

Abstract
Oxo-iron(IV) species are implicated as key intermediates in the catalytic cycles of heme and nonheme oxygen activating iron enzymes that selectively functionalize aliphatic C–H bonds. Ferryl complexes can exist in either quintet or triplet ground states. Density functional theory calculations predict that the quintet oxo-iron(IV) species is more reactive toward C–H bond activation than its corresponding triplet partner, however; the available experimental data on model complexes suggests that both spin multiplicities display comparable reactivities. To clarify this ambiguity, a detailed electronic structure analysis of alkane hydroxylation by an oxo-iron(IV) species on different spin-state potential energy surfaces is performed. According to our results, the lengthening of the Fe–oxo bond in ferryl reactants, which is the part of the reaction coordinate for H-atom abstraction, leads to the formation of oxyl-iron(III) species that then perform actual C–H bond activation. The differential reactivity stems from the fact that the two spin states have different requirements for the optimal angle at which the substrate should approach the (FeO)2+ core because distinct electron acceptor orbitals are employed on the two surfaces. The H-atom abstraction on the quintet surface favors the “σ-pathway” that requires an essentially linear attack; by contrast a “π-channel” is operative on the triplet surface that leads to an ideal attack angle near 90°. However, the latter is not possible due to steric crowding; thus, the attenuated orbital interaction and the unavoidably increased Pauli repulsion result in the lower reactivity of the triplet oxo-iron(IV) complexes.
Keywords: density functional calculation, nonheme iron, reaction mechanism
Oxo-iron(IV) intermediates have attracted much interest in bioinorganic chemistry because they are implicated as key intermediates in the catalytic cycles of heme and nonheme oxygen activating iron enzymes that selectively functionalize unactivated C–H bonds (1). Detailed experimental and theoretical studies on the hydroxylation of saturated hydrocarbons by ferryl species in heme systems, foremost cytochrome P450, have been performed (2). On the other hand a number of nonheme enzymes are able to activate molecular dioxygen to modify alkane or arene substrates as well. So far, nonheme ferryl species have been spectroscopically characterized in four mononuclear iron enzymes and were found to feature high-spin (HS) (S = 2) electronic ground state configurations (3). In parallel, a wide range of synthetic Fe(IV) = O complexes were synthesized and characterized (4). In almost all cases they contain intermediate-spin (IS) (S = 1) rather than HS ferryl centers (4). The only exceptions are [Fe(IV)(O)(H2O)5]2+ (5) and the recently reported model complex [Fe(IV)(O)(TMG3tren)]2+ (TMG3tren = N[CH2CH2N = C(NMe2)2]) (6). Presumably because the (FeO)2+ core is sheltered by the sterically bulky supporting ligand in [Fe(IV)(O)(TMG3tren)]2+, its reactivity toward C–H bond cleavage is only comparable with triplet ferryl analogues (7). Recently, an unmasked HS Fe(IV) = O unit was identified in a valence-localized open core diiron (Fe(III)/Fe(IV)) complex (8). The reaction of the C–H bond cleavage by this complex turned out to be approximately 100 times faster than the corresponding mononuclear IS oxo-iron(IV) complex (8). This experimental finding is in accord with the theoretical prediction that quintet ferryl species are more aggressive oxidants than the corresponding triplet counterparts (9, 10, 11). However, there is a long-term debate on how to rationalize the differential reactivity between quintet and triplet oxo-iron(IV) species. De Visser, Shaik, and coworkers argued that the enhanced exchange interaction upon approaching the transition state (TS) on the quintet surface flattens the potential energy surface (PES) and hence lowers the barrier (12, 13). Baerends, Solomon, and coworkers proposed that the greater degree of exchange stabilization in the HS ferryl reactant relative to the IS analogues significantly stabilizes the Fe-dz2 based σ-antibonding molecular orbital (MO) (14–18). Therefore, this orbital is able to serve as the electron acceptor on the quintet surface, whereas the energy of the corresponding MO in the triplet reactant is too high to be an electron acceptor. Thus, the lower energy π-antibonding MOs that mainly consist of the Fe-dxz/yz and the O-px/y fragment orbitals act as electron acceptors on the triplet surface (17). As a consequence, a σ-pathway is often suggested to be operative on the quintet surface, whereas the triplet reaction proceeds via a π-channel (18). However, under certain circumstances quintet π- and triplet σ-pathways were also reported in the process of C–H bond activation (19, 20). Recent studies on the reaction of alkane hydroxylation by ferryl model compounds demonstrated that the reaction can take place through both the σ- and the π-pathways on the quintet and triplet surfaces with barrier heights displaying the order 5σ > 5π ≈ 3π > 3σ (21). The results indicated that quintet oxo-iron(IV) species are not always more reactive toward C–H bond cleavage than the corresponding triplet species and confirmed that of all possible pathways the quintet σ-pathway is the most effective one. More importantly, a careful analysis of the electronic structure changes along the quintet pathway revealed that the reaction requires a preparatory stage in which an oxyl-iron(III) species is formed en route to the TS that then functions as the active species in the actual C–H bond activation process (22). Thus far all interpretations about which spin multiplicity is more reactive toward C–H bond activation have been based on the notion that the “real” oxidant is oxo-iron(IV) rather than oxyl-iron(III). Therefore, to address this question one needs to understand how the real oxyl-ferric reactant is formed and how it then reacts with the substrate on the quintet and triplet surfaces, respectively.
In the present contribution we address the above formulated problem through an analysis of the electronic structure changes that occur upon alkane C–H bond hydroxylation by the taurine:α-ketoglutarate dioxygenase (TauD) oxo-iron(IV) intermediate (23) along the septet (hypothetical), quintet, and triplet reaction channels. This allows us to gain electronic structure insight into the differential reactivity of the distinct spin states and draw conclusions that might have a wider range of applicability.
Results
The energy profile for H-atom abstraction by the TauD oxo-iron(IV) intermediate is shown in Fig. 1. The reaction follows the consensus mechanism for the activation of aliphatic C–H bonds by oxo-iron(IV) compounds (24).The reaction is initiated by the rate-determining H-atom abstraction reaction via TS1 to yield a hydroxo-iron(III) complex with a nearby alkyl radical intermediate (IN). This step is followed by an OH-rebound process leading to the final product ethanol. Interestingly, for H-atom transfer the hypothetical septet reaction pathway is calculated to show the lowest barrier (Fig. 1 and Table 1). However, because the singlet product ethanol cannot be formed on the septet surface, the reaction on the septet surface stops at intermediate 7IN. In addition, the reaction cannot start on the septet surface, because the septet reactant is much higher in energy than the quintet oxo-iron(IV) reactant. In fact, a low-lying septet state is only possible for a mononuclear 3d transition metal center if at least one ligand carries an unpaired electron.
Fig. 1.

The energy profile of H-atom abstraction from ethane by the TauD oxo-iron(IV) intermediate (B3LYP/def2-TZVPP).
Table 1.
The relative enthalpy, entropy, and free energy for the key local minima and transition states on the septet, quintet, and triplet surface
| ΔH (kcal/mol) | −TΔS kcal/mol) | ΔG (kcal/mol) | |
| 5R | 0 | 0 | 0 |
| 5TS1 | 14.0 | 10.6 | 24.6 |
| 5IN | 0.9 | 9.0 | 9.8 |
| 7R | 16.3 | −1.3 | 15.1 |
| 7TS1 | 19.1 | 10.0 | 29.0 |
| 7IN | 2.7 | 7.8 | 10.5 |
| 3R | 6.8 | 2.4 | 9.2 |
| 3TS1 | 28.5 | 13.7 | 42.2 |
| 3IN | 14.1 | 8.9 | 23.0 |
Septet.
Due to the effective C4v symmetry of the complex, the lowest-energy septet state of the (FeO)2+ core is 7E with the orbital occupation pattern (σ(O-pz))2(π(O-px,y))3(Fe-dxy)1(π∗(Fe-dxz,yz))2(σ∗(Fe-dx2-y2))1(σ∗(Fe-dz2))1 (Fig. 2, Left). This corresponds to a HS ferric center (SFe = 5/2) that is ferromagnetically coupled to an oxyl-radical (SO = 1/2) thereby yielding an overall septet state. Relative to the quintet ground state of the (FeO)2+ core (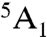 ), the septet state represents an oxo-to-iron π(O-px,y) → σ∗(Fe-dz2) charge transfer excited state that is accompanied by a spin flip. The orbital occupation pattern implies a Fe–Ooxobond order of 1 compared to a Fe–Ooxo bond order of 2 that exists in the quintet ground state. The reduction in the bond order results in a significant lengthening of the Fe–Ooxo bond (1.62 Å in 5(FeO)2+ vs 1.91 Å in 7(FeO)2+). B3LYP calculations predict the septet species to be ∼16 kcal/mol higher in energy than the corresponding quintet ferryl complex. Hence, one can safely rule out the possibility that the septet species can be trapped as an intermediate in enzymes or model systems. However, among the three alternative spin-state surfaces, the septet species is the most reactive toward H-atom abstraction. Thus, a discussion of its electronic structure still provides valuable clues to understanding the reaction mechanism on the other two spin-state surfaces that will be investigated below.
), the septet state represents an oxo-to-iron π(O-px,y) → σ∗(Fe-dz2) charge transfer excited state that is accompanied by a spin flip. The orbital occupation pattern implies a Fe–Ooxobond order of 1 compared to a Fe–Ooxo bond order of 2 that exists in the quintet ground state. The reduction in the bond order results in a significant lengthening of the Fe–Ooxo bond (1.62 Å in 5(FeO)2+ vs 1.91 Å in 7(FeO)2+). B3LYP calculations predict the septet species to be ∼16 kcal/mol higher in energy than the corresponding quintet ferryl complex. Hence, one can safely rule out the possibility that the septet species can be trapped as an intermediate in enzymes or model systems. However, among the three alternative spin-state surfaces, the septet species is the most reactive toward H-atom abstraction. Thus, a discussion of its electronic structure still provides valuable clues to understanding the reaction mechanism on the other two spin-state surfaces that will be investigated below.
Fig. 2.

Schematic MO diagrams for the septet reactant (Left) and 7TS1 (Right).
The geometry of 7TS1 exhibits a slight contraction of the Fe–Ooxo bond relative to the reactant (1.89 Å in 7TS1 (Table 2) vs. 1.91 Å in the reactant). The electronic structure of 7TS1 is best interpreted as featuring a HS ferric ion (SFe = 5/2) interacting with a three-center C–H-O σ-radical (SCHO = 1/2) in a ferromagnetic fashion (Fig. 2, Right). In comparison to the electronic structure of the reactant, one may envision that during the approach of the substrate toward the 7(FeO)2+ core, the singly occupied O-px orbital interacts with the C–H σ-bonding orbital in the substrate to form a pair of three-centered MOs (σCHO and  ). Because the electron acceptor for H-atom abstraction is a π-bonding orbital (O-px), the septet reaction mechanism employs a π-pathway.
). Because the electron acceptor for H-atom abstraction is a π-bonding orbital (O-px), the septet reaction mechanism employs a π-pathway.
Table 2.
The important geometric features of the key local minima and transition states on the septet, quintet, and triplet surfaces
| 7TS1 | 7IN | 5TS1 | 5IN | 3TS1 | 3IN | |
| O–Fe (Å) | 1.890 | 1.847 | 1.765 | 1.838 | 1.756 | 1.793 |
| O–H (Å) | 1.279 | 0.981 | 1.224 | 0.986 | 1.164 | 0.987 |
| C–H (Å) | 1.262 | 2.368 | 1.289 | 2.231 | 1.388 | 2.442 |
| Fe–O–H (degree) | 113.1 | 123.0 | 165.0 | 122.0 | 119.2 | 107.0 |
| O–H–C (degree) | 178.7 | — | 177.9 | — | 173.5 | — |
After passing through the very low barrier for H-atom abstraction, the system evolves to a HS Fe(III) (SFe = 5/2) center that is ferromagnetically coupled to a substrate alkyl radical (SC = 1/2). At this point the first electron transfer has been accomplished. However, ferromagnetic coupling between the HS ferric center and the substrate radical renders the second electron transfer impossible; thus, the final products cannot be produced on the septet surface.
Quintet.
The most noticeable geometric feature in 5TS1 is a fairly long Fe–Ooxo bond in comparison with that in the reactant (1.76 Å in 5TS1 (Table 2) vs. 1.62 Å in the reactant). As shown in the left column of Fig. 3, the electronic structure of 5TS1 is best interpreted as a HS Fe(III) ion (SFe = 5/2) antiferromagnetically coupled to a three-center C–H–O radical (SCHO = 1/2). Unexpectedly, the Fe-dz2 based MO is the bonding combination between the Fe-dz2 and the O-pz fragment orbitals rather than the antibonding combination that would be expected from the electronic structure of the quintet oxo-iron(IV) reactant. As discussed elsewhere (22), this signifies that dramatic changes in the electronic structure of the quintet ferryl species must have occurred as the Fe–Ooxo bond is lengthened to approach 5TS1. The evolution of the electronic structure of the quintet oxo-iron(IV) complex was investigated more closely through a relaxed surface scan at a series of fixed Fe–Ooxo bond distances (Fig. 3, Right). When the Fe–Ooxo bond is elongated to 1.80 Å similar to that found in 5TS1, the σ-bonding MO that is mainly of O-pz character at the equilibrium geometry splits into a spin-coupled pair. One can identify that the MO in the spin-up manifold becomes Fe-dz2 based (Fe 61% vs O 36%) and that the MO in the spin-down set acquires more O-pz character (Fe 24% vs O 68%). Thus, the elongation of the Fe–Ooxo bond leads to a crossing of the ground state [best described as oxo-iron(IV)] with a state that is best characterized as a HS ferric center (SFe = 5/2) antiferromagnetically coupled to an oxyl-radical (SO = 1/2). Accordingly, the lowest unoccupied molecular orbital in the spin-up manifold that is the Fe-dz2 based σ*-orbital at the equilibrium geometry evolves to an O-pz based orbital (Fig. 3, Right). As the Fe–Ooxo bond is lengthened, the energy of this orbital is significantly lowered from -2.4 eV (Fe–Ooxo 1.65 Å) to -3.0 eV (Fe–Ooxo 1.80 Å). Moreover, the O-pz based orbital overlaps more efficiently with the electron donor σC-H than the Fe-dz2 based orbital that has limited O-pz character. Therefore, the oxyl species that is generated possesses higher electrophilicity than the oxo-form. The oxyl oxygen may more easily accept an α-electron from the substrate C–H bond. In this electron transfer process the bonding nature of the Fe-dz2 based MO is retained and the interaction of the singly occupied O-pz orbital with the electron donor affords a pair of three-centered MOs (σCHO and  ). Thus, one may visualize the entire C–H bond activation process as consisting of a preparatory stage [the formation of the oxyl-iron(III) species] and the actual H-atom abstraction that is accomplished by the highly reactive oxyl-radical. It is important to point out that the oxyl-iron(III) species does not represent a minimum on the PES and hence is not a true reaction intermediate. This way of looking at the reaction is simply an approach to rationalize the electronic structure changes that occur en route to the H-atom abstraction TS. As a consequence of this interpretation, the electron acceptor for the quintet σ-pathway is best viewed to be the O-pz based σ*-orbital rather than the Fe-dz2 orbital as proposed before (10, 12, 14). 5IN and 7IN are essentially energetically degenerate, because they have the same orbital occupation pattern except for the different spin coupling between the alkyl-radical and the HS ferric center.
). Thus, one may visualize the entire C–H bond activation process as consisting of a preparatory stage [the formation of the oxyl-iron(III) species] and the actual H-atom abstraction that is accomplished by the highly reactive oxyl-radical. It is important to point out that the oxyl-iron(III) species does not represent a minimum on the PES and hence is not a true reaction intermediate. This way of looking at the reaction is simply an approach to rationalize the electronic structure changes that occur en route to the H-atom abstraction TS. As a consequence of this interpretation, the electron acceptor for the quintet σ-pathway is best viewed to be the O-pz based σ*-orbital rather than the Fe-dz2 orbital as proposed before (10, 12, 14). 5IN and 7IN are essentially energetically degenerate, because they have the same orbital occupation pattern except for the different spin coupling between the alkyl-radical and the HS ferric center.
Fig. 3.

Schematic MO diagrams for 5TS1 (Left) and the evolution of the quintet oxo-iron(IV) species as a function of the Fe–Ooxo bond distance (Right).
Triplet.
Given the rather high barrier for H-atom abstraction, the triplet channel has no catalytic relevance for C–H bond activation. In analogy to 5TS1, an elongated Fe–Ooxo bond was also identified in 3TS1 (see Table 2). The cleaving C–H bond distance in 3TS1 is 0.1 Å longer than that in 5TS1. The relative orientation of the substrate with respect to the Fe–Ooxo core is similar to that in 7TS1 but significantly different from that calculated for 5TS1. As depicted in the left column of Fig. 4, the electronic structure of 3TS1 is best rationalized as consisting of a low-spin (LS) Fe(III) ion (SFe = 1/2) ferromagnetically coupled to a three-center C–H–O radical (SCHO = 1/2). Surprisingly, the Fe-dxz based MO is the bonding combination of the Fe-dxz and O-px fragment orbitals, rather than the expected antibonding combination in the triplet oxo-iron(IV) reactant. This suggests that, like in the quintet pathway, a preparatory stage may also be needed in the triplet reaction channel.
Fig. 4.

Schematic MO diagrams for 3TS1 (Left) and the constraint model of the triplet oxo-iron(IV) species with the Fe–Ooxo bond length at 1.80 Å (Right).
To probe the changes in the electronic structure of the 3(FeO)2+ core as the Fe–Ooxo bond is lengthened, a constrained geometry optimization was performed with a Fe–Ooxo bond distance kept fixed at 1.8 Å. The constraint model is destabilized by 8.1 kcal/mol relative to the triplet reactant. As shown in the right column of Fig. 4, the elongation of the Fe–Ooxo bond results in qualitative changes in the electronic structure. The description changes from an IS ferryl (S = 1) to a LS ferric ion (SFe = 1/2) that is ferromagentically coupled to an oxyl-radical (SO = 1/2). At the same time one of the π-bonding MOs becomes dominantly Fe-dxz in character. Accordingly the corresponding π*-MO changes to an O-px based orbital. Comparison of the electronic structure of the triplet oxyl-ferric species with that of 3TS1 provides a coherent picture on how the first electron transfer from the substrate to the iron center takes place in the triplet reaction channel. Upon approach of 3TS1 the singly occupied O-px orbital, formed by the elongation of the Fe–Ooxo bond, interacts with the σC-H bonding orbital thus yielding a pair of three-centered MOs (σCHO and  ) similar to what is found on the septet surface. Hence, the triplet reaction mechanism employs a π-pathway using the O-px based π*-orbital as the electron acceptor. If no electronic structure rearrangement of the triplet ferryl complex occurred en route to 3TS1, the Fe-dxz based MO would be the antibonding combination of the Fe-dxz and O-px fragmental orbitals in 3TS1. These results confirm the necessity of the preparatory stage to form an oxyl-ferric species that then performs the actual H-atom abstraction.
) similar to what is found on the septet surface. Hence, the triplet reaction mechanism employs a π-pathway using the O-px based π*-orbital as the electron acceptor. If no electronic structure rearrangement of the triplet ferryl complex occurred en route to 3TS1, the Fe-dxz based MO would be the antibonding combination of the Fe-dxz and O-px fragmental orbitals in 3TS1. These results confirm the necessity of the preparatory stage to form an oxyl-ferric species that then performs the actual H-atom abstraction.
Discussion
Formation of Oxyl-Ferric Species on Quintet and Triplet Surfaces.
If the interaction between the iron center and the oxo ligand were dominantly ionic, as is the case for classical Werner-type transition metal complexes, lengthening of the Fe–Ooxo bond would tend to lower the covalency of the Fe-oxo interaction; hence, the bonding MO that is mainly ligand based would acquire more O-p character, and accordingly the magnitude of the O-p fragment orbital in the antibonding partner would be decreased. As a result, if the Fe-3d based antibonding MOs acted as electron acceptors for C–H bond activation, the overlap between the substrate σC-H and the O-p fragment orbitals would gradually decrease upon approaching the TS. This would be counterproductive.
In fact, the lengthening of the Fe–Ooxo bond on the quintet surface leads to the homolytic cleavage of the Fe–oxo σ-bond. In analogy to the elongation of the H–H bond in H2 yielding a singlet diradical, the doubly occupied σ-bonding MO splits into a spin-coupled pair. Due to the incurred exchange stabilization, the α-electron resides on the iron center, whereas the oxygen atom carries the β-electron. As such, the Fe-dz2 based MO in the spin-up manifold is the bonding rather than the antibonding combination as observed in Fig. 3. In line with this discussion, in σCHO and  the interaction between the Fe-dz2 and O-pz fragment orbitals is of bonding nature as well.
the interaction between the Fe-dz2 and O-pz fragment orbitals is of bonding nature as well.
However, on the triplet surface elongation of the Fe–Ooxo bond results in the heterolytic cleavage of the Fe–oxo π-bond. Due to the fact that the Fe–oxo interaction is predominantly covalent, it follows that these two fragments have comparable electronegativity; thus, one cannot determine a priori eventually whether the O-atom obtains two electrons or the iron center acquires two electrons. By inspection of Fig. 4, one can anticipate that upon lengthening of the Fe–Ooxo bond on the triplet surface the iron center will obtain the two electrons in the π-bonding MO (referred to as Fe-dxz in the right column of Fig. 4), whereas the O-atom will receive the electron in the π-antibonding singly occupied molecular orbital (SOMO) (referred to as O-px). This results in the observation that the Fe-dxz based MO is of bonding character and the corresponding antibonding combination is found in σCHO and  in Fig. 4. If the electron distribution patterns were opposite during the heterolysis of the Fe–oxo π-bond, the electron acceptor would be the Fe-dxz based π*-SOMO. Upon approaching the TS, the O-px contribution in this SOMO would be decreased and hence so would be the overlap between the electron donor and the acceptor.
in Fig. 4. If the electron distribution patterns were opposite during the heterolysis of the Fe–oxo π-bond, the electron acceptor would be the Fe-dxz based π*-SOMO. Upon approaching the TS, the O-px contribution in this SOMO would be decreased and hence so would be the overlap between the electron donor and the acceptor.
Intrinsic Electron Acceptors for H-Atom Abstraction.
From the consensus mechanism one is readily aware that the hydroxylation of aliphatic C–H bonds by ferryl complexes is a two-electron oxidation process and that the σC–H bonding orbital serves as the electron donor. However, a number of mechanisms can be envisioned that differ in the acceptor orbital on the reactive metal-oxo unit.
Due to the steric hindrance provided by the ligand framework, the substrate can only interact with the oxo group rather than directly with the iron center. It follows that direct electron transfer from the substrate to the iron center is very unlikely, and that all possible electron transfer pathways (9) have to pass through the O-atom. In other words, during the reaction the O-atom relays the electron from the substrate to the final electron acceptor that is the iron center. Therefore, viable electron acceptor orbitals should contain significant contributions from the O-atom. The Fe-dxy and Fe-dx2-y2 orbitals can be excluded, because they are located in the plane perpendicular to the line of the substrate attack and hence have no significant interactions with the oxo ligand or the substrate. As discussed above, the quintet and triplet reaction pathways require a preparatory stage to produce an oxyl-ferric species. As a consequence, in the relay pathway the oxo group first delivers one of its electrons to the iron center and then accepts an electron from the σC-H orbital. Hence, the oxyl-p orbitals act as intrinsic electron acceptors in the actual H-atom abstraction. More importantly, upon approaching 3,5TS1 the oxyl-p based MOs have lower energy and better overlap with the σC-H orbital than the Fe-dz2 and Fe-dxz,yz based orbitals.
Differential Reactivity of H-Atom Abstraction for Quintet and Triplet Ferryl Species.
To address the question of why the quintet oxo-iron(IV) is more reactive toward C–H bond cleavage than the corresponding triplet species, one may loosely divide the barrier for the H-atom abstraction process into three contributions: (i) the geometric rearrangements, which mainly consists of elongation of the Fe–Ooxo bond in the (FeO)2+ core and the cleaving C–H bond in the substrate, (ii) Pauli repulsion between the incoming substrate and the high-valent iron species, and (iii) the orbital interactions between the electron donor and acceptor orbitals. Which of these factors is dominant in determining the differential reactivity of the quintet and triplet reaction pathways?
In the preparatory stage the quintet pathway involves the partial breaking of the Fe–oxo σ-bond to generate a hole in the O-pz based σ*-orbital, whereas only half of the π-bond is broken in the triplet channel (Fig. 3, 4). Moreover, the TSs for H-atom abstraction (5TS1 and 3TS1) exhibit essentially the same Fe–Ooxo bond distance. Therefore, one may simply argue that in terms of the Fe–Ooxo bond lengthening, the triplet pathway should be energetically favored. In fact, the energy required for the elongation of the Fe–Ooxo bond in the quintet oxo-iron(IV) intermediate from its equilibrium geometry (1.65 Å) to 1.80 Å in 5TS1 is 9.4 kcal/mol, whereas the corresponding energy increment is 8.1 kcal/mol for the triplet species. Thus, unexpectedly the quintet channel is only marginally energetically disfavored. This may be ascribed to the much stronger spin-polarization stabilization created by five unpaired electrons in the HS ferric center in 5TS1 compared to only one unpaired electron in the LS iron(III) in the corresponding 3TS1 (12, 13). Hence, the formation of oxyl-ferric species is not the key factor for the differential reactivity.
In the quintet σ-mechanism where the O-pz based σ*-orbital serves as the electron acceptor, the system arranges a vertical attack of the cleaving C–H bond toward the (FeO)2+ core such that the overlap between the O-pz and the σC–H orbital is maximized. This requires the Fe–O–H arrangement to be approximately collinear. More importantly, the linear geometry significantly reduces the Pauli repulsion between the substrate and the oxo-iron(IV) reactant in comparison with the π-pathway (see below) (16). For the π-mechanism the O-px based π*-orbital acts as the electron acceptor. Thus, the optimum Fe–O–H angle should be 90° to afford the maximum overlap between the O-px and the σC–H orbitals. One readily appreciates that such a strictly horizontal approach of the substrate would result in a large Pauli repulsion. The compromise between the orbital interactions and Pauli repulsion yields a final Fe–O–H angle close to 120° as found in 3TS1. This may rationalize the observation that 3TS1 is a “later” TS as suggested by a longer C–H bond length identified in 3TS1 than that in 5TS1. Taken together, the present analysis implies that the synergy of the minimal Pauli repulsion and the favorable orbital interactions render the quintet σ-pathway more effective for H-atom abstraction reactions than the triplet π-pathway.
Based on the above analysis one may anticipate that the quintet π-channel may encounter a comparable barrier to that calculated on the triplet surface due to the unavoidably increased Pauli repulsion and the reduced orbital overlap relative to the quintet σ-pathway. This is in agreement with our previous studies on the relative reactivity of all viable reaction channels for alkane hydroxylation by oxo-iron(IV) complexes (21). For the triplet σ-pathway, in the preparatory stage the triplet ferryl species needs to evolve into an IS ferric ion (SFe = 3/2) that would be antiferromagnetically coupled to an oxyl radical (SO = 1/2). In fact, the strength of the Fe–oxo σ-bond is identical irrespective of the spin multiplicity of the oxo-iron(IV) species (25). The energy penalty for accomplishing this transformation should be much higher on the triplet surface than for the corresponding process on the quintet surface because of the substantially reduced exchange stabilization generated by three rather than five unpaired electrons in the IS ferric center. Hence, the π-pathway is the energetically more favored mechanism on the triplet surface, and our previous calculations suggested that among the four reaction pathways for H-atom abstraction the triplet σ-pathway has the highest barrier (21). The observation that the septet channel involves the lowest barrier for H-atom abstraction although it employs a π-pathway may be rationalized as follows: (i) no preparatory stage is needed; (ii) more importantly, the driving force is quite large because the final electron acceptor is a bonding rather than an antibonding orbital with respect to the Fe–oxo interaction as evidenced by a slightly contracted Fe–Ooxo bond in 7TS1 compared to that in the septet reactant. This is in sharp contrast with what is found on the other two spin-state surfaces where the Fe–Ooxo bond is dramatically lengthened in 3,5TS1; (iii) it involves reduced Pauli repulsion caused by the significantly longer Fe–Ooxo bond in 7TS1 (Table 2).
As elaborated above, in terms of the two-electron oxidation of alkanes by oxo-iron(IV) intermediates several orbitals may act as electron acceptors. Different electron acceptors require different optimal attack geometries for the substrate to maximize the orbital interactions between the electron donor and the electron acceptor. Hence, the already inherently complex nature of the reactivity of open-shell transition metal complexes is further complicated by the fact that different substrate orientations lead to different preferred reaction channels depending on the angle at which the substrate approaches the reactive core. This is clearly a more general feature that is not limited to high-valent oxo-iron(IV) sites. Obviously, it is possible for enzyme active sites to direct the actual substrate attack geometry through steric hindrance. Thus, the outcome of the enzymatic reaction may depend in a subtle way on the substrate orientation and this adds another dimension of control to catalysis by metalloenzyme active sites.
In conclusion, the quintet oxo-iron(IV) species is inherently more reactive toward C–H bond activation than its corresponding triplet partner. This conclusion is in agreement with previous density functional theory studies (10, 12, 14). Our detailed analysis of the electronic structure changes along the reaction coordinate suggests that the real C–H bond cleaving agent is an oxyl-ferric species that is generated by lengthening of the Fe–oxo bond in the ferryl reactant en route to TS. This enables us to approach the question of which spin multiplicity of oxo-iron(IV) species has higher reactivity in a completely unique perspective in this work. The main factor contributing to the differential reactivity is that the two spin states have different requirements for the optimal angle at which the substrate should approach the (FeO)2+ core, because distinct electron acceptors are employed on the two surfaces. The H-atom abstraction on the quintet surface favors the σ-pathway thus requiring an essentially linear attack. By contrast, a π-channel is operative on the triplet surface that leads to an optimal attack angle near 90°. However, the latter is not possible due to a very large energy penalty if the substrate interacts too closely with the supporting ligands of the ferryl center. This effect is the dominant factor because the energy required for partially breaking the Fe–oxo σ-bond (quintet surface) in comparison to the π-bond (triplet surface) is very similar. Whereas this is at first glance surprising, it can be explained by the fact that the spin polarization effect stabilizes the HS ferric center (formed on the quintet surface) much more strongly than the LS ferric center that obligatorily evolves on the triplet surface.
Methods
A truncated model of the ferryl intermediate is employed in the present work where the iron center is coordinated by two imidazole ligands (representing His), a monodentate acetate ligand (representing Asp), a bidentate acetate ligand (representing succinate), and an oxo group. The substrate taurine 2-aminoethane- 1-sulfonic acid) is simply mimicked by ethane.
We used the hybrid B3LYP density functional (26, 27) in combination with triple-ζ quality TZVP basis sets (28) on Fe, O, and N as well as SV(P) basis sets (29) on the remaining atoms. The RIJONX approximation (30) was used to accelerate the calculations in combination with the auxiliary basis sets TZV/J (Fe, O, and N) and SV/J (rest) (31, 32). Geometry optimizations were performed without constraints. The subsequent frequency calculations verified that all local minima had only real frequencies and that transition states (TSs) were distinguished by a single imaginary frequency. The zero-point energies, thermal corrections, and entropy terms for the optimized geometries were obtained from these frequency calculations.
Final energy calculations were also performed with the hybrid B3LYP density functional using the extensively polarized basis sets of triple-ζ quality including high angular momentum polarization functions (def2-TZVPP) (33) for all atoms. The density fitting and chain of spheres (RIJCOSX) approximations (34) have been employed together with the def2-TZVPP/J auxiliary basis set (35). Following Siegbahn (36), the protein environment was crudely modeled by the COSMO model by taking dielectric constant as 4.0.
All calculations were performed with the ORCA program package (37). For an analysis of the changes in the electronic structure of the OH-rebound step, see SI Text.
Supplementary Material
Acknowledgments.
The authors gratefully acknowledge a grant from the German Science Foundation (NE 690/7-1) and financial support from the special research unit SFB 813 (Chemistry at Spin Centers).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1008411108/-/DCSupplemental.
References
- 1.Ghosh A, editor. High-valent iron intermediates in biology. J Inorg Biochem. 2006;100:419–880. doi: 10.1016/j.jinorgbio.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Ortiz de Montellano PR, editor. Cytochrome P450: Structure, mechanism and biochemistry. 3rd ed. New York: Kluwer; 2004. [Google Scholar]
- 3.Krebs C, Fujimori DG, Walsh CT, Bollinger JM., Jr Non-heme Fe(IV)-oxo intermediates. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Que L., Jr The road to non-heme oxoferryls and beyond. Acc Chem Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 5.Pestovsky O, et al. Aqueous FeIV = O: Spectroscopic identification and oxo-group exchange. Angew Chem Int Ed. 2005;44:6871–6874. doi: 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]
- 6.England J, et al. The crystal structure of a high-spin oxoiron(IV) complex and characterization of its self-decay pathway. J Am Chem Soc. 2010;132:8635–8644. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.England J, et al. A synthetic high-spin oxoiron(IV) complex: generation, spectroscopic characterization, and reactivity. Angew Chem Int Ed. 2009;48:3622–3626. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xue G, De Hont R, Münck E, Que L., Jr Million-fold activation of the [Fe2(μ-O)2] diamond core for C–H bond cleavage. Nature Chem. 2010;2:400–405. doi: 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirao H, Kumar D, Que L, Jr, Shaik S. Two-state reactivity in alkane hydroxylation by non-heme iron-oxo complexes. J Am Chem Soc. 2006;128:8590–8606. doi: 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- 10.Hirao H, Que L, Jr, Nam W, Shaik S. A two-state reactivity rationale for counterintuitive axial ligand effects on the C–H activation reactivity of non-heme FeIV = O oxidants. Chem Eur J. 2008;14:1740–1756. doi: 10.1002/chem.200701739. [DOI] [PubMed] [Google Scholar]
- 11.Shaik S, Hirao H, Kumar D. Reactivity of high-valent iron-oxo species in enzymes and synthetic reagents: A tale of many states. Acc Chem Res. 2007;40:532–542. doi: 10.1021/ar600042c. [DOI] [PubMed] [Google Scholar]
- 12.de Visser SP. Propene activation by the oxo-iron active species of taurine/α-ketoglutarate dioxygenase (TauD) enzyme. How does the catalysis compare to heme-enzyme? J Am Chem Soc. 2006;128:9813–9824. doi: 10.1021/ja061581g. [DOI] [PubMed] [Google Scholar]
- 13.Janardanan D, Wang Y, Schyman P, Que L, Jr, Shaik S. The fundamental role of exchange-enhanced reactivity in C–H activation by S = 2 oxo iron(IV) complexes. Angew Chem Int Ed. 2010;49:3342–3345. doi: 10.1002/anie.201000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernasconi L, Louwerse ML, Baerends EJ. The role of equatorial and axial ligands in promoting the activity of non-heme oxidoiron(IV) catalysts in alkanehydroxylation. Eur J Inorg Chem. 2007:3023–3033. [Google Scholar]
- 15.Michel C, Baerends EJ. Oxidative properties of FeO2+: electronic structure and solvation effects. Phys Chem Chem Phys. 2007;9:156–166. doi: 10.1039/b613182d. [DOI] [PubMed] [Google Scholar]
-
16.Michel C, Baerends EJ. What singles out the FeO2+ moiety? A density-functional theory study of the methane-to-methanol reaction catalyzed by the first row transition-metal oxide dications
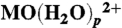 , M = V - Cu. Inorg Chem. 2009;48:3628–3638. doi: 10.1021/ic802095m. [DOI] [PubMed] [Google Scholar]
, M = V - Cu. Inorg Chem. 2009;48:3628–3638. doi: 10.1021/ic802095m. [DOI] [PubMed] [Google Scholar] - 17.Decker A, et al. Spectroscopic and quantum chemical studies on low-spin FeIVO complexes: Fe–O bonding and its contributions to reactivity. J Am Chem Soc. 2007;129:15983–15996. doi: 10.1021/ja074900s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solomon EI, Wong SD, Liu LV, Decker A, Chow MS. Peroxo and oxo intermediates in mononuclear nonheme iron enzymes and related active sites. Curr Opin Chem Biol. 2010;13:99–113. doi: 10.1016/j.cbpa.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neidig ML, et al. Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen-atom abstraction by non-heme iron enzymes. Proc Natl Acad Sci USA. 2006;103:12966–12973. doi: 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Han K. Steric hindrance effect of the equatorial ligand on Fe(IV)O and Ru(IV)O complexes: A density functional study. J Biol Inorg Chem. 2010;15:351–359. doi: 10.1007/s00775-009-0607-4. [DOI] [PubMed] [Google Scholar]
- 21.Geng C-Y, Ye S, Neese F. Analysis of reaction channels for alkane hydroxylation by nonheme iron(IV)-oxo complexes. Angew Chem Int Ed. 2010;49:5717–5720. doi: 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]
- 22.Ye S, Neese F. Quantum Chemical studies of C–H activation reactions by high-valent nonheme iron centers. Curr Opin Chem Biol. 2009;13:89–98. doi: 10.1016/j.cbpa.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: A high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 24.Groves JT, McClusky GA. Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J Am Chem Soc. 1976;98:859–861. [Google Scholar]
- 25.Berry JF, DeBeer GS, Neese F. Electronic structure and spectroscopy of “superoxidized” iron centers in model systems: theoretical and experimental trends. Phys Chem Chem Phys. 2008;10:4361–4374. doi: 10.1039/b801803k. [DOI] [PubMed] [Google Scholar]
- 26.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 27.Lee CT, Yang WT, Parr RG. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 28.Schäfer A, Huber C, Ahlrichs R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 29.Schäfer A, Horn H, Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Lito Kr. J Chem Phys. 1992;97:2571–2577. [Google Scholar]
- 30.Neese F, Olbrich G. Efficient use of the resolution of the identity approximation in time-dependent density functional calculations with hybrid density functional. Chem Phys Lett. 2002;362:170–178. [Google Scholar]
- 31.Eichkorn K, Treutler O, Ohm H, Häser M, Ahlrichs R. Auxiliary basis sets to approximate Coulomb potentials. Chem Phys Lett. 1995;242:652–660. [Google Scholar]
- 32.Eichkorn K, Treutler O, Ohm H, Häser M, Ahlrichs R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor Chem Acc. 1997;97:119–124. [Google Scholar]
- 33.Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence, and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 34.Neese F, Wennmohs F, Hansen A, Becker U. Efficient, approximate, and parallel Hartree–Fock and hybrid DFT calculations. A “chain-of-spheres” algorithm for the Hartree–Fock exchange. Chem Phys. 2009;356:98–109. [Google Scholar]
- 35.Weigend F. Accurate coulomb-fitting basis sets for H to Rn. Phys Chem Chem Phys. 2006;8:1057–1065. doi: 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- 36.Siegbahn PEM. Mechanism of metalloenzymes studied by quantum chemical methods. Q Rev Biophys. 2003;36:91–145. doi: 10.1017/s0033583502003827. [DOI] [PubMed] [Google Scholar]
- 37.Neese F. ORCA—An ab Initio, Density Functional, and Semiempirical Program Package. Bonn, Germany: Universität Bonn; 2008. Verson 2.6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.