

Abstract
The ATP-binding cassette transporter associated with antigen processing (TAP) plays a key role in the adaptive immune defense against infected or malignantly transformed cells by translocating proteasomal degradation products into the lumen of the endoplasmic reticulum for loading onto MHC class I molecules. The broad substrate spectrum of TAP, rendering peptides from 8 to 40 residues, including even branched or modified molecules, suggests an unforeseen structural flexibility of the substrate-binding pocket. Here we used EPR spectroscopy to reveal conformational details of the bound peptides. Side-chain dynamics and environmental polarity were derived from covalently attached 2,2,5,5-tetramethylpyrrolidine-1-oxyl spin probes, whereas 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid spin-labeled peptides were used to detect backbone properties. Dependent on the spin probe’s position, striking differences in affinity, dynamics, and polarity were found. The side-chains’ mobility was strongly restricted at the ends of the peptide, whereas the central region was flexible, suggesting a central peptide bulge. In the end, double electron electron resonance allowed the determination of intrapeptide distances in doubly labeled peptides bound to TAP. Simulations based on a rotamer library led to the conclusion that peptides bind to TAP in an extended kinked structure, analogous to those bound to MHC class I proteins.
Keywords: adaptive immune system, antigenic peptide binding, peptide conformation, double electron electron resonance EPR, site-directed spin labeling
The transporter associated with antigen processing (TAP) plays a pivotal role in the antigen-processing pathway via MHC class I molecules (1). A fraction of proteasomal degradation products is translocated from the cytosol into the lumen of the endoplasmic reticulum by the TAP complex for loading onto MHC class I molecules. Subsequently, these peptide–MHC complexes traffic to the cell surface where they display their antigenic cargo to cytotoxic T-lymphocytes, which can thereby efficiently recognize and eliminate infected or malignantly transformed cells.
TAP belongs to the superfamily of ATP-binding cassette (ABC) transporters, which are found in all kingdoms of life. These transport machineries translocate a very broad spectrum of solutes across biological membranes by hydrolysis of ATP, which is critical for a variety of cellular functions, such as signal transduction, protein secretion, ion homeostasis, multidrug resistance, or antigen presentation (2, 3). The heterodimeric TAP complex is composed of the two half-transporters TAP1 (ABCB2) and TAP2 (ABCB3), each containing a hydrophobic transmembrane domain (TMD) followed by a cytosolic nucleotide-binding domain. Each subunit can be subdivided into a unique extra N-terminal domain (TMD0), which is required for the assembly of the peptide loading complex composed of TAP, tapasin, MHC class I, and the chaperones ERp57 and calreticulin, followed by a core transport unit of six transmembrane helices, which is essential and sufficient for peptide transport (4). The core TMD harbors the peptide-binding site, which was mapped by photo-cross-linking studies to the last cytosolic loop between the helices TM4 and TM5 and a region of 15 amino acids following TM6 of each TAP core subunit (5). Notably, both TAP1 and TAP2 are required for peptide binding (6). TAP transports most efficiently peptides with a length of 8–12 amino acids. However peptides with a length of 8–16 amino acids are bound with comparable affinity (6, 7). Strikingly, even peptides of 40 amino acids in length and peptides with bulky side chains are bound and transported, thus underlining the structural flexibility of the substrate-binding pocket (8–10). Peptide binding is an ATP-independent, two-step process accompanied by large structural rearrangements (6, 9, 11, 12). For peptide binding, the first three N-terminal residues and the C-terminal residue were identified as critical, whereas the amino acids in between these “anchor” positions appear to form only minor contacts to the binding site and may even protrude into the solvent in the case of long or sterically restricted peptides (10, 13–17).
Knowing the conformation of bound substrates in an enzyme is often the first step for the design of new drugs. In the field of ABC exporters, this seems to be a challenging task. For resolving the structure of bound peptides and the mechanism of peptide binding, we used EPR spectroscopy in combination with site-directed spin labeling of antigenic peptides to (i) probe the spatial arrangement of the peptide-binding pocket of TAP and (ii) to elucidate the conformation of the bound peptide.
Results
Experimental Strategies.
EPR spectroscopy was applied on singly and doubly spin-labeled peptides to probe peptide–TAP interactions and to obtain insights into the structure of peptides in complex with the antigen translocation machinery. The properties of the side chains and of the backbone were probed by iodoacetamido–2,2,5,5-tetramethylpyrrolidine-1-oxyl (proxyl) spin labels covalently attached to cysteines and by the amino acid 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid (TOAC) inserted at each position of the high affinity, HLA-B27 restricted epitope RRYQKSTEL, respectively (Fig. 1A). TOAC lacks the rotatable bonds found in most conventional spin probes as the nitroxide ring is rigidly attached to the α-carbon of the amino acid, thus it is more sensitive to peptide backbone dynamics than to the orientational freedom of the side chain (18). Distance constraints were derived from doubly spin-labeled 9- and 15-mer peptides (RRYQKSTEL and RRYQKLTSAVNSTEL, respectively) via continuous wave (CW) and pulse EPR to obtain the conformation of the peptide bound to the TAP complex. For brevity, an N-mer peptide with a proxyl label or a TOAC amino acid at position n will be designated as 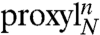 or
or 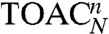 , respectively.
, respectively.
Fig. 1.

Mobility of spin-labeled peptides bound to TAP. (A) Structure of 9-mer peptides modified by proxyl or TOAC spin probes at position 4. Arrows indicate the possible rotations around different bonds in the proxyl spin probe. (B) Spin-labeled 9-mer peptides bind specifically to TAP. Spin-normalized CW derivative spectra of 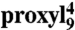 free in solution (orange line), bound to TAP (black line, multiplied by two for clarity, 10 μM
free in solution (orange line), bound to TAP (black line, multiplied by two for clarity, 10 μM 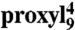 after incubation with 48 μM active TAP at 277 K for 15 min), and competed with a 165-fold molar excess of unlabeled competitor peptide RRYQKSTEL for 2 min at 310 K. The spectrum of free peptide is scaled (dotted orange line) and superimposed to the spectra of bound and competed spin-labeled peptides to reveal the fraction of free peptide or residual bound peptide after competition, respectively (indicated by arrows). (C) EPR derivative spectra of bound proxyl and TOAC-labeled peptides. Peptides were incubated with TAP for 15 min at 4 °C. For proxyl-labeled peptides, the fraction of free peptide was subtracted. Asterisks indicate small artifacts due to subtraction. The red dotted line highlights displacement of the low-field peak, and the green dotted lines show an exemplary central line width (ΔHpp). (D) Mobility parameters extracted from the spectra shown in panel C versus residue numbers: the low-field peak positions for the proxyl and TOAC probes are shown in red and black, respectively; the inverse of the line width is shown only for the proxyl probes in green.
after incubation with 48 μM active TAP at 277 K for 15 min), and competed with a 165-fold molar excess of unlabeled competitor peptide RRYQKSTEL for 2 min at 310 K. The spectrum of free peptide is scaled (dotted orange line) and superimposed to the spectra of bound and competed spin-labeled peptides to reveal the fraction of free peptide or residual bound peptide after competition, respectively (indicated by arrows). (C) EPR derivative spectra of bound proxyl and TOAC-labeled peptides. Peptides were incubated with TAP for 15 min at 4 °C. For proxyl-labeled peptides, the fraction of free peptide was subtracted. Asterisks indicate small artifacts due to subtraction. The red dotted line highlights displacement of the low-field peak, and the green dotted lines show an exemplary central line width (ΔHpp). (D) Mobility parameters extracted from the spectra shown in panel C versus residue numbers: the low-field peak positions for the proxyl and TOAC probes are shown in red and black, respectively; the inverse of the line width is shown only for the proxyl probes in green.
Spin-Labeled Peptides Bind Specifically to TAP.
The CW spectra detected for proxyl- or TOAC-labeled peptides freely tumbling in solution were clearly distinguishable from those of peptides bound to TAP (Fig. 1B and Fig. S1). As shown for example for  , the spectrum of the free peptide is characterized by the presence of three narrow lines, typical for nitroxides in the fast motional regime. In contrast, the spectrum of the bound peptide displays a broad powder-like pattern exhibiting a characteristic positive peak at low-B fields and a negative one at high field. In the spectrum of the bound peptides, two narrow peaks are still present, which represent the low- and high-field line of a small fraction of residual unbound peptide in solution. Addition of an excess of unlabeled peptide restores almost completely the spectral features of free peptide, demonstrating the reversible and specific binding of the labeled peptide. Analysis of the spectra obtained upon titration of the spin-labeled peptide to TAP allowed the determination of the peptide affinities by second integral determination of the fractions of free versus bound peptide. In addition, the concentration of active TAP was revealed by this method, which was in good accordance with protein concentration determination demonstrating a peptide-to-TAP stoichiometry of 1∶1 (Fig. S2). Dissociation constants derived from EPR and filter binding assays were found to be in agreement (Table S1). Notably, the binding pattern reflects the selectivity pattern of TAP, in which anchor residues are localized at both ends of the peptide and are sensitive to modifications whereas the region in between is highly promiscuous in sequence and length (9, 13–15, 19). The same effects were observed for peptides containing TOAC amino acids, however with lower binding affinities than the proxyl-labeled peptides (Fig. S1; Table S1). In conclusion, proxyl- and TOAC-labeled peptides bind specifically to TAP and the relevant EPR spectral changes from the free to the bound state allow monitoring of their conformational change in terms of side-chain and backbone flexibility.
, the spectrum of the free peptide is characterized by the presence of three narrow lines, typical for nitroxides in the fast motional regime. In contrast, the spectrum of the bound peptide displays a broad powder-like pattern exhibiting a characteristic positive peak at low-B fields and a negative one at high field. In the spectrum of the bound peptides, two narrow peaks are still present, which represent the low- and high-field line of a small fraction of residual unbound peptide in solution. Addition of an excess of unlabeled peptide restores almost completely the spectral features of free peptide, demonstrating the reversible and specific binding of the labeled peptide. Analysis of the spectra obtained upon titration of the spin-labeled peptide to TAP allowed the determination of the peptide affinities by second integral determination of the fractions of free versus bound peptide. In addition, the concentration of active TAP was revealed by this method, which was in good accordance with protein concentration determination demonstrating a peptide-to-TAP stoichiometry of 1∶1 (Fig. S2). Dissociation constants derived from EPR and filter binding assays were found to be in agreement (Table S1). Notably, the binding pattern reflects the selectivity pattern of TAP, in which anchor residues are localized at both ends of the peptide and are sensitive to modifications whereas the region in between is highly promiscuous in sequence and length (9, 13–15, 19). The same effects were observed for peptides containing TOAC amino acids, however with lower binding affinities than the proxyl-labeled peptides (Fig. S1; Table S1). In conclusion, proxyl- and TOAC-labeled peptides bind specifically to TAP and the relevant EPR spectral changes from the free to the bound state allow monitoring of their conformational change in terms of side-chain and backbone flexibility.
Side-Chain and Backbone Mobility.
The specific binding of spin-labeled peptides to TAP provides an optimal starting point to determine the side-chain and the backbone mobility of the bound peptide, which is encoded in the width of the EPR lines and in the position of the low magnetic field peak. For restricted spin labels the first positive peak shifts to lower B-field values (dotted red lines in Fig. 1C) and the width of the central peak (ΔHpp in Fig. 1C) is broader with respect to mobile spin labels. To compare differences in the side-chain dynamics of the bound proxyl-labeled peptides, we subtracted the residual free peptide fraction when present (Fig. 1C). The plot of the two mobility parameters, extracted from the spectra of the proxyl-labeled 9-mer peptides versus the spin-label positions correlate nicely with each other (Fig. 1D) and demonstrate that the N- and C-terminal regions of the peptide are strongly immobilized, whereas the central region features a higher degree of freedom, with the exception of position 4.
To disentangle the mobility of the side chains, reflected by the proxyl probes, from the intrinsic backbone flexibility, we employed the TOAC-labeled peptides. The rigid spin-label TOAC, built into the peptide backbone, has only one degree of freedom, which is the flip of the six-membered ring. Despite being generally more restricted in motion than the proxyl-labeled side chains, the highest mobility is again observed for the backbone positions 5, 6, and 7 (Fig. 1D), in line with the proxyl probes. The mobility of  and
and 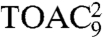 could not be derived because of low affinity of these peptides. Interestingly, comparing positions 4 and 6 labeled with proxyl or TOAC revealed that in both cases position 4 is anchored to TAP, whereas position 6 is indeed more flexible, with the large degree of freedom mainly dominated by the side-chain flexibility.
could not be derived because of low affinity of these peptides. Interestingly, comparing positions 4 and 6 labeled with proxyl or TOAC revealed that in both cases position 4 is anchored to TAP, whereas position 6 is indeed more flexible, with the large degree of freedom mainly dominated by the side-chain flexibility.
To generalize this pattern of increased side-chain flexibility going from the N- and C-terminal residues to the central region of the peptide, side-chain dynamics of spin labels attached at selected positions in 11- and 15-mer peptides were also investigated. The 11- and 15-mer peptides labeled at the middle positions (6 and 8, respectively) and a 15-mer peptide doubly labeled at positions 2 and 14 were compared to 9-mer peptide carrying the spin labels at analogous positions (Fig. S3). Remarkably, the middle positions showed a high flexibility whereas those close to the anchoring residues showed a more restricted mobility perfectly in agreement with the findings in the 9-mer peptides.
In conclusion, the data revealed that upon TAP association the central region of the peptide is flexible, with the exception of position 4, whereas the N- and C-terminal residues are tightly bound to the binding pocket of TAP.
Microenvironment of TOAC- and Proxyl-Labeled Peptides.
To determine the polarity of the peptide-binding pocket of TAP, the spectra of 9-mer peptides labeled with proxyl or TOAC at the “restricted” position 4 and the “mobile” position 6 were recorded at 160 K (Fig. 2). At this temperature, the motional averaging of the anisotropic hyperfine tensor is avoided, thus the mobility of the spin label is frozen out, and the obtained so-called powder spectrum contains only information about the polarity of the spin-label microenvironment. The distance between the low and high field peaks of a powder spectrum is 2Azz, which directly correlates with the polarity of the spin-label microenvironment. High Azz values (3.6–3.7 mT) characterize polar water-exposed spin labels, whereas lower values (down to 3.3 mT) indicate apolar surroundings. Upon binding of 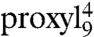 to TAP, the Azz value decreases (ΔAzz - 0.07 mT; Table S2) indicating that in the bound state the spin probe is partially shielded from the bulk water (Fig. 2; Table S2). Interestingly, the low-temperature spectrum of
to TAP, the Azz value decreases (ΔAzz - 0.07 mT; Table S2) indicating that in the bound state the spin probe is partially shielded from the bulk water (Fig. 2; Table S2). Interestingly, the low-temperature spectrum of  is very similar to the one obtained at 277 K (Fig. 2A, Inset), demonstrating that the spin probe at this position is extremely restricted in motion. In contrast, the polarity at position 6 differs only slightly between the free and the bound peptide state (ΔAzz - 0.04 mT), implying that the spin probe at this position is still exposed to the water in the bound state (Fig. 2 and Table S2). The pronounced changes visible in the low-field peak of
is very similar to the one obtained at 277 K (Fig. 2A, Inset), demonstrating that the spin probe at this position is extremely restricted in motion. In contrast, the polarity at position 6 differs only slightly between the free and the bound peptide state (ΔAzz - 0.04 mT), implying that the spin probe at this position is still exposed to the water in the bound state (Fig. 2 and Table S2). The pronounced changes visible in the low-field peak of  between 277 and 160 K indicate that the spectrum is dominated by the spin-label dynamics and not by polarity (Fig. 2B, Inset).
between 277 and 160 K indicate that the spectrum is dominated by the spin-label dynamics and not by polarity (Fig. 2B, Inset).
Fig. 2.

Polarity of the spin probe microenvironment. The 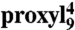 (A) and
(A) and  (B) peptides free in solution (black, 30 μM) or bound to TAP (10 μM, red) were shock frozen in liquid nitrogen and the spectra were recorded at 160 K. For
(B) peptides free in solution (black, 30 μM) or bound to TAP (10 μM, red) were shock frozen in liquid nitrogen and the spectra were recorded at 160 K. For 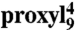 , the spectrum after addition of 165 molar excess unlabeled competitor is also shown. The dotted lines represent simulated spectra fitted to the experimental ones using the program DIPFIT (25). The 2Azz separation between the low- and high-field peaks is indicated by vertical dotted lines. As determined by CW EPR spectra at 277 K (gray spectra in the insets), the fraction of free peptide was smaller than 5%. The two insets also show the effect of lowering the temperature on the spectral shape of the bound peptides.
, the spectrum after addition of 165 molar excess unlabeled competitor is also shown. The dotted lines represent simulated spectra fitted to the experimental ones using the program DIPFIT (25). The 2Azz separation between the low- and high-field peaks is indicated by vertical dotted lines. As determined by CW EPR spectra at 277 K (gray spectra in the insets), the fraction of free peptide was smaller than 5%. The two insets also show the effect of lowering the temperature on the spectral shape of the bound peptides.
Unlike the proxyl-labeled peptides, the TOAC spin probes at positions 4 and 6 were found to be water accessible in both free and bound states (Table S2), demonstrating that the proxyl and TOAC spin probes can be accommodated into slightly different binding pockets at peptide position 4. The very similar Azz values detected for 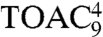 and
and 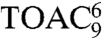 at 160 K confirm that the spectral changes observed at 277 K again depend only on dynamics. In summary, the data revealed that upon TAP association both the backbone and the side chain at position 6 of the peptide are water exposed, whereas the water accessibility at position 4 is sampled differently by the proxyl and TOAC probes.
at 160 K confirm that the spectral changes observed at 277 K again depend only on dynamics. In summary, the data revealed that upon TAP association both the backbone and the side chain at position 6 of the peptide are water exposed, whereas the water accessibility at position 4 is sampled differently by the proxyl and TOAC probes.
Conformation of Bound Peptides.
The high mobility detected in the central region of the bound 9-mer peptide poses the question as to which conformation of the peptide can allow such flexibility. Doubly spin-labeled variants of the 9-mer epitope were engineered in order to obtain interspin distances from which the conformation of the bound peptide could be revealed. One proxyl label was placed at position 8 and the second at positions 2, 4, or 6. The interspin distances were determined by CW EPR spectra recorded at 160 K, sensitive in the 0.8–2-nm range, and by double electron electron resonance (DEER) at 50 K, which for most applications is sensitive in the 1.7–6-nm range.
The free 9-mer peptide labeled at positions 2 and 8 (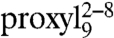 ) displayed a broad interspin distance distribution centered at 2.1 nm (FWHM 1 nm). Upon binding to TAP, the interspin distance distribution was found to be narrowed (FWHM 0.6 nm) and centered at 2.2 nm (Fig. 3A). The CW EPR spectrum detected on the same sample at 277 K showed 97% of spin-labeled peptide bound to TAP (Fig. 3A, Upper, Inset), therefore the narrow 2.2-nm distance describes the bound conformation.
) displayed a broad interspin distance distribution centered at 2.1 nm (FWHM 1 nm). Upon binding to TAP, the interspin distance distribution was found to be narrowed (FWHM 0.6 nm) and centered at 2.2 nm (Fig. 3A). The CW EPR spectrum detected on the same sample at 277 K showed 97% of spin-labeled peptide bound to TAP (Fig. 3A, Upper, Inset), therefore the narrow 2.2-nm distance describes the bound conformation.
Fig. 3.

Interspin distances in the 9- and 15-mer peptides bound to TAP. (A) DEER analysis of 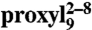 free in solution (black, 30 μM) and bound to TAP (red, ∼15 μM). (Upper, Left) Intensity normalized DEER time traces V(t) and exponentially decaying background signals arising from a 3D distribution of remote spins (black dotted lines). The inset shows the CW EPR spectrum of the peptide bound to TAP detected at 277 K (red), with the spectrum of the 3% residual free peptide (light gray) superimposed. (Lower, Left) Background corrected experimental data F(t) and fit by a Gaussian distribution of distances (black dotted lines). (Right) Distance distribution P(r) obtained with DeerAnalysis 2009. The inset shows the spin-normalized CW EPR spectra detected at 160 K and the DIPFIT simulations (dotted) for the three doubly labeled 9-mer peptides. (B) Analogous DEER analysis of
free in solution (black, 30 μM) and bound to TAP (red, ∼15 μM). (Upper, Left) Intensity normalized DEER time traces V(t) and exponentially decaying background signals arising from a 3D distribution of remote spins (black dotted lines). The inset shows the CW EPR spectrum of the peptide bound to TAP detected at 277 K (red), with the spectrum of the 3% residual free peptide (light gray) superimposed. (Lower, Left) Background corrected experimental data F(t) and fit by a Gaussian distribution of distances (black dotted lines). (Right) Distance distribution P(r) obtained with DeerAnalysis 2009. The inset shows the spin-normalized CW EPR spectra detected at 160 K and the DIPFIT simulations (dotted) for the three doubly labeled 9-mer peptides. (B) Analogous DEER analysis of 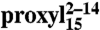 free in solution (black, 30 μM) and bound to TAP (red, ∼15 μM). (Right, Inset) Background corrected DEER trace detected for
free in solution (black, 30 μM) and bound to TAP (red, ∼15 μM). (Right, Inset) Background corrected DEER trace detected for 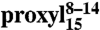 bound to TAP with the fit and the obtained distance distribution centered at 1.7 nm. The model-free Tikhonov analysis for the 9- and 15-mer peptides is presented in the Fig. S4.
bound to TAP with the fit and the obtained distance distribution centered at 1.7 nm. The model-free Tikhonov analysis for the 9- and 15-mer peptides is presented in the Fig. S4.
Additionally, distances were determined for all three 9-mer doubly labeled peptides by analysis of the line broadening of the CW EPR spectra recorded at 160 K. The EPR spin-normalized spectra of the three doubly labeled 9-mer peptides bound to TAP show a gradual decrease in intensity with the second spin label moving closer to the spin label at position 8, indicative of increased dipolar coupling because of shorter distances (Fig. 3A, Right, Inset). Fitting of simulated spectra to the experimental ones gave interspin distances of 2.1, 1.9, and 1.4 nm for the positions 2–8, 4–8, and 6–8, respectively (Table S3). The interspin distances in 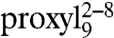 determined by continuous wave and pulsed techniques are in agreement showing the accuracy of both methods.
determined by continuous wave and pulsed techniques are in agreement showing the accuracy of both methods.
To generalize these findings, based on the fact that TAP preferentially binds peptides with a length of 8–16 residues with similar affinity (6), doubly labeled variants of the 15-mer peptides were also engineered and interspin distances investigated by DEER. A broad interspin distance distribution centered at 2.7 nm (FWHM 2.5 nm) was found between positions 2 and 14 in the free state (Fig. 3B). Upon binding to TAP, the distance decreased to 2.0 nm (FWHM 1.5 nm). Integration of the CW EPR spectrum recorded on the same sample at 277 K showed that 93% of 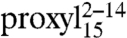 was bound to TAP (Fig. 3B, Upper, Inset). An interspin distance of 1.7 nm (FWHM 1.8 nm) was also determined by DEER for
was bound to TAP (Fig. 3B, Upper, Inset). An interspin distance of 1.7 nm (FWHM 1.8 nm) was also determined by DEER for 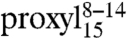 (Fig. 3B, Right, Inset).
(Fig. 3B, Right, Inset).
Based on the fact that the interspin distances between spin probes attached next to the 9- and 15-mer peptide anchoring sites (positions 2 and N-1) are found to be very similar and do not depend on the peptide length, specific binding sites for the N- and C-terminal residues of any peptide in the binding pocket of TAP are suggested.
Discussion
The spatial arrangement of the peptide-binding pocket and the structure of bound peptides are required to understand the mechanism of epitope selection and TAP-dependent antigen translocation in adaptive immunity. The lack of a high-resolution structure of an ABC exporter in complex with its substrate called for the use of alternative biophysical techniques, in this case EPR, to get information about the structure and conformational dynamics of the bound substrate. By the concomitant use of proxyl and TOAC spin-labeled peptides, the dynamics and polarity of the side chains and of the peptide backbone could be disentangled. Additionally, with the help of a set of doubly spin-labeled 9- and 15-mer peptides, intrapeptide distances could be derived to propose the conformation of the peptide in the TAP binding pocket.
The N- and C-terminal positions are highly restricted in motion upon TAP association. This immobilization could result from pockets in the TAP binding site, which define the side-chain specificity. Because the backbone is also immobile at both termini, we propose additional stabilization of the peptide backbone induced by TAP. Spin probes at positions 3, 5, and 6 show the highest mobility for the side chain as well as for the backbone. Because positions 5 and 6 also have the highest variability in peptide sequence (13), we speculate that these side chains stick out of the binding pocket and are not in contact with TAP. This assumption is supported by the polar microenvironment of residue 6, which does not significantly change between the free and bound state. Additionally, from the mobility data obtained with the TOAC reporter molecule at position 6, we also suggest a loose contact of the backbone with TAP. The high flexibility at position 3 of the peptide is unexpected because this position is involved in peptide recognition. However, labeling position 3 with TOAC or proxyl drastically increased the peptide dissociation constant that was also observed after labeling with fluorescein or a small chemical protease (9, 17). Therefore, we assume a disturbed peptide conformation in which position 3 is no longer in contact with the binding pocket, and thus it is characterized by a higher mobility of the spin probe. The mobility at position 4 is strongly restricted and comparable with the mobility of the N- and C-terminal residues. The reduction in mobility seems to result from backbone as well as side-chain contacts. Interestingly, the TOAC probe is water accessible, whereas the proxyl side chain at the same position is shielded and seems to be buried in the binding site. Despite this strong mobility restriction, this position is not involved in peptide specificity as reflected by the high variability of amino acids and the tolerance of bulky modifications at this position (9, 13, 17). Therefore, we propose peptide stabilization by backbone interactions and an immobilization of bulky side chains by spatial constraints in the binding pocket of TAP.
To derive information on the structure of the peptide bound to TAP based on the interspin distances experimentally obtained, we analyzed in silico the orientational freedom of two proxyl labels at positions 2 and N-1 in peptides of different lengths in an extended structure in vacuum (Fig. 4A). In  , the labels can reorient to result in an interspin distance of 2.0 nm. In contrast, the simulated distance of
, the labels can reorient to result in an interspin distance of 2.0 nm. In contrast, the simulated distance of  peaks at 4.5 nm. The simulations clearly suggest that the formation of an internal kink in the longer peptide would fit the experimentally observed 2-nm distance between positions 2 and N-1. An α-helical conformation for the 9- and 15-mer was also tested in silico, and we found that it agrees equally with the experimental values. However such a conformation in the binding pocket of TAP can be excluded for several reasons: (i) Peptide library studies showed that a proline residue in the body of the peptide does not disturb peptide binding, although it functions as a helix breaker (13); (ii) there is no preference for peptides containing amino acids with high propensity to form an α-helix; and (iii) in an α-helical conformation, the relative orientation of the anchoring side chains would change according to the helical pattern. Thus, the binding affinities should display a periodic pattern, whereas in fact peptides of 8–16 amino acids in length have the same affinity (6).
peaks at 4.5 nm. The simulations clearly suggest that the formation of an internal kink in the longer peptide would fit the experimentally observed 2-nm distance between positions 2 and N-1. An α-helical conformation for the 9- and 15-mer was also tested in silico, and we found that it agrees equally with the experimental values. However such a conformation in the binding pocket of TAP can be excluded for several reasons: (i) Peptide library studies showed that a proline residue in the body of the peptide does not disturb peptide binding, although it functions as a helix breaker (13); (ii) there is no preference for peptides containing amino acids with high propensity to form an α-helix; and (iii) in an α-helical conformation, the relative orientation of the anchoring side chains would change according to the helical pattern. Thus, the binding affinities should display a periodic pattern, whereas in fact peptides of 8–16 amino acids in length have the same affinity (6).
Fig. 4.

Coevolution of TAP and MHC class I with regard to the conformation of the bound antigenic peptide. (A, Left) Isolated peptides (15-, 9-, and 7-mer) in an extended structure spin-labeled in silico with iodoacetamido-proxyl probes at positions 2 and N-1. The resulting spin-label rotamers are represented in ball and stick models, with the position of each NO group highlighted by a violet cloud. (Right) Comparison between experimental (violet) and simulated (gray) interspin distance distributions. (B, Left) In silico spin-labeled peptides bound to MHC class I proteins. The 13-mer peptide bound to rat MHC class I RT1-A [Protein Data Bank (PDB) ID 1ED3] and two peptides (9- and 10-mer) crystallized with HLA-B27 (PDB ID 2BSR, 2BSS) were spin-labeled at position 2 and N-1. (Right) Comparison between experimental (violet) and simulated (gray) interspin distance distributions.
Based on these considerations, we propose an extended kinked structure for the bound peptides in the TAP binding pocket, with an extrusion that becomes more evident and is a prime feature for longer peptides. Such an extrusion would also be in agreement with the measured 1.7-nm distance between positions 8 and 14 of the 15-mer peptide. Together with high water accessibility of position 6 of the 9-mer peptide, it is tempting to speculate that these peptide extrusions bulge out of the TAP binding pocket or are arranged in a large water-filled cavity, as is observed for the periplasmic peptide-binding protein OppA of the peptide translocase of Lactococcus lactis (20).
Remarkably, the distance distribution between the labels of the 15-mer peptides is larger than that obtained in the 9-mer, which could reflect the mobility of the extrusion with the shorter peptides being less dynamic because they are clamped between both binding sides. The drop in affinity of longer peptides could result from the high mobility of the bulging residues. The minimal length of peptides recognized by TAP is restricted to 8-mer peptides (6, 7). Importantly, the simulated spin-label distance between positions 2 and 7 of an 8-mer extended peptide still fits the experimental 2-nm distance, whereas the distance between equivalent residues in a 7-mer peptide is smaller than 2 nm, thus it does not conform to the minimal required length (Fig. 4A). Therefore, we postulate that the pockets for the anchor residues (1 and N) in TAP are separated by approximately 2.5 nm, which can only be bridged by peptides of 8 residues and longer.
Strikingly, this extended structure of peptides bound to TAP is reminiscent of the structure of peptides bound to MHC class I molecules. In general, the reported structures show the bound peptides to be anchored at the ends of the MHC class I binding groove (so-called A and F pockets) via their N and C termini, respectively (21). The distance of ∼2.0–2.5 nm from the A to F pockets is consistent with the distance detected here between the second and penultimate positions of the TAP-bound peptides. To further exploit this correlation, the experimental distance constraints obtained by EPR for peptides bound to TAP were compared with the expected distances in crystal structures of peptides bound to MHC I molecules (Fig. 4B). The peptides were in silico spin labeled with the proxyl label, and the interspin distances between positions 2 and N-1 simulated taking into account the steric contacts with the residues of the protein binding pocket. We selected two structures of peptides bound to HLA-B27, the allele, which also binds the 9-mer peptide used in this study (22), and of an extra-long 13-mer peptide bound to rat MHC class I RT1-A, which provided evidence that the central residues between the A- and F-bound termini are highly exposed and bulged (23). The good agreement of the simulated DEER traces and distance distributions of the MHC class I bound peptides with the experimental data of TAP-bound peptides confirms the similarities between the conformations of the peptides bound to the two classes of proteins (Fig. 4B). It is noteworthy that the spin label at position 2 in all crystallized peptides bound to MHC is more sterically hindered than at position 8 (having fewer rotamers populated; see Fig. 4B, Left) and the same was observed based on the mobility analysis also for the spin-labeled 9-mer peptide in this study. Interestingly, the coevolution of TAP and MHC class I molecules seems to be reflected not only in the same preference for the C-terminal anchor residues and the phylogenetic appearance, but also in the conformation of the bound peptides, despite the absence of structural homology between ABC exporters and MHC class I molecules. In summary, this study reports a remarkable coevolution of antigen recognition of unrelated but key machineries in antigen processing by selecting epitopes based on overlapping conformation and anchor positions.
Materials and Methods
Expression and Purification of TAP.
Human TAP containing a His10 tag at the C terminus of TAP1 was expressed with the baculovirus expression system in Sf9 insect cells or was expressed in Pichia pastoris. Preparation of crude membranes and TAP purification using digitonin as detergent were performed as described (24). After purification, TAP was stored at -80 °C in digitonin buffer (20 mM Hepes, 140 mM NaCl, 15% glycerol, 0.1% digitonin, pH 7.4) containing 200 mM histidine.
EPR of Spin-Labeled Peptides.
Spin labeling of peptides is described in SI Text. All experiments were performed at X-band frequencies (9.3–9.4 GHz) with a Bruker Elexsys 580 spectrometer equipped with a Bruker Elexsys super high sensitive probe head (CW EPR) or a Bruker Flexline split-ring resonator ER 4118X-MS3 (pulsed EPR) using a continuous flow He cryostat (ESR900; Oxford Instruments) controlled by an Oxford Instruments temperature controller ITC 503S.
Fitting of simulated dipolar broadened EPR powder spectra to the experimental ones detected at 160 K was performed with the software DIPFIT according to the method described previously (25). The line width parameters for the fitting of the doubly labeled peptides were determined by fitting of spectra of singly labeled peptides in buffer or bound to TAP.
Dipolar time evolution data were acquired using the four-pulse DEER experiment (26). Data analysis of the DEER traces was performed with the software DeerAnalysis 2009 (27). Further details are provided in SI Text. The simulation of the possible spin-label rotamers attached at positions 2 and N-1 peptides was performed using the Matlab program package MMM (freely available at http://www.epr.ethz.ch/software/index) based on a rotamer library approach (28). Further details are provided in SI Text. The peptides were built in a fully extended structure or α-helical conformation using the software B (http://casegroup.rutgers.edu/).
Supplementary Material
Acknowledgments.
E.B. especially thanks G. Jeschke for stimulating discussions. The German Research Foundation (SFB807—Transport and Communication Across Biological Membranes, TA157/7 and AB149/1) as well as the European Drug Initiative on Channels and Transporters funded by the European Council Seventh Framework Program supported the work of R.A. and R.T.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012355108/-/DCSupplemental.
References
- 1.Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev. 2005;207:145–157. doi: 10.1111/j.0105-2896.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 2.Schmitt L, Tampé R. Structure and mechanism of ABC transporters. Curr Opin Struct Biol. 2002;12:754–760. doi: 10.1016/s0959-440x(02)00399-8. [DOI] [PubMed] [Google Scholar]
- 3.Hollenstein K, Dawson RJP, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Koch J, Guntrum R, Heintke S, Kyritsis C, Tampé R. Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP) J Biol Chem. 2004;279:10142–10147. doi: 10.1074/jbc.M312816200. [DOI] [PubMed] [Google Scholar]
- 5.Nijenhuis M, Hammerling GJ. Multiple regions of the transporter associated with antigen processing (TAP) contribute to its peptide binding site. J Immunol. 1996;157:5467–5477. [PubMed] [Google Scholar]
- 6.van Endert PM, et al. A sequential model for peptide binding and transport by the transporters associated with antigen processing. Immunity. 1994;1:491–500. doi: 10.1016/1074-7613(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 7.Koopmann JO, Post M, Neefjes JJ, Hammerling GJ, Momburg F. Translocation of long peptides by transporters associated with antigen processing (TAP) Eur J Immunol. 1996;26:1720–1728. doi: 10.1002/eji.1830260809. [DOI] [PubMed] [Google Scholar]
- 8.Uebel S, et al. Requirements for peptide binding to the human transporter associated with antigen-processing revealed by peptide scans and complex peptide libraries. J Biol Chem. 1995;270:18512–18516. doi: 10.1074/jbc.270.31.18512. [DOI] [PubMed] [Google Scholar]
- 9.Neumann L, Tampé R. Kinetic analysis of peptide binding to the TAP transport complex: evidence for structural rearrangements induced by substrate binding. J Mol Biol. 1999;294:1203–1213. doi: 10.1006/jmbi.1999.3329. [DOI] [PubMed] [Google Scholar]
- 10.Gromme M, et al. The rational design of TAP inhibitors using peptide substrate modifications and peptidomimetics. Eur J Immunol. 1997;27:898–904. doi: 10.1002/eji.1830270415. [DOI] [PubMed] [Google Scholar]
- 11.Neumann L, Abele R, Tampé R. Thermodynamics of peptide binding to the transporter associated with antigen processing (TAP) J Mol Biol. 2002;324:965–973. doi: 10.1016/s0022-2836(02)01148-8. [DOI] [PubMed] [Google Scholar]
- 12.Reits EA, Vos JC, Gromme M, Neefjes J. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature. 2000;404:774–778. doi: 10.1038/35008103. [DOI] [PubMed] [Google Scholar]
- 13.Uebel S, et al. Recognition principle of the TAP transporter disclosed by combinatorial peptide libraries. Proc Natl Acad Sci USA. 1997;94:8976–8981. doi: 10.1073/pnas.94.17.8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heemels MT, Schumacher TN, Wonigeit K, Ploegh HL. Peptide translocation by variants of the transporter associated with antigen processing. Science. 1993;262:2059–2063. doi: 10.1126/science.8266106. [DOI] [PubMed] [Google Scholar]
- 15.Momburg F, et al. Selectivity of MHC-encoded peptide transporters from human, mouse and rat. Nature. 1994;367:648–651. doi: 10.1038/367648a0. [DOI] [PubMed] [Google Scholar]
- 16.van Endert PM, et al. The peptide-binding motif for the human transporter associated with antigen processing. J Exp Med. 1995;182:1883–1895. doi: 10.1084/jem.182.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herget M, et al. Mechanism of substrate sensing and signal transmission within an ABC transporter: Use of a trojan horse strategy. J Biol Chem. 2007;282:3871–3880. doi: 10.1074/jbc.M608480200. [DOI] [PubMed] [Google Scholar]
- 18.Wright K, et al. 4-Amino-1-oxyl-2,2,6,6-tetramethylpiperidine-3-carboxylic acid ([beta]-TOAC), the first spin-labelled, cyclic, chiral [beta]-amino acid resolved in an enantiomerically pure state. Tetrahedron Lett. 2003;44:3381–3384. [Google Scholar]
- 19.van Endert PM, et al. The peptide-binding motif for the human transporter associated with antigen processing. J Exp Med. 1995;182:1883–1895. doi: 10.1084/jem.182.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berntsson RP, et al. The structural basis for peptide selection by the transport receptor OppA. EMBO J. 2009;28:1332–1340. doi: 10.1038/emboj.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madden DR. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol. 1995;13:587–622. doi: 10.1146/annurev.iy.13.040195.003103. [DOI] [PubMed] [Google Scholar]
- 22.Stewart-Jones Guillame BE, et al. Crystal structures and KIR3DL1 recognition of three immunodominant viral peptides complexed to HLA-B*2705. Eur J Immunol. 2005;35:341–351. doi: 10.1002/eji.200425724. [DOI] [PubMed] [Google Scholar]
- 23.Speir JA, Stevens J, Joly E, Butcher GW, Wilson IA. Two different, highly exposed, bulged structures for an unusually long peptide boundto rat MHC class I RT1-Aa. Immunity. 2001;14:81–92. doi: 10.1016/s1074-7613(01)00091-7. [DOI] [PubMed] [Google Scholar]
- 24.Herget M, et al. Purification and reconstitution of the antigen transport complex TAP: A prerequisite for determination of peptide stoichiometry and ATP hydrolysis. J Biol Chem. 2009;284:33740–33749. doi: 10.1074/jbc.M109.047779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinhoff HJ, et al. Determination of interspin distances between spin labels attached to insulin: Comparison of electron paramagnetic resonance data with the x-ray structure. Biophys J. 1997;73:3287–3298. doi: 10.1016/S0006-3495(97)78353-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. Dead-time free measurement of dipole-dipole interactions between electron spins. J Magn Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- 27.Jeschke G, et al. DeerAnalysis2006–a comprehensive software package for analyzing pulsed ELDOR data. Appl Magn Reson. 2006;30:473–498. [Google Scholar]
- 28.Polyhach Y, et al. Rotamer libraries of spin labelled cysteines for protein studies. Phys Chem Chem Phys. 2011 doi: 10.1039/c0cp01865a. 10.1039/C0CP01865A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.