

Abstract
Many fatal neurodegenerative diseases such as Alzheimer’s, Parkinson, the prion-related diseases, and non-neurodegenerative disorders such as type II diabetes are characterized by abnormal amyloid fiber aggregates, suggesting a common mechanism of pathogenesis. We have discovered that a class of systematically designed natural tri- to hexapeptides with a characteristic sequential motif can simulate the process of fiber assembly and further condensation to amyloid fibrils, probably via unexpected dimeric α-helical intermediate structures. The characteristic sequence motif of the novel peptide class consists of an aliphatic amino acid tail of decreasing hydrophobicity capped by a polar head. To our knowledge, the investigated aliphatic tripeptides are the shortest ever reported naturally occurring amino acid sequence that can adopt α-helical structure and promote amyloid formation. We propose the stepwise assembly process to be associated with characteristic conformational changes from random coil to α-helical intermediates terminating in cross-β peptide structures. Circular dichroism and X-ray fiber diffraction analyses confirmed the concentration-dependent conformational changes of the peptides in water. Molecular dynamics simulating peptide behavior in water revealed monomer antiparallel pairing to dimer structures by complementary structural alignment that further aggregated and stably condensed into coiled fibers. The ultrasmall size and the dynamic facile assembly process make this novel peptide class an excellent model system for studying the mechanism of amyloidogenesis, its evolution and pathogenicity. The ability to modify the properties of the assembled structures under defined conditions will shed light on strategies to manipulate the pathogenic amyloid aggregates in order to prevent or control aggregate formation.
Keywords: self-assembly mechanism, ultrasmall peptides, supramolecular peptide scaffolds, fiber diffraction, molecular dynamics simulation
During self-assembly, compounds autonomously organize to functional stable entities by formation of order from disorder (1). The self-organization of biological molecules to supramolecular structures by noncovalent interactions is an important organizational process that is crucial for the existence of life. It constitutes, for example, an indispensible role in the storage of genetic information through the pairing of nucleic acids strands. Protein folding is another crucial process for proper cellular regulation (2). Biological aggregates such as cellular membranes, micelles, and vesicles utilize amphiphilic molecules as self-assembling building blocks (3–5).
Amyloid fiber aggregates are further examples of self-assembly. They are found in microorganisms (such as bacteria and fungi) and in storage granules with biologically meaningful functions in the endocrine system (6). It remains a mystery how amyloid structures terminate in a pathogenic state as found in threatening neurodegenerative diseases such as Alzheimer’s, Parkinson, prion-related diseases, and non-neurodegenerative disorders such as type II diabetes (7, 8). It has been speculated that the essence of pathogenic amyloidosis is intimately connected with the capability of the involved peptides to take more than one conformation. This feature has earned the notation of chameleon proteins (9). It is assumed that these pathogenic conditions occur due to misfolding, by assembly of unstructured peptides, intrinsically disordered proteins, or unfolded and misfolded fragments of generally folded proteins (7). Also pathogenic systemic amyloidoses such as familial amyloid polyneuropathy, light chain and lysozyme amyloidoses seem to be associated with partial unfolding prior to amyloid formation. As a conclusion, the formulation of the conformational change hypothesis has been proposed (10). Because it is integral to unequivocally understand the driving forces needed to form and maintain self-assembled amyloid structures to reverse or change amyloid formation, appropriate model systems are indispensable. The peptide class introduced in this study might offer new insights toward amyloidogenesis because its use for assembly studies is facilitated by the small size of the peptides and facile assembly process. Here we report on a peptide class that demonstrates the shortest reported aliphatic peptides (consisting of natural amino acids) to form transitional α-helices in aqueous solutions and to self-assemble into supramolecular structures exerting amyloid β-structures. We propose a specified hypothesis of amyloid formation that describes an at least threefold assembly process from unstructured random coil to structured α-helical intermediates terminating in cross-β peptide structures. Different from the idea of unfolding as a crucial prerequisite prior to amyloid assembly we want to stress the importance of existing short α-helical structures as key intermediates, probably as antiparallel dimer structures, for amyloid formation.
Results
Novel Class of Rationally Designed Ultrasmall Peptides.
A specific peptide motif enables ultrasmall peptides with 3–6 amino acids to self-assemble to helical fibers in supramolecular structures (Table 1). The amphiphilic peptide motif consists of a tail of aliphatic nonpolar amino acids (N terminus) with decreasing hydrophobicity and a hydrophilic head group of acidic, neutral, or basic nonaromatic polar amino acids (C terminus). The N terminus was acetylated in order to keep it uncharged. We observed that the length of the hydrophobic tail and the polarity of the head group were integral elements that supported facile hydrogel formation. Hexamers typically formed gels more readily than pentamers, tetramers, and trimers. Stronger gels were derived from head groups with acidic (D and E), followed by neutral (S and T) and basic (K) polar, nonaromatic amino acids. The sequence motif started from N terminus to C terminus in the order of leucine (L) [or isoleucine (I)], followed by isoleucine (or leucine), valine (V), alanine (A), and glycine (G) to guarantee the favorable decrease of nonpolar character toward the polar C terminus. This arrangement of amino acids gave rise to cone-like structures that were prone to assemble noncovalently by molecular recognition in a parallel–antiparallel stacked fashion.
Table 1.
Group of peptides that self-assemble to ordered supramolecular networks, constituting in hydrogels
| Head group | Hexamer | Pentamer | Tetramer | Trimer |
| Aspartic acid (D) | LIVAGD | LIVAD | IVAD | IVD |
| ILVAGD | LIVGD | IID | ||
| LIVAAD | ||||
| LAVAGD | ||||
| AIVAGD | ||||
| Glutamic acid (E) | LIVAGE | |||
| Lysine (K) | LIVAGK | |||
| Serine (S) | LIVAGS | |||
| ILVAGS | ||||
| AIVAGS | ||||
| Threonine (T) | LIVAGT | |||
| AIVAGT |
All peptides were acetylated at the N terminus while the carboxyl group at the C terminus was unchanged, except for LIVAGK, which was amidated to suppress the charge at the C terminus. The more extensively investigated peptides are shown in bold; namely, LIVAGD (also named Ac-LD6), AIVAGD (also named Ac-AD6), and IVD (also named Ac-ID3).
During condensation the helical fibers are visible to the naked eye. As the condensation proceeds, the visible fiber structures disappear within solid-like hydrogels. According to our current understanding, the ease of self-assembly, from small aggregates and unstable fibers to firm meshes of stably assembled supramolecular fibrils, depends strongly on the decline in the hydrophobic character of the hydrophobic amino acid tail. This goes along with a favorable structure fitted for complementary pairing. We verified this assumption by an alanine scan of the peptide sequence Ac-LIVAGD (Table 1). By sequentially exchanging each amino acid of the hydrophobic tail to alanine, we evaluated the altered peptides on their ease of fiber formation and hydrogel strength.
Hypothesis of Self-Assembly Through Structural Transition as Confirmed by Circular Dichroism (CD).
We hypothesize that peptide network formation requires at least three steps, depending on increasing peptide concentrations: Step I: antiparallel pairing of two peptide monomers and structural transition to α-helical conformation. Step II: assembly of peptide pairs to fibers and nanostructures. Step III: condensation of peptide fibers to fibrils (Fig. 1). We believe that the initiation of peptide assembly to pairs and fibers by antiparallel stacking is the nucleation step needed for further processing of fibers to fiber aggregates. This step is governed by a change of the peptide’s secondary structure from random coil to α-helical conformation.
Fig. 1.

Hypothesis of self-assembly from peptide monomers to supramolecular networks of condensed fibers. (A) Self-assembly is initiated with antiparallel pairing of two peptide monomers by changing to α-helical conformation. Subsequently, peptide pairs assemble to fibers and nanostructures and condense to fibrils resulting in hydrogel formation.
It is commonly thought that small linear peptides of 3–6 amino acids would not show α-helical propensities due to size restrictions. To our surprise, our observations proved otherwise: Ultrasmall peptides (3–6 mers) changed conformation from random coil to α-helical structures upon crossing a threshold concentration (Fig. 2 A and B). The CD spectra demonstrated that at low concentrations all investigated peptides were stable up to temperatures of 90 °C. At low concentrations, hexamer Ac-LD6 slightly and gradually changed its random coil structure during a temperature scan from 25 °C to 90 °C (Fig. 2C). It reverted to its individual original structure when stepwise cooled to 25 °C. The observed peaks corresponded well with the reported random coil transitions of a very small positive n - π∗ transition near 217 nm and a single large negative transition near 190 nm (11). Slightly below its critical gel forming concentration of 1 mg/mL, transitions occurred similar to those found in α-helices. Specifically, a negative n - π∗ transition near 222 nm, a split π - π∗ transition with a negative peak near 208 nm, and a positive peak near 192 nm (peak at 192 nm not shown) were observed (Fig. 2 A and B). Our CD spectra pointed to a combination of α-helix and poly-L-proline-like II conformations previously observed in amyloidogenic peptides prior to assembly (12). The α-helical content increased when the critical gel forming concentration was exceeded. The CD spectrum of subsequent fiber structures was similar to short prion peptides and amyloid fiber aggregates that are β-turn structures (Fig. 2D) (13). Interestingly, the β-turn fibers of our hydrogels were stable structures. When the fiber-containing hydrogels were heated stepwise from 25 °C to 90 °C, their secondary structure remained unchanged, while further condensation occurred (Fig. 2E). After cooling, there was no significant change of the scaffold structure observable. All investigated scaffold-forming peptides displayed similar behavior, indicating a motif-dependent condensation process.
Fig. 2.

CD spectra of peptide solution and scaffold. (A) CD spectra demonstrate the transition of Ac-LD6 peptide conformation from random coil (below threshold concentration) to α-helical (peaks at 208 and 222 nm) and finally β-type (negative band at 218 nm) structures, increased by heating. (B) Far-UV CD spectra of Ac-ID3 at different concentrations showing the conformational transition from random coil via α-helical to β-type structures. All spectra were measured at 25 °C. (C) Random coil conformations of Ac-LD6 (0.2 mg/mL) were reversibly affected by stepwise temperature increases (solid lines) from 25 °C to 90 °C and temperature decreases (dotted lines) to 25 °C. (D) Temperature increases of Ac-LD6 gels (1 mg/mL) stabilized the β-type structures irreversibly. (E) Subsequent cooling did not alter the conformation.
Evaluation of Fiber Structures by Field-Emission Scanning Electron Microscopy (FESEM) and X-ray Diffraction.
We confirmed the fiber formation of the peptide scaffolds by low-temperature FESEM (Fig. 3 A and B). Undissolved lyophilized peptide powder already contained short assembled fibers. Each peptide had a critical minimum concentration to form fibrous scaffolds in water. When the critical peptide concentration was exceeded, the resulting hydrogel consists of water entrapped by the whole fibrous network.
Fig. 3.

Morphological and structural characterization of the peptide scaffolds by FESEM and X-ray fiber diffraction. (A and B) Condensed fibers of Ac-ID3 hydrogels of 15 mg/mL (A) and 20 mg/mL (B). (C and D) Averaged microdiffraction patterns after background subtraction of β-turn (with α-helical propensities) secondary structures of Ac-ID3 (C) and Ac-LD6 (D). (E and F) After drying the gels on a hydrophobic surface, the Ac-ID3 residuals attached to glass capillaries were scanned. Orientation of the local fiber axis shows single diffraction patterns of Ac-ID3 (G) and Ac-LD6 (H) (dark arrows m1-3: meridional, e1-3: equatorial reflections). In particular, the Ac-ID3 residual skin showed a strong fiber texture. Azimuthally averaged patterns of Ac-ID3 (I) and Ac-LD6 (J) residuals at room temperature. The patterns have been fitted by Gaussian profiles for the Bragg peaks and a 0-order polynomial for the residual background (in blue). Positions of the individual peaks are indicated by arrows. The part of the pattern excluded to the fit is in red. Small amounts of residual ice are indicated. Errors indicated correspond to ± σ values of the fits.
We performed X-ray microdiffraction experiments on two peptide hydrogels, the trimer Ac-ID3 and hexamer Ac-LD6. Despite the low volume concentration of scattering material, we were able to record powder diffraction data from Ac-ID3 and Ac-LD6 peptide films, flash-frozen to 100 K in MiTeGen Kapton MicroMounts by raster microdiffraction (Fig. 3 C–J). The main features of the patterns were two powder rings at d1 ∼ 9.8 Å, d2 ∼ 4.6 Å and a diffuse powder ring at d3 ∼ 4.0 Å (Fig. 3 C and D). To relate these peaks to the fibrils visible in the FESEM images, we generated a fibrous texture material by controlled evaporation of drops from the same hydrogel solutions on a superhydrophobic surface. The residuals were gently detached from the surface and attached to a glass capillary support. Trimer Ac-ID3 formed hollow, flattened spherical residuals (Fig. 3E). Fiber texture was observed with the beam in normal orientation to the skin of the Ac-ID3 residual (Fig. 3F). We observed a transition to a random crystallite orientation in the center of the dried drop, as expected for diffraction when the beam orients along the fiber axis. The Ac-LD6 residual collapsed upon drying to an irregular structure (Fig. S1C) but also showed fiber texture at the skin (Fig. S1 C and D). The fiber texture formation appeared to be independent from the structuring of the polymethylmethacrylate (PMMA) surface. From the X-ray microdiffraction patterns, the residuals and the hydrogels had similar general scattering features. However, for the residuals it was possible to distinguish the equatorial and meridional reflections (Fig. 3 I and J and Fig. S1 E and F). Azimuthally averaged patterns of the peptide residuals revealed two prominent reflections that were too narrow for an amorphous material such as α-helical keratin (14). The d ∼ 4.6 Å meridional reflection did not completely match an expected d = 5.15 Å reflection corresponding to the 5.4-Å helical repeat of an α-helix supercoiled within a coiled coil (15, 16), as reported for 28-mer α-helical peptide hydrogel (15). While we cannot completely exclude α-helical propensities, the appearance of a meridional d ∼ 4.6 Å reflection is, however, characteristic for cross-β-type bonding as experimentally observed in amyloid-like structures for ≥4-mer peptide crystallization (17). In the cross-β-model, the equatorial (e) reflections relate to the fibril structure perpendicular to the fiber direction (Fig. 3 G and H). The d ∼ 10 Å reflection was attributed to the structure of the fibrils projected down the fiber axis. This reflection has been identified to represent the spacing of the β-sheets in amyloid fibrils. The variation in d spacing could be related to the side-chain composition of the β-sheets (18, 19). It is interesting to note that model simulations for high concentrations of short peptides suggest the formation of assembled intermediate structures retaining α-helical conformations, prior to β-sheet transition (20). The circular dichroism experiments on the current peptides also suggest α-helical intermediate structures, followed by β-type-structures at higher concentrations.
Molecular Dynamics Simulations of Ultrasmall Peptide Self-Assembly.
Further investigating the assembly behavior, particularly the pair formation (Fig. 1, Step I), we conducted molecular dynamics simulations of 4 hexamer Ac-LD6 peptides in water (Fig. 4A). At t = 0, the peptides were placed apart from each other in the simulation box. By t = 1 ns, the peptides formed pairs, and by t = 6 ns, the two pairs aggregated into a cluster of four peptides. Similar dimer formation was observed from extensive molecular dynamics simulations of several β-sheet forming peptides in implicit solvent (21). Our simulation results support the hypothesis of pair formation preceding fiber formation and eventual condensation into supramolecular structures (20). Simulations with the shorter trimer Ac-ID3 in water revealed similar pairing behavior, although the dynamics of pair formation was slower compared to the longer Ac-LD6 peptides. This could be attributed to the lower hydrophobicity of the trimer compared to the hexamer. However, the tendency to form pairs in water was consistent in both peptides, highlighting pair formation as a general mechanism for self-assembly. Additional simulations of the pairing behavior and investigation of the peptide orientation are discussed in SI Text.
Fig. 4.

Molecular dynamics simulations of hexapeptide Ac-LD6 in water. (A) Simulation snapshots of initially separated four Ac-LD6 peptides in water. By t = 1 ns, the peptides formed two pairs that assembled to form a cluster of fourpeptides. (B) Simulation snapshots of a fiber consisting of 72 Ac-LD6 peptides. Peptides were initially stacked horizontally in antiparallel manner. Over time, the fiber turned along its axis, forming bends. (C) Final structure from a stacked simulation of an Ac-LD6 fiber consisting of 216 peptides. After 20 ns of simulation time, the fiber remained intact and coiled around the fiber axis. (D) Water molecules are viewed down the fiber axis without the fiber. Very few water molecules exist within the fiber; instead, the water forms a layer surrounding the fiber.
The formation of fiber structures was probed using molecular dynamics simulations of stacked peptides. We investigated horizontal versus vertical stacking configurations. The latter resulted in much more disordered structures (Fig. S2A). Initial simulations involved a single strand of 18 horizontally stacked peptides in water. Periodic boundary conditions applied in all directions resulted in an effectively infinite peptide strand that was unstable and broke apart within 5 ns of simulation time. We then assembled four peptide strands in a 2 × 2 configuration, resulting in a fibrous structure of 72 peptides in water. After 5 ns of simulation time, the fiber remained stable without breaking apart (Fig. 4B). This supports our hypothesis that the individual peptide fibers condense to larger supramolecular structures. It is also consistent with known biological behavior in nature where DNA and collagen form double and triple helices, respectively, rather than single strands. This suggests that the fibril aggregates represent a lower free energy state than the individual separate protofibril strands, leading to assembly of larger aggregates. However, further simulations would be necessary to calculate a free energy change associated with the assembly process. Although we were unable to observe helix formation in our fiber, the fiber formed bends and turns along the fiber axis. Increasing simulation time and length scale to 20 ns with 216 horizontally stacked Ac-LD6 peptides, the fiber remained intact and formed a coil along the fiber axis (Fig. 4C). It is evident from our simulations that these peptide fibers do not form linear structures. However, the observation of the helix formation in the fibers is probably beyond the limits of our present simulations. To further understand the hydrogel’s amazing water retention ability, we investigated the position of water molecules relative to peptide fibers. There was very little water within the peptide fibers (Fig. 4D). This was not surprising considering the hydrophobic nature of the peptide tail. Instead, the water formed a layer around the fiber, suggesting that the bulk of the water in the hydrogel will be stored between the fibers and not within.
Discussion
Amphiphilic peptides have a strong tendency to self-assemble. We noticed that all investigated peptides from an investigated group exceeding 100 peptides were able to self-assemble. Interestingly, only the minority (∼25%), designated with a distinct cone-shaped peptide motif, exerted the potential of further assembly to ordered supramolecular amyloid structures, constituting in hydrogels. The majority of peptides remained in solution as nanomolecular aggregates or precipitates. A minimum concentration was needed for gelation, while elevated temperatures accelerated self-assembly.
The general assembly process toward amyloid aggregates presumably is a complex stepwise process including conformational peptide or protein changes (20). The “folding energy landscape theory” proposes a funnel-like pathway with conformational intermediates that lead to the final species (22). It includes the conversion of the native protein or peptide structure into a predominantly β-sheet secondary structure, where N and C terminus are oriented in antiparallel fashion. Our observation of a stepwise assembly process of the investigated peptides to amyloid structures strikingly resembles the proposed pathological mechanism of amyloidosis. Following the structural conversion of our peptides toward supramolecular structures (fibril formation), we propose that the transition from random coil to an intermediate α-helical conformation is the most critical of the multistep process. We believe that structural conversion is necessary for processing of fibers or nanostructures toward stable β-turn fibrils. A recent review discusses the increasing evidence that a variety of natively unfolded polypeptides, prone to amyloid formation, show enriched α-helical intermediates during the crucial lag phase of aggregate formation (23). The authors conclude that, according to the published data, peptide–membrane or peptide–solvent interactions are probably the reasons for the appearance of α-helical intermediates. In our study we are able to show that formation of α-helical intermediates only needs a critical peptide concentration. The structural transformation probably serves as a signaling command. It is conceivable that in biological systems conformational changes are necessary signals or communication tools, either for catalysis or for the assembly process of functional or toxic aggregates. With this communication tool, the surrounding molecules would receive instructions for actions such as assembly. The decision to proceed to supramolecular aggregates, as observed with our hydrogel-forming peptides, relies on the efficiency of the transition to a different secondary conformation, which in turn depends on the peptide sequence. Examples of functional protein aggregates (6) and toxic amyloid aggregates (7, 8) are abundant in nature. Because it is not understood when, why, or how a protein or peptide self-assembles to amyloidogenic structures and eventually proceeds to a pathogenic state, model systems for detailed studies are needed. We propose our unique peptide class as an excellent model system. Nonetheless, our peptide motifs do not resemble the lately presented amylomes—proteins capable of forming amyloid-like fibrils (24). Performing a blast search of the Protein Data Bank revealed that the hexapeptides Ac-LIVAGD, Ac-AIVAGD, and Ac-ILVAGD (Table 1) did not correspond to any putative conserved domains. Of the 83% or 100% matches obtained, the peptide sequences did not appear more than once in each of the database proteins. Because these hexapeptides are the best examples of the gelating peptides, evolution might have well decided not to favor these motifs in natural occurring protein structures, thereby reducing the risk of predictable toxic fiber assembly.
The presented class of ultrasmall peptides offers a plethora of interesting aspects for future investigations. There is the general view that short peptides, in particular linear tri- to hexapeptides, are restricted by size to form α-helical structures under aqueous conditions. The smallest ever reported α-helices in water are adopted by cyclic pentapeptides (25). Although these peptide structures are of interest as biological probes or therapeutics, they do not resemble any natural occurring peptide. Alanine-rich linear tri- to hexapeptides are reported to adopt mixed β-turn(310)/α-helix(413) conformations, though not in water. They adopt their mixed conformation only in aprotic environments, such as in the protected cavity of a hydrophobic porphyrin-prism host (26). In contrast, our peptides do not require specific environments or conditions but simply aqueous or physiological conditions and a specific concentration to turn into α-helical conformation. We would like to point out that other ultrashort peptides such as Ala-Phe-Ala (27) have been reported to adopt stable non-α-helical structures in aqueous solution. This implies that ultrashort peptides can adopt defined secondary structures, even under physiological conditions, increasing the evidence for a reassessment of the structure–function paradigm (28). Because our class of peptides can adopt different conformations, we can imagine using them as building blocks for proteins with predetermined secondary and tertiary structures (29).
We are especially intrigued by the driving forces that enable distinct ultrasmall peptides to stably self-assemble to macromolecular structures. The ability to modify the properties of the assembled structures under defined conditions will shed light on strategies to manipulate particularly the pathogenic amyloid aggregates or will prevent or control aggregate formation.
Materials and Methods
Peptide-Based Hydrogel Preparation.
All peptides (GL Biochem, ≥98% purity) were freshly prepared in order to avoid premature peptide assembly. The peptides were dissolved in water and left at room temperature to form hydrogels. Depending on the peptide concentration, the self-assembly process occurred immediately, within hours or even within days (experimental time frame for gelation). For higher peptide concentrations peptides were dissolved in milliQ water by vortexing. If an accelerated hydrogel preparation was needed, the peptide solution was subjected to sonication in a water bath at 35 kHz (Barnstead Labline 9319 UltrasonicLC60H). No significant structural differences were observed between hydrogels produced via self-assembly and those whose assembly was facilitated by sonication.
FESEM Studies.
Samples were frozen at -80 °C and then vacuum dried. Subsequently they were fixed onto a sample holder using conductive tape and sputtered with platinum from both the top and the sides in a JEOL JFC-1600 High Resolution Sputter Coater. The coating current used was 30 mA, and the process lasted for 60 sec. The surface of interest was then examined with a JEOL JSM-7400F field-emission scanning electron microscopy system using an accelerating voltage of 5–10 kV.
CD Spectroscopy.
CD spectra were collected with an Aviv 410 CD spectrophotometer fitted with a Peltier temperature controller, using a rectangular quartz cuvette with a fitted cap and an optical path length of 1 mm. For higher peptide concentration (10–20 mg/ml), quartz cuvettes with optical path lengths of 0.01 mm were used. Data acquisition was performed in steps of 0.5 nm at a wavelength range from 185–270 nm with a spectral bandwidth of 1.0 nm. Spectra were obtained in a stepwise fashion up and then down. The investigated temperatures ranged over 25 °C–90 °C (in the following steps: 25 °C, 37 °C, 50 °C, 70 °C, 90 °C, 70 °C, 50 °C, 37 °C, 25 °C). To ensure reproducibility of the CD spectra, three samples of each peptide were prepared and characterized spectroscopically, but the spectra were not averaged. All spectra were corrected in baseline with milli Q water as the blank. The signals were normalized to mean residue ellipticity (MREs) based on the peptide concentration
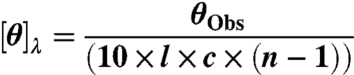 |
with [θ]λ as the MRE at wavelength λ in degcm2 dmol-1, l as the pathlength in cm, c as the concentration in M and (n - 1) as the number of peptide bonds in the studied polymer (30).
X-ray Fiber Diffraction.
Experiments were performed under cryoconditions (100 K) on a trimer Ac-ID3 (L) slurry with a glycol/water (20/80 v/v%) solution as cryoprotectant. Experiments on an “air dry” Ac-ID3 (L) powder were performed without cryoprotectant. No significant amount of ice formation was observed in either case. A small quantity of each sample was picked-up with a MiTeGen MicroMount Kapton mount with 100 μm aperture. Precipitated solute residues were examined at room temperature (RT). The residues were attached to tapered glass capillaries by a small amount of rapid glue.
Precipitation Experiments.
A superhydrophobic surface, based on a micro- or nanopatterned polymethylmethacrylate (PMMA) surface (31, 32) was used to compact the peptides by precipitation. Hydrogel powders were dissolved in deionized water (2.15 mg peptide/mL water) by vortex-induced vibration. A drop of solute was deposited by a syringe on the PMMA surface and allowed to dry. The solid residue was gently detached from the surface and attached by a small amount of fast glue to a glass capillary.
Synchrotron Radiation Experiments.
Microdiffraction experiments were performed in transmission geometry using a 1hor ∗ 0.8vert μm2 monochromatic X-ray beam from crossed mirrors at a wavelength of λ = 0.09947 nm. Sample supports were attached by a magnetic base to the ID13 scanning goniometer (33) and aligned normal to the beam by an on-axis Olympus microscope, which was calibrated to the beamline focal spot. An Oxford Cryosystems cryoflow system was used for experiments at 100 K. A FreLon CCD detector (34) with 2K × 2K pixels (binned to 512 × 512 pixels) and 16-bit readout was used for data collection. The detector-to-sample distance was calibrated with an Al2O3 powder pattern to 94.3 mm. Raster scans were performed with the ID13 scanning goniometer (33) with variable step-resolution down to 1 μm. The data collection time was up to 5 sec /pattern at 100 K and 0.5 sec /pattern at RT. Patterns recorded outside the sample were used for background correction. Data analysis was performed with the FIT2D program (www.esrf.fr/computing/scientific/FIT2D/).
Computer Simulation.
The peptides were described using the OPLS force field (35) and the water molecules were described using the SPC/E model (36). Molecular dynamics simulations were performed using GROMACS version 4.0.5 (37). Simulations were performed at time steps of 2 fs. Periodic boundary conditions were applied in all three directions. Cut-off radii were set at 0.9 nm for electrostatic interactions and 1.4 nm for Lennard–Jones interactions. Long-range electrostatic interactions were treated using the particle-mesh Ewald (PME) method (38). Simulations in the isothermal-isobaric (NPT) ensemble have been carried out. Temperature coupling was done with a Berendsen thermostat but with an additional stochastic term to ensure a correct kinetic energy distribution and produce a correct canonical ensemble (39). Pressure coupling was achieved with the Berendsen barostat (40). Relaxation times of 1 ps and 2 ps were used for the thermostat and barostat, respectively. For all investigated peptides the total simulation time was in the ns-range. For the largest system of 216 peptides in water, 10 million simulation steps were performed to obtain a total time of 20 ns. The calculations were performed at a rate of about 11 ns/day on 32 Intel Xeon 2.27 Ghz CPUs.
Supplementary Material
Acknowledgments.
We thank Enzo Di Fabrizio (Italian Institute of Technology, Genova, and BIOMEMS, University Magna Græcia Catanzaro, Catanzaro, Italy) for helpful discussions and laboratory facilities for the production of micro- and nano-structured PMMA surfaces. We thank Norma Greenfield (Department of Neuroscience and Cell Biology, Robert Wood Johnson Medical School) for helpful comments on CD spectra. This work was supported by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1014796108/-/DCSupplemental.
References
- 1.Whitesides GM, Grzybowski B. Self-assembly at all scales. Science. 2002;295:2418–2421. doi: 10.1126/science.1070821. [DOI] [PubMed] [Google Scholar]
- 2.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Zhang S, Holmes T, Lockshin C, Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc Natl Acad Sci USA. 1993;90:3334–3338. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehn JM. Toward self-organization and complex matter. Science. 2002;295:2400–2403. doi: 10.1126/science.1071063. [DOI] [PubMed] [Google Scholar]
- 5.Hauser CAE, Zhang S. Designer self-assembling peptide nanofiber biological materials. Chem Soc Rev. 2010;39:2780–2790. doi: 10.1039/b921448h. [DOI] [PubMed] [Google Scholar]
- 6.Maji SK, et al. Amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 8.Lansbury PT, Lashuel HA. A century old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 9.Perutz MF. Amyloid fibrils. Mutations make enzyme polymerize. Nature. 1997;385:773–775. doi: 10.1038/385773a0. [DOI] [PubMed] [Google Scholar]
- 10.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Nat Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 11.Venyaminov SY, Baikalov IA, Shen ZM, Wu CSC, Yang JT. Circular dichroic analysis of denatured proteins—inclusion of denatured proteins in the reference set. Anal Biochem. 1993;214:17–24. doi: 10.1006/abio.1993.1450. [DOI] [PubMed] [Google Scholar]
- 12.Starck CS, Sutherland-Smith AJ. Cytotoxic aggregation and amyloid formation by the myostatin precursor protein. PloS One. 2010;5:e9170. doi: 10.1371/journal.pone.0009170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kayed R, et al. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J Mol Biol. 1999;287:781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 14.Busson B, Engstrom P, Doucet J. Existence of various structural zones in keratinous tissues revealed by X-ray microdiffraction. J Synchrotron Radiat. 1999;6:1021–1030. [Google Scholar]
- 15.Banwell EF, et al. Rational design and application of responsive alpha-helical peptide hydrogels. Nat Mater. 2009;8:596–600. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraser RDB, Macrae TP. Structure of alpha-keratin. Polymer. 1973;14:61–67. [Google Scholar]
- 17.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 18.Arnott S, Dover SD, Elliott A. Structure of beta-poly-L-alanine—redefined atomic coordinates for an anti-parallel beta-pleated sheet. J Mol Biol. 1967;30:201–208. doi: 10.1016/0022-2836(67)90252-5. [DOI] [PubMed] [Google Scholar]
- 19.Geddes AJ, Parker KD, Atkins EDT, Beighton E. Cross-beta comformation in proteins. J Mol Biol. 1968;32:343–358. doi: 10.1016/0022-2836(68)90014-4. [DOI] [PubMed] [Google Scholar]
- 20.Auer S, Dobson CM, Vendruscolo M. Characterization of the nucleation barriers for protein aggregation and amyloid formation. HFSP J. 2007;1:137–146. doi: 10.2976/1.2760023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang W, Zhang S, Kamm RD, Karplus M. Kinetic control of dimer structure formation in amyloid fibrillogenesis. Proc Natl Acad Sci USA. 2004;101:12916–12921. doi: 10.1073/pnas.0402634101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobson CM, Karplus M. The fundamentals of protein folding: Bringing together theory and experiment. Curr Opin Struct Biol. 1999;9:92–101. doi: 10.1016/s0959-440x(99)80012-8. [DOI] [PubMed] [Google Scholar]
- 23.Abedini A, Raleigh DP. A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys Biol. 2009;6:015005. doi: 10.1088/1478-3975/6/1/015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shepherd NE, Hoang HN, Abbenante G, Fairlie DP. Single turn peptide alpha helices with exceptional stability in water. J Am Chem Soc. 2005;127:2974–2983. doi: 10.1021/ja0456003. [DOI] [PubMed] [Google Scholar]
- 26.Hatakeyama Y, Sawada T, Kawano M, Fujita M. Conformational preferences of short peptide fragments. Angew Chem Int Edit. 2009;48:8695–8698. doi: 10.1002/anie.200903563. [DOI] [PubMed] [Google Scholar]
- 27.Motta A, Reches M, Pappalardo L, Andreotti G, Gazit E. The preferred conformation of the tripeptide Ala-Phe-Ala in water is an inverse γ-turn: Implications for protein folding and drug design. Biochemistry. 2005;44:14170–14178. doi: 10.1021/bi050658v. [DOI] [PubMed] [Google Scholar]
- 28.Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-funtion paradigm. J Mol Biol. 1999;294:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 29.Mutter M. The construction of new proteins and enzymes—a prospect for the future. Angew Chem Int Edit. 1985;24:639–653. [Google Scholar]
- 30.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Accardo A, et al. In situ X-ray scattering studies of protein solution droplets drying on micro- and nanopatterned superhydrophobic PMMA surfaces. Langmuir. 2010;26:15057–15064. doi: 10.1021/la102958w. [DOI] [PubMed] [Google Scholar]
- 32.Gentile F, et al. Ultra low concentrated molecular detection using superhydrophobic surface based biophotonics devices. Microelectron Eng. 2010;87:798–801. [Google Scholar]
- 33.Riekel C, Burghammer M, Davies R, Gebhardt R, Popov D. In: Applications of Synchrotron Light to Non-Crystalline Diffraction in Materials and Life Sciences. García-Gutiérrez M, Nogales A, Gómez M, Ezquerra TA, editors. Heidelberg: Springer; 2008. [Google Scholar]
- 34.Labiche JC, et al. The fast readout low noise camera as a versatile X-ray detector for time resolved dispersive extended x-ray absorption fine structure and diffraction studies of dynamic problems in materials science, chemistry, and catalysis. Rev Sci Instrum. 2007;78:091301. doi: 10.1063/1.2783112. [DOI] [PubMed] [Google Scholar]
- 35.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 36.Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- 37.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 38.Essmann UL, et al. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 39.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 40.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.