, and the cellular debris was removed by centrifugation at 18000g for 45 min. The supernatant was loaded by gravity flow onto a Ni-NTA column pre-equilibrated with the resuspension buffer. The column was washed by gravity flow with 5 column volumes of wash buffer [50 mM Tris, pH 8.0, containing 250 mM NaCl and 60 mM imidazole]. Bound protein was eluted by gravity flow of 1.5–2 column volumes of 50 mM Tris pH 8.0, containing 250 mM NaCl and 250 mM imidazole. The eluted protein was >95% pure as judged by SDS PAGE, and further purification was judged unnecessary. The eluted protein was dialyzed overnight against 2l of 30 mM HEPES, pH 7.5 containing 50 mM NaCl and stored at −20°C.
Preparation of H57D mutant
Site directed mutagenesis of the pET-28a(+) plasmid by standard PCR techniques was performed utilizing the primers 5’-GACCCGGGTGA-CGACTTCTCCGGCACA-3’ and 5’-TGTGCCGGAGAAGTCGTCACCCGGCTC-3’. The mutated plasmid was expressed and purified as done above. DNA sequencing analysis was performed by Genewiz (South Plainfield NJ)
Protein Estimation
The enzyme concentration was determined using ε280nm = 19940 M−1 cm−1 for native PncA. The concentration was also estimated by the Bio-Rad protein assay method using bovine serum albumin as a standard, and the two methods agreed favorably.
Measurement of Enzyme Activity by Coupled Assay
Initial velocities for the reaction of MtPncA were determined by monitoring the loss of absorbance at 340nm associated with oxidation of NADH to NAD+ by L-glutamic dehydrogenase (GDH) to produce glutamate from α-ketoglutarate. This reaction consumes one molecule of ammonia; therefore, the production of ammonia from the catalytic hydrolysis of nicotinamide or pyrazinamide by PncA is stoichiometric with NADH oxidation (Scheme 2). In a typical assay, 100 mM HEPES, pH 7.5 containing 5 mM α-ketoglutarate, 8 units of GDH, 200 μM NADH, and various concentrations of substrate (nicotinamide or pyrazinamide) were combined in a 1 mL cuvette. The reaction was initiated by addition of PncA, typically 37.5 nM final concentration, and the decrease in absorbance at 340 nm was monitored at 25°C for 8–10 minutes. Enzyme activities were calculated using the molar extinction coefficient of NADH (ε340 = 6220 M−1cm−1).
Scheme 2.

The coupled enzymatic activity assay used for spectrophotometric observation of steady state PncA kinetics.
Measurement of Enzyme Activity with HPLC
PncA activity was determined by HPLC by applying the reaction mixture directly to a Mono Q anion exchange column and separating the substrate and product using a linear NaCl gradient. In a typical assay, 100 mM HEPES, pH 8.0, containing 100 μM nicotinamide, and 37.5 nM PncA were combined at 25°C. Addition of enzyme initiated the reaction, and at a specified time point, the reaction was directly applied to an anion exchange column. A 100 ml NaCl gradient (0 – 1.2 M) afforded the separation of substrate and product, and the amount of conversion of nicotinamide by PncA was calculated by comparing peak areas to authentic standards.
Dependence of PncA activity on pH
The pH dependence of the kinetic parameters of PncA catalytic activity was determined using nicotinamide as the variable substrate. Activity was monitored every 0.5 pH unit from 5.0 – 9.5, using the following buffers, from pH 5.0–5.5, Acetate; pH 5.5–6.7, MES; pH 6.7–8.0, HEPES; pH 8.0–9.5, TAPS. The kinetic parameters were determined using both spectrophotometric and HPLC assays.
pH dependence of inactivation by iodoacetamide
A solution (100 μL final volume) containing 6 μM PncA in 100 mM phosphate buffer at various pH values was treated at 25°C with 150 μM of iodoacetamide. Aliquots (6 μL) were removed at various time points and added to a 1.0 ml solution containing 100 mM HEPES, pH 7.5, 5 mM nicotinamide, 5 mM α-ketoglutarate, 8 units of GDH, and 200 μM NADH. The enzyme activity was measured by monitoring the change in absorbance at 340 nm as described above. The rate of inactivation was measured over a pH range of 5.5–8.0, and the data was fit to the following equation using Prism
| (1) |
where V is the rate of inactivation, YL and YH are the pH-independent plateau values for the lower and upper region, respectively, K is the ionization constant, and H is the hydrogen ion concentration.
Solvent Kinetic Isotope Effects
Solvent kinetic isotope effects on V and V/K were determined by measuring the initial velocity of amide cleavage by PncA at various concentrations of nicotinamide in either H2O or 98% D2O. The mixtures contained 100 mM HEPES, pH 8.0, nicotinamide (2.5–250 μM), 5 mM α-ketoglutarate, 8 units of GDH, 200 μM NADH, and 37.5 nM PncA. Assays were performed at 25°C and were initiated by addition of PncA. Solvent deuterium isotope effects were fitted to the following equation:
| (2) |
where EV and EV/K are the isotope effects on kcat − 1 and kcat/Km − 1, respectively. F1 represents the fraction of isotope (0.98), V is the maximal velocity, and A is the concentration of substrate.
Inhibition studies of PncA
Several substrate analogues were tested as inhibitors of PncA using the coupled assay described above. In the case of the reversible inhibitors, initial velocities were measured while keeping various concentrations of inhibitor constant and varying the concentration of nicotinamide (250 μM – 10 μM). Time courses for each inhibitor concentration were determined five times and an average was calculated and used for subsequent data analysis. The time courses were fit to equation 3 for competitive inhibition kinetics:
| (3) |
where appKm is the apparent Michaelis-Menten constant, Km is the true Michaelis-Menten constant, I is the concentration of the inhibitor, and KI is the inhibitors dissociation constant.
In case of the irreversible inhibitor pyrazinecarbonitrile (PCN), PncA (5 μM) was incubated in 30 mM HEPES, pH 7.5 containing 50 mM NaCl with various concentrations of PCN (10–25 μM) and at 0.5, 1, and 2 minutes an aliquot was taken (6 μL) and the remaining PncA activity was determined spectrophotometrically with the coupled assay as described above. All measurements were performed in triplicate and compared to a control with no inhibitor. A Kitz-Wilson replot of the inactivation time course data was used to calculate the inactivation kinetic parameters.
RESULTS AND DISCUSSION
Cloning, Expression, and Purification
PCR amplification of the pncA gene of M. tuberculosis yielded a single fragment, and DNA sequencing confirmed the expected sequence and the absence of mutations introduced during PCR amplification. Overexpression of the PCR product yielded a protein with an apparent molecular mass in agreement with the expected mass (21 kDa) for MtPncA, as judged by SDS-PAGE. Approximately 10 mgs of purified enzyme was obtained per liter of culture. The enzyme was monomeric as judged by gel filtration of the purified protein.
Coupled Assay Development
In order to efficiently observe PncA catalytic activity, we utilized the catalytic activity of glutamate dehydrogenase (GDH), which converts α-ketoglutarate to glutamate, consuming one molecule of ammonia and oxidizing one molecule of NADH to NAD+. The oxidation of NADH to NAD+ produces a loss of absorbance at 340 nm, which can be easily monitored spectrophotometrically (Scheme 2). By using saturating concentrations of α-ketoglutarate and NADH, and high concentrations of GDH, we were able to directly monitor the steady-state production of ammonia from the conversion of nicotinamide or pyrazinamide to their respective acids by PncA. In order to ensure that this assay was reliable, we compared the spectrophotometric, coupled assay with the fixed-time HPLC assay, and demonstrated that similar kinetic constants were obtained by the two methods.
Determination of the kinetic parameters
Utilizing the coupled enzymatic assay, we determined the kinetic parameters of MtPncA with both pyrazinamide and nicotinamide. With the natural substrate nicotinamide, we determined a Km value of 14 ± 1 μM and a kcat value of 3.1 ± 0.1 s−1, corresponding to a kcat/Km value of 2.2 × 105 M−1s−1. In the case of pyrazinamide, we determined a Km value of 300 ± 40 μM and a kcat value of 3.8 ± 0.1 s−1, corresponding to a kcat/Km value of 1.3 × 104 M−1s−1. It is important to note that these values are similar to those we determined with the previously mentioned end-point HPLC assay. Also, PncA is extremely specific for nicotinamide and pyrazinamide, as we did not observe any enzymatic activity with benzamide or 6-aminonicotinamide.
Metal Ion content
PncA from a number of sources is a divalent cation-dependent metalloenzyme reported to contain Mn+2, Fe+2 or Zn+2 (8,10). Our homogeneous preparations of the enzyme exhibited a distinct blue-green color at high concentrations (>2 mg/ml). The metal ion in the MtPncA appears to be tightly-bound, and neither the addition of 5 mM EDTA, nor reported inhibitory metals (Zn+2 for MtPncA) resulted in any decrease in activity, although extensive dialysis versus EDTA has previously been reported to generate a metal ion-reconstitutable apo enzyme. In this report, the metal ion content of the MtPncA was reported to include 0.44 and 0.47 moles of Mn+2 and Fe+2, respectively, per mole of enzyme (8), and either Mn+2 or Fe+2 added to apo-MtPncA could restore activity. When we performed EPR spectroscopy on our preparations, we observed a typical six-line spectrum indicating the presence of Mn+2 bound to the enzyme (data not shown). Based on the three-dimensional structures of the related Pyrococcus horikoshii and Acinetobacter baumanii nicotinamidases (10,11), and multiple sequence alignment, the side chains of Asp49, His57 and His71 are the likely metal ion ligands in the MtPncA active site. These nitrogen and oxygen containing ligands are commonly observed in metalloenzymes that bind either manganese or ferrous iron.
pH Dependence of the Kinetic Parameters
The pH dependence of the deamidase reaction was measured over the pH range of 5.0–9.5 using nicotinamide as the substrate. We did not observe any significant effect of pH on either the maximal velocity or the V/Km value of nicotinamide. It is worth noting that care was taken to insure the coupling enzyme, GDH, was present at high enough concentrations to not be limiting across the pH range studied. Further, at several points across the pH range, the kinetic parameters were determined using the end point HPLC assay, and the results were qualitatively equal at every point tested, as seen in Figure 1.
Figure 1.
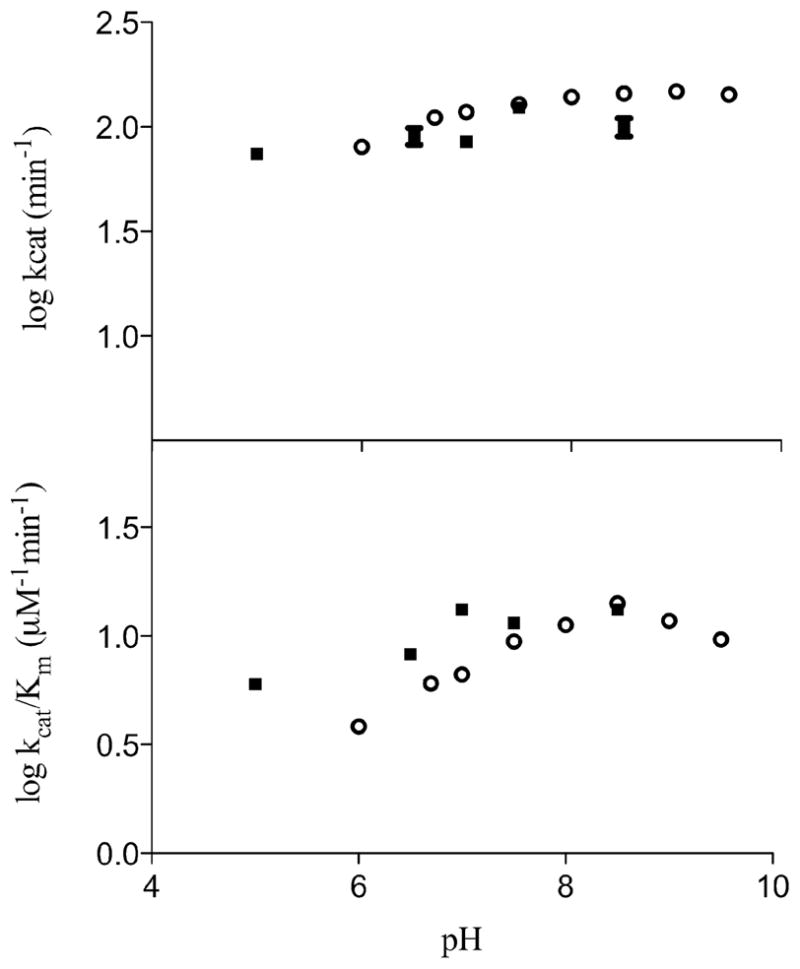
The pH profile of PncA with nicotinamide as the substrate. Log kcat vs pH (top), Log kcat/Km vs pH (below). All data points represent the mean of at least three separate sets of experiments. Black squares are values collected from the end point HPLC assay method, open circles represent data collected with the enzyme coupled assay method.
Lack of pH dependence on the amidase reaction is an intriguing result, as we had expected to see a pH dependence involving deprotonation of the active site cysteine (see below). Catalytic mechanisms proposed for PncA in the literature involve base-assisted attack by a nucleophilic cysteine on the carbonyl carbon of the substrate. This mechanism resembles the mechanisms proposed for the nitrilase superfamily of enzymes (12). The catalytic mechanism of nitrilases involves a nucleophilic cysteine, a lysine that stabilizes the tetrahedral intermediate, and a glutamate that acts as a general base catalyst (12). These residues most likely correspond to the conserved, and essential Cys138, Lys96, and Asp8 residues in M. tuberculosis PncA and other nicotinamidases, where the glutamate is replaced by an aspartate. Therefore, the enzyme catalytic mechanism most likely involves a proton transfer from the cysteine thiol to the aspartate group, as has been previously proposed (10, 11).
pH Dependence of the Inactivation by Iodoacetamide
Since PncA catalysis most likely involves a nucleophilic cysteine residue, we wanted to probe if the thiol-specific labeling agent, iodoacetamide, was an inactivator of PncA. Indeed, iodoacetamide was found to be an inactivator of PncA, and furthermore, incubation of PncA with iodoacetamide yielded a time and concentration dependent lost of enzymatic activity that was first order over more than 30 minutes. The inactivation proved to be pH-dependent, as can be seen in Figure 2. The inactivation rate was increased as the pH of the solution was increased from about 6 to 8, but above pH 8, the rate of inactivation reached a plateau. Fitting the data to Equation 1 revealed the change in the inactivation rate was due to a single ionizable group, with a pK value of 6.6, reflecting the pK of the active site, nucleophilic Cys138 in the free enzyme.
Figure 2.
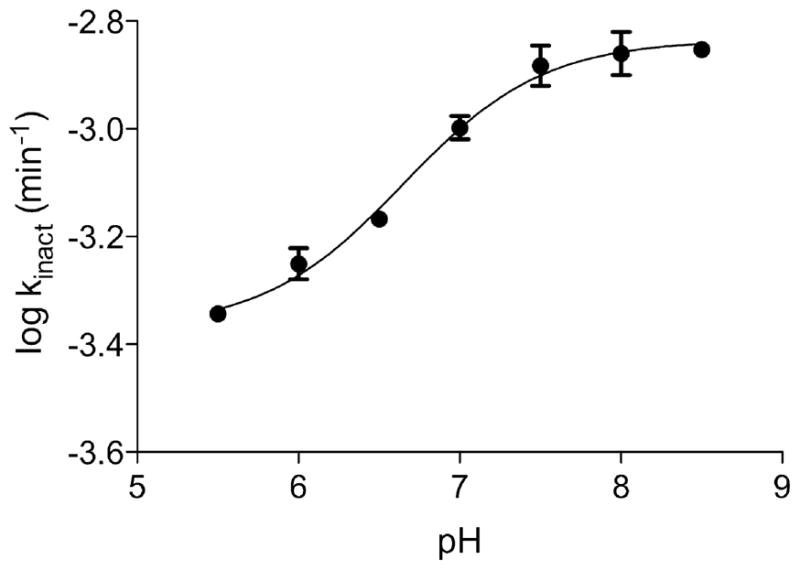
The pH dependence of iodoacetamide inactivation of PncA. Dots represent experimentally determined values; line represents calculated fit as detailed in the materials and methods section.
Solvent Kinetic Isotope Effects
Solvent kinetic isotope effects were determined by measuring the initial velocities in both H2O and 98% D2O at 7 different concentrations of nicotinamide. These experiments were performed at pH 8.0, where both V and V/K were judged to be maximal and independent of pH. As Figure 3 reveals, D2Okcat was inverse, with a value of 0.64 ± 0.02. However, the solvent isotope effect did not have a statistically significant effect on kcat/Km, as it was determined to be 1.1 ± 0.1. Fractionation factors of most enzymatic groups that participate in acid/base chemistry are normal, or above 1. However, inverse solvent kinetic isotope effects are often associated with the participation of thiol groups in acid/base chemistry (13, 14). Thiols are known to exhibit inverse fractionation factors (solvent equilibrium isotope effects) in the range of 0.4–0.6, (13) and we suggest the inverse fractionation factor of the catalytic cysteine of MtPncA is responsible for the inverse solvent kinetic isotope effect on kcat, as the nucleophilic thiolate anion is required for attack on the carbonyl carbon of nicotinamide.
Figure 3.
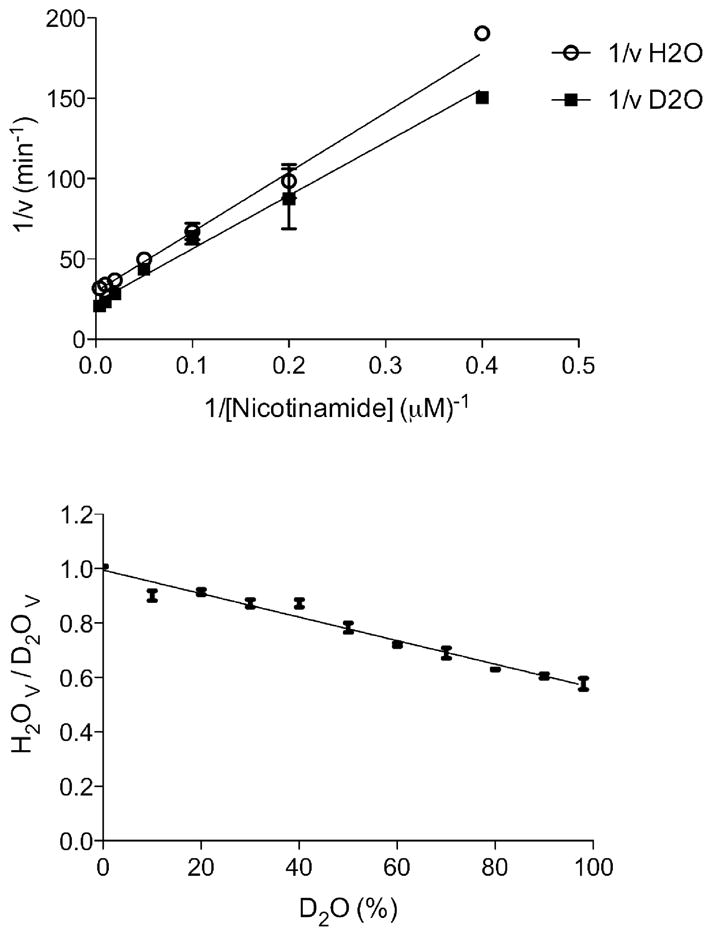
Solvent deuterium isotope effects. Circles and squares represent experimentally determined values, open circles in the presence of H2O, squares, in the presence of 98% D2O (top). Proton inventory of the solvent kinetic isotope effects. Nicotinamide was used as the substrate under saturating conditions and the amount of D2O was varied from 0 to 98%. (bottom)
A proton inventory was performed to determine the proton multiplicity that is present in the solvent kinetic isotope effect. The proton inventory was determined under saturating conditions of nicotinamide and the percent of D2O was varied from 0–98% in increments of 10%. As shown in Figure 3 (lower panel), the linearity exhibited by the proton inventory demonstrates that one proton is being transferred in the isotopically sensitive step, and the negative slope confirms the inverse nature and magnitude of the solvent kinetic isotope effect observed on kcat.
Comparing Mycobacterium bovis PncA
Mycobaterium bovis expresses a pncA gene that contains a single mutation resulting in an aspartic acid replacing His57 of PncA (H57D). Further, M. bovis has been shown to be innately resistant to treatment by pyrazinamide. His57 has been proprosed to be directly involved in metal binding of the Mn2+ in several PncA’s for which three-dimensional structures are known, and literature reports that this mutant is unable to bind metal. This results in a substantial decrease in enzymatic activity, although to our knowledge, the kinetic properties of M. bovis PncA have not yet been reported. Toward this end, we employed site-directed mutagenesis to express and purify the H57D mutant as detailed in the materials and methods section. As expected, we observed a loss of the Mn2+ signal in the EPR spectrum of the H57D mutant (data not shown). The mutant is not, however, completely inactive. With nicotinamide as the substrate, we determined the Km value to be 13 ± 2 μM and the kcat value to be 0.50 ± 0.01 s−1, corresponding to a kcat/Km value of 3.7 × 104 M−1s−1. This represents a 6-fold decrease in the observed catalytic turnover of the substrate by the mutant, but interestingly, we did not observe a change in the Km for nicotinamide. Using the substrate pyrazinamide, we determined a Km value to be 16 ± 4 μM and a kcat value of 0.10 ± 0.01 s−1, corresponding to a kcat/Km value of 6.2 × 103 M−1s−1. For pyrazinamide, both the Km value and the kcat value have been decreased 20- and 38-fold, respectively, for the H57D mutant. The 38-fold decrease in kcat for pyrazinamide is likely enough to account for the resistance of M. bovis to the bactericidal effects of pyrazinamide.
Catalytic Mechanism of PncA
There has been some disagreement in the literature over the catalytic mechanism of PncA. The earliest mechanistically relevant report came from Du et al., who reported the crystal structure of the Pyrococcus horikoshii nicotinamidase (PhPncA) (10). The overall structure was most similar to N-carbamoylsarcosine amidohydrolase from Arthrobacter sp., and contained the active site catalytic triad of conserved residues of Cys133, Arg84, and Asp19. Further, they found the protein was a monomer and the active site contained a Zn2+ ion. Based on the crystal structure, Du et al. proposed a mechanism that involves nucleophilic attack by the active site Cys to form a tetrahedral intermediate and subsequent loss of ammonia yields an acyl intermediate. Hydrolysis was proposed to occur by the subsequent attack from a Zn2+-bound hydroxide ion to give the free enzyme and product.
Following this initial report, Zhang et al. reported a characterization of Mycobacterium tuberculosis nicotinamidase (MtPncA) (8). They found MtPncA to be a monomer and the protein contained Mn2+ or Fe2+ in an approximate 0.5 to 0.5 molar ratio. Zinc did not restore amidase activity when incubated with the apoenzyme, and was also inhibitory to the wild-type enzyme. Site directed mutagenesis of several residues of MtPncA showed that the triad of Cys138, Asp8, and Lys96 were essential for catalytic activity, as the mutants C138A, D8A, and K96A completely lost amidase activity but retained the ability to bind metal. Further, Asp49, His51, His57, and His71 were deemed likely to be directly involved in metal binding as mutation of any of these residues caused complete loss of metal from the protein.
Most recently, Fyfe et al. reported the crystal structure of Acinetobacter baumanii PncA (AbPncA), and found it to be a dimer that contained either Fe2+ or Zn2+ in a roughly 0.5 to 0.5 molar ratio (11). They proposed a mechanism of catalysis that involves nucleophilic attack of the active site cysteine that is aided through stabilization of the tetrahedral intermediate by an oxyanion hole formed from the backbone amides of Cys159 and Ala155 (AbPncA numbering). Asp16 was proposed to deprotonate Cys159 to initiate catalysis and to donate a proton to aid the release of NH3 during the tetrahedral intermediate collapse to form the thioester acyl intermediate. Water is then activated by proton abstraction by Asp16 for nucleophilic attack on the covalent acyl-enzyme intermediate to yield a thiolate and nicotinic acid. Transfer of a proton from Asp16 to the active site cysteine completes the catalytic cycle.
Based on our data, we propose the catalytic mechanism of Mycobacterium tuberculosis PncA is most similar to the mechanism recently proposed for AbPncA. As can be seen in Scheme 3, the catalytic mechanism involves an initial proton equilibrium between Cys138 and the ionized Asp8, yielding the nucleophilic thiolate that attacks the carbonyl of the amide substrate to produce a tetrahedral intermediate. The ionization state of Asp8 is very likely to be strongly influenced by the adjacent Lys96. This first step is clearly the one that is reported on by the inverse solvent kinetic isotope effect on kcat. In the free enzyme, the pK value of Cys138 (or more appropriately the Cys138-Asp8 pair) is close to 6.6. However, upon binding the substrate, the ionization behavior of these residues is substantially altered, causing the pK value to shift downwards, and out of the experimentally accessible range. Even at the lower pH extremes, only a small decrease in kcat is observed.
Scheme 3.

Proposed mechanism of catalysis of MtPncA.
In the second step, the productive collapse of the tetrahedral intermediate is assisted by protonation of the amine to generate ammonia and the covalent Cys138 thioester. The departing ammonia is replaced by a water molecule, which is activated for attack on the thioester by the ionized carboxylate of Asp8. Reaction of water with the thioester generates the second tetrahedral intermediate, which in the final step, decomposes to generate the final aryl carboxylic acid product. Productive decomposition is again facilitated by proton transfer from the protonated carboxyl of Asp8, regenerating the active site residues in their original ionization state.
Inhibition of PncA
It has been well established that current antitubercular drugs have either reduced or minimal effect on latent, nonreplicating bacilli which leads to long treatment therapy as well as contributing to the appearance of multiple drug-resistant strains. (1, 15) It has been reported that the NAD+ salvage pathway is upregulated in Mycobacterium tuberculosis under conditions of hypoxia. (1) Inhibition or disruption of both the NAD+ salvage and de novo pathways is an interesting option for targeting both active and latent tuberculosis.
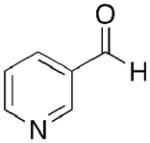
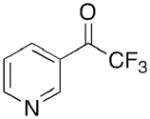
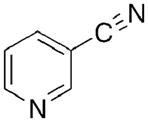
Toward this end, we performed a screen of nicotinamide analogues to search for possible inhibitors of PncA. As shown in Table 1, the screen resulted in several competitive inhibitors with inhibition constants in the micromolar to submicromolar range. The most potent inhibitor, 3-pyridine carboxaldehyde exhibited a Ki value of 290 nM. All of the inhibitors tested were reversible, competitive inhibitors, with one exception. Unlike the other inhibitors, pyrazinecarbonitrile (PCN) is an irreversible inactivator of PncA. Incubation of the enzyme with PCN resulted in a time and concentration dependent loss of enzymatic activity (Figure 4, left), and enzymatic activity is not restored upon removal of excess inactivator or prolonged dialysis. A Kitz-Wilson replot of the inactivation data (Figure 4, right) gave a kinact of 0.06 ± 0.01 sec−1, and a Ki of 61 ± 3 μM. This gives an apparent second-order rate constant for the inactivation of the enzyme by PCN, kinact/Ki of 975 M−1s−1. Interestingly, the nicotinamide analogue of PCN, 3-pyridinecarbonitrile, is a reversible, competitive inhibitor of PncA with a Ki of around 100 μM. Neither compound is a substrate for the enzyme. Given the conserved nature of the catalytic residues in deamidases, such as nicotinamidase, and bona fide nitrilases in the nitrilase superfamily, it is curious that PCN is an irreversible inactivator of PncA. Nitrilases use the catalytic cysteine to attack the carbon atom of the cyano group, generating initially the thioimine, which upon hydrolysis yields the thioester, an intermediate in both reactions. The mechanism by which PCN inactivates the enzyme is currently under investigation, as is the compound’s ability to inhibit NAD+ biosynthesis is microorganisms.
Table 1.
Inhibition constants for PncA of several structural analogues.
| Inhibitor | Ki (μm) |
|---|---|
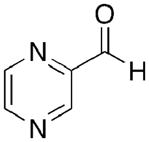 |
1.5 ± 0.7 |
 |
0.3 ± 0.1 |
 |
420 ± 100 |
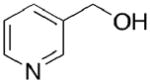 |
63 ± 31 |
 |
140 ± 14 |
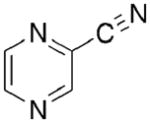 |
Kinact = 0.06 ± 0.01 sec−1 Ki = 61 ± 3 μM |
Figure 4.

Semi log plot of the time courses for the inactivation of PncA with various concentrations of pyrazine carbonitrile (left). The Kitz-Wilson replot of the inactivation data (right).
Conclusion
Mycobacterium tuberculosis is responsible for more deaths worldwide than any other bacterial infections disease.(16). It is therefore important to expand our understanding of the activation of the front line antibiotic, pyrazinamide, as well as to increase our understanding of the NAD+ salvage pathway. This work provides a detailed examination of the chemical and kinetic mechanism of PncA, the amidase that is serendipitously responsible for the activation of a major TB therapeutic drug, pyrazinamide. The NAD+ salvage and de novo pathways are essential to both latent and active TB, and efforts to develop new therapeutics that targets enzymes in these pathways in M. tuberculosis are emerging.
Acknowledgments
We sincerely thank Dr. Gary J. Gerfen for help with the EPR experiments.
This work was supported by NIH grant AI33696 (to J.S.B.)
Abbreviations
- DNA
deoxyribonucleic acid
- EDTA
ethylenediaminetetraacetic acid
- GDH
L-glutamic dehydrogenase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high performance liquid chromatography
- IPTG
isopropyl β-D-thiogalactopyranoside
- LB
Luria broth
- MES
2-(N-morpholino)ethanesulfonic acid
- NADH
reduced nicotinamide adenine dinucleotide
- Ni-NTA
nickel nitriloacetic acid
- PCR
polymerase chain reaction
- PncA
Nicotinamidase
- PZA
pyrazinamide
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TAPS
N-Tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid
- TB
tuberculosis
- Tris
tris(hydroxymethyl)aminomethane
References
- 1.Boshoff HIM, Xu X, Tahlan K, Dowd CS, Pethe K, Camacho LR, Park TH, Yun CS, Schnappinger D, Ehrt S, Williams KJ, Barry CE. Biosynthesis and Recycling of Nicotinamide Cofactors in Mycobacterium tuberculosis. Journal of Biological Chemistry. 2008;283:19329–19341. doi: 10.1074/jbc.M800694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheen P, Ferrer P, Gilman R, Lopez-Llano J, Furentes P, Valencia E, Zimic M. Effect of pyrazinamidase activity on pyrazinamide resistance in Mycobacterium tuberculosis. Tuberculosis. 2009;89:109–113. doi: 10.1016/j.tube.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Mitchison D. The curious characteristics of pyrazinamide: a review. Int J Tuberc Lung Dis. 2003;7:6–21. [PubMed] [Google Scholar]
- 4.Lemaitre N, Sougakoff W, Truffot-Pernot C, Jarlier V. Characterization of new mutations in pyrazinamide-resistant strains of Mycobacterium tuberculosis and identification of conserved regions important for the catalytic activity of the pyrazinamidase PncA. Antimicrobial Agents and Chemotherapy. 1999;43:1761–1763. doi: 10.1128/aac.43.7.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuksel P, Tansel O. Characterizaiton of pncA mutations in pyrazinamide-resistant Mycobacterium tuberculosis in Turkey. New Microbiol. 2009;32:153–158. [PubMed] [Google Scholar]
- 6.Pandey S, Newton S, Upton A, Roberts S, Drinkovic D. Characterisation of pncA mutations in clinical Mycobacterium tuberculosis isolates in New Zealand. Pathology. 2009;41:582–584. doi: 10.1080/00313020903071587. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H, Bi L, Li C, Sun Z, Deng J, Zhang X. Mutations found in the pncA gene of Mycobacterium tuberculosis in clinical pyrzainamid-eresistant isolates from a local region of China. J Int Med Res. 2009;37:1430–1435. doi: 10.1177/147323000903700517. [DOI] [PubMed] [Google Scholar]
- 8.Zhang H, Deng J, Bi L, Zhou Y, Zhang Y, Zhang C, Zhang Y, Zhang X. Characterization of Mycobacterium tuberculosis nicotinamidase/pyrazinamidase. FEBS Journal. 2008;275:753–762. doi: 10.1111/j.1742-4658.2007.06241.x. [DOI] [PubMed] [Google Scholar]
- 9.Zimhony O, Cox J, Welch J, Vilcheze C, Jacobs W. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nature Medicine. 2000;6:1043–1047. doi: 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- 10.Du X, Wang W, Kim R, Yakota H, Nguyen H, Kim SH. Crystal Structure and Mechanism of Catalysis of a Pyrazinamidase from Pyrococcus horikoshii‚A†. Biochemistry. 2001;40:14166–14172. doi: 10.1021/bi0115479. [DOI] [PubMed] [Google Scholar]
- 11.Paul KF, Vincenzo AR, Aleksandra Z, Scott C, William NH. Specificity and Mechanism of Acinetobacter baumanii Nicotinamidase: Implications for Activation of the Front-Line Tuberculosis Drug Pyrazinamide. Angewandte Chemie International Edition. 2009;48:9176–9179. doi: 10.1002/anie.200903407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thuku RN, Brady D, Benedik MJ, Sewell BT. Microbial nitrilases: versatile, spiral forming, industrial enzymes. Journal of Applied Microbiology. 2009;106:703–727. doi: 10.1111/j.1365-2672.2008.03941.x. [DOI] [PubMed] [Google Scholar]
- 13.Quinn DM, Sutton LD. In: Enzyme Mechanism from isotope effects. Cook PF, editor. CRC Press; Boca Raton, FL: 1991. pp. 73–126. [Google Scholar]
- 14.Sikora AL, Frankel BA, Blanchard JS. Kinetic and Chemical Mechanism of Arylamine N-Acetyltransferase from Mycobacterium tuberculosis. Biochemistry. 2008;47:10781–10789. doi: 10.1021/bi800398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez JE, Mckinney JD. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis. 2003;84:29–44. doi: 10.1016/j.tube.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC, Monitoring P for the WHOGS. Global Burden of Tuberculosis: Estimated Incidence. Prevalence, and Mortality by Country. JAMA. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]