

Abstract
Mercuric Hg(II) species form complexes with natural dissolved organic matter (DOM) such as humic acid (HA), and this binding is known to affect the chemical and biological transformation and cycling of mercury in aquatic environments. Dissolved elemental mercury, Hg(0), is also widely observed in sediments and water. However, reactions between Hg(0) and DOM have rarely been studied in anoxic environments. Here, under anoxic dark conditions we show strong interactions between reduced HA and Hg(0) through thiolate ligand-induced oxidative complexation with an estimated binding capacity of ~3.5 μmol Hg/g HA and a partitioning coefficient >106 mL/g. We further demonstrate that Hg(II) can be effectively reduced to Hg(0) in the presence of as little as 0.2 mg/L reduced HA, whereas production of Hg(0) is inhibited by complexation as HA concentration increases. This dual role played by DOM in the reduction and complexation of mercury is likely widespread in anoxic sediments and water and can be expected to significantly influence the mercury species transformations and biological uptake that leads to the formation of toxic methylmercury.
Keywords: Hg-dissolved organic matter complex, environmental factors, methylation, redox
Mercury (Hg) is well known to bioaccumulate and biomagnify as neurotoxic methylmercury (CH3Hg+) in organisms, particularly fish (1–3). Biologically mediated production of CH3Hg+ predominantly occurs under anaerobic conditions (4–8). However, the environmental factors that determine Hg availability to methylating bacteria and its transformation under these conditions remain poorly understood (1, 9–12). In particular, the coupled reactions between Hg redox transformation and complexation with natural dissolved organic matter (DOM) remain unclear, yet this process may critically control the speciation, biological uptake, and methylation of aqueous Hg in aquatic environments (9–11, 13–17). DOM occurs in all natural sediments and water, usually at concentrations much higher than Hg (1, 9). It is known to form exceptionally strong complexes with the oxidized mercuric species, Hg(II), due to its coordination with reduced sulfur (−S) or thiol (−SH) functional groups in DOM at relatively high DOM:Hg(II) ratios (11, 18–21). Such complexation has been shown to limit Hg(II) availability for bacterial methylation (9, 22, 23); however, facilitated uptake and methylation are also reported, especially when Hg(II) is complexed with small molecular-weight thiol compounds such as cysteine (5, 24).
Although a large body of literature is now available on the interactions of oxidized Hg(II) species with DOM, reactions between reduced gaseous Hg(0) and DOM have rarely been examined in natural sediments and water where dissolved Hg(0) is also observed (16, 17, 25–31). Hg(0) has a solubility of ~56 μg/L in water (32). Its formation can be mediated biologically (25, 26, 33), chemically (34, 35), or photochemically in the aquatic environment (15–17, 27–31). However, the role played by DOM in Hg(0) production remains a subject of debate. It has been observed that photoreduction of Hg(II) is inversely correlated to DOM levels in water (16, 29). Other studies, however, have shown that the presence of DOM corresponds with significant increase in Hg(II) photoreduction and that reduction rates increase with DOM concentration (15, 17, 30). Similarly, whereas in some studies DOM is shown to chemically reduce Hg(II) to Hg(0) (34, 35), in others it appears to have an inhibitory effect on Hg(II) reduction (36, 37). These apparently contradictory results suggest an incomplete mechanistic understanding of DOM and Hg interactions and the likely involvement of additional factors.
In this report, we show that under anoxic dark conditions, DOM can rapidly convert Hg(II) to Hg(0) at very low DOM concentrations (0.2 mg/L), but that Hg(0) production diminishes to undetectable levels as DOM concentration increases. The reduced DOM strongly interacts with Hg(0), likely through thiolate–ligand-induced oxidative complexation, with a measured binding affinity >106 mL/g, leading to an apparently low Hg(0) generation or Hg(II) reduction even under strongly reducing conditions. However, DOM shows little tendency to react with Hg(0) under conditions where DOM has been oxidized. Our results demonstrate that ambient redox conditions, the redox state of sulfur in DOM, and the DOM:Hg ratio are linked critical factors influencing the effects that DOM can have on Hg species transformation.
Results and Discussion
Hg(II) Reduction by Reduced Humic Acids.
To investigate Hg(II) reduction to Hg(0) by reduced DOM, solutions containing ≈10 nM Hg(II) [as Hg(NO3)2] were allowed to react with a reference Elliot soil humic acid (HA) (obtained from the International Humic Substances Society) at varying concentrations in an anaerobic glove chamber. To mimic the natural reducing conditions under which both bacterial methylation and HA reduction can occur, the HA was first reduced using established biological reduction techniques (38–41) detailed in Materials and Methods. Following equilibration (4 h) between HA and Hg(II), the free, purgeable Hg(0) in solution was quantified directly (without pretreatments) by cold-vapor atomic absorption spectrometry under a flow of ultra-high-purity nitrogen. Parallel experiments were performed using bacterial culture solutions (without HA) as controls because of concerns that media components and bacterial metabolites (e.g., secreted flavins) might contribute to Hg(II) reduction (42, 43). We note that this contribution is insignificant (<1%) in the final diluted samples, even assuming a secreted concentration of 5 μM flavins/g cellular protein by Shewanella cells (43). Fig. 1 shows that, at relatively low HA concentrations, Hg(II) is effectively reduced to Hg(0) by the reduced HA. Up to ~70% of Hg(II) was converted to purgeable Hg(0) in the presence of as little as ~0.2 mg/L reduced HA, a concentration well below that commonly observed in natural aquatic environments (9, 14, 17), equivalent to a calculated molar ratio of Hg(II):DOC of ~10−3. At this low HA concentration, we therefore find direct reduction of Hg(II) by reduced HA in the dark (34, 35) and Hg(0) production under reducing environments.
Fig. 1.

Reduction of mercuric Hg(II) by reduced humic acid (HA). Elliott soil HA was obtained from the International Humic Substances Society (IHSS) and prereduced by incubating with washed cells of S. putrefacien CN32 anaerobically (38–41). Cells were removed by filtering through a 0.2-μm filter. After appropriate dilutions, the filtered HA solution was used for reaction with Hg(II) as Hg(NO3)2 at 10 nM, and the purgeable Hg(0) was determined by direct purging and analysis after a 4-h period of equilibration. All data represent an average of duplicate measurements, with an estimated experimental error within ±14%. The bacterial culture blank was prepared identically to samples but without adding HA.
With further increase in HA concentration beyond 0.2 mg/L, however, purgeable Hg(0) decreased and was practically undetectable at HA concentration >5 mg/L (Fig. 1). This decrease in purgeable Hg(0) with increasing HA concentration indicates that reduction of Hg(II) is hindered by HA as observed previously (36, 37). Similar observations have been reported in photoreduction studies, where production of Hg(0) has been found to be inversely correlated to DOM levels in water (16, 29). This inverse relationship suggests that Hg(II) reduction is likely outcompeted by DOM complexation. It has been reported that strong complexation between Hg(II) and reduced-S or thiol functional groups in DOM prevents reduction of Hg(II) by SnCl2 (14, 44), particularly in the presence of excess DOM. On the basis of a total S content of 0.44% of the Elliott HA (source: International Humic Substances Society, http://www.ihss.gatech.edu/elements.html), of which ~1–15% may exist as reactive thiols (18, 21, 45), we estimate that in the presence of 0.2 mg/L HA ~0.3–4.1 nM of reactive thiols is available for complexation. However, on the basis of a reducing capacity of 0.34–0.53 mmol/g HA (40, 46), HA contains sufficient reducing moieties such as semiquinones in HA (~68–106 nM equivalents) to completely reduce Hg(II) (10 nM) in solution. Therefore, the prereduced HA has more reducing than binding equivalents for Hg(II). As HA was increased from 0 to 0.2 mg/L [~50 μmol Hg(II)/g HA], there were only a limited number of available binding equivalents in HA. Hg(II) was present primarily as non-HA-bound species and thus was subject to reduction, leading to the generation of a large percentage of purgeable Hg(0) (~70%) in solution (Fig. 1).
With increasing HA concentration, the binding equivalent also increased, and so more Hg(II) was complexed. In our specific case, when >0.2 mg/L HA was added, decreased production of Hg(0) occurred. Thus reduced HA plays a dual role in mercury redox reactions: At low HA concentrations it reduces Hg(II) whereas at high HA concentrations it competitively binds Hg(II). This process results in the optimal Hg(II):HA ratio for Hg(II) reduction observed under our experimental conditions (Fig. 1) although this ratio can vary with the redox state, the chemical and structural properties of DOM of different origins. The competition between Hg(II) reduction and complexation by DOM therefore offers a plausible explanation for the decrease in purgeable Hg(0) with increasing HA (Fig. 1), but the question remains: Would reduced Hg(0) also interact with HA to become stabilized and nonpurgeable in the solution?
Interactions Between Hg(0) and Reduced Humic Acids.
Thiol compounds and DOM are also reported to have a high affinity to sorb elemental Hg(0) (47–50). Therefore, direct interaction and stabilization of dissolved Hg(0) by HA cannot be ruled out in our experiment (Fig. 1). To elucidate the interactions between Hg(0) and HA, solutions containing 1.2 nM dissolved Hg(0) were first allowed to react with the reduced HA at varying concentrations in an anoxic glove chamber. The Hg(0) stock solution was prepared by equilibrating a small droplet of elemental Hg(0), sealed in silicon tubing, with deoxygenated water (51). The initial Hg(0) concentration was analyzed and validated for every batch of the experiment. Following 4-h equilibration, the free, purgeable Hg(0) concentration in solution was quantified as described earlier. Results show that purgeable Hg(0) decreased rapidly with increasing addition of HA (Fig. 2A). In the presence of > ~2 mg/L HA, no purgeable Hg(0) could be detected, demonstrating strong interactions between Hg(0) and HA. Because only reduced HA was used, and anoxic conditions were maintained throughout the experiments, direct oxidation of Hg(0) (as opposed to ligand-induced oxidation discussed below) does not appear to account for the observed retention or stabilization of Hg(0) by HA. This evaluation is also supported by parallel experiments, in which the HA solution was added together with 100 nM of reducing agent, either stannous chloride (SnCl2) or ascorbic acid, to ensure that strongly reducing conditions were maintained before introducing Hg(0) for reaction with HA. Results showed no change with or without the addition of SnCl2 or ascorbic acid (Fig. 2A); purgeable Hg(0) could not be detected at >2 mg/L HA, indicating that Hg remained stabilized in solution.
Fig. 2.

Interactions between dissolved gaseous Hg(0) (1.2 nM) and reduced or oxidized Elliott soil HA. (A) Purgeable Hg(0) measured at 4 h at varying HA concentrations; the open triangles and asterisks represent experiments performed in the presence of 100 nM SnCl2 and ascorbic acid, before the addition of Hg(0). (B) Reaction kinetics performed at a fixed HA concentration of 5 mg/L.
To further elucidate the mechanisms involved in Hg(0) and HA interaction, experiments were conducted to determine whether oxidized HA reacts with Hg(0), leading to increased oxidation of Hg(0) to Hg(II) for subsequently increased complexation with HA. Here “oxidized HA” refers to the same HA that was prepared under ambient conditions without the microbial reduction step (38–41). Whereas results show somewhat decreased purgeable Hg(0) (~20%) by using the oxidized HA (Fig. 2), ~80% of the Hg(0) remained free and purgeable, even during an extended reaction period of up to 48 h (Fig. 2B). Direct oxidation of Hg(0) by oxidized HA may thus account for the observed 20% Hg(0) loss in the experiment, but direct oxidation of Hg(0) by HA alone cannot explain the large percentage of decrease in Hg(0) in the presence of reduced HA. If direct oxidation of Hg(0) were the main removal mechanism, then, greater loss in Hg(0) would be expected when oxidized HA is used, which was not the case (Fig. 2).
In addition, we examined whether HA stabilizes Hg(0) through hydrophobic interactions, because natural humic substances are known to possess both hydrophobic and hydrophilic organic moieties. To do this, a solution of Hg(0) was allowed to react with two commonly used surfactants, polyoxyethylene nonylphenylether (Igepal CO-520) and cetyl trimethylammonium, with varying hydrophobic alkyl tail lengths and functional head groups (Fig. S1). No interactions between these surfactants and Hg(0) were observed. Potential loss of Hg(0) from the reaction vessel during experiments is discounted because good recovery of the total Hg (>90%) was obtained following complete oxidation of the HA or Hg–HA complexes by bromine chloride (14, 44).
These several lines of evidence suggest that specific interactions, including physicochemical sorption and ligand-induced oxidative complexation between reduced HA and Hg(0), are likely responsible for the decrease in purgeable Hg(0) with increasing HA in solution, as expressed in Reactions 1 and 2:
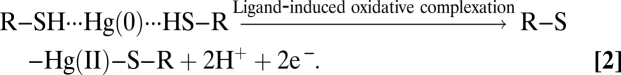 |
We therefore infer that the reaction may involve initial physical sorption (reaction 1), followed by S–H bond cleavage or charge transfer from Hg(0) to HA, leading to the formation of Hg(II)–HA complexes via Hg–thiolate bonds (reaction 2). Reactions and high affinities of thiols for Hg(0) are well documented (32, 47–50), although direct bond formation between Hg(0) and thiols (–SH) is unlikely due to both species being electron rich and Hg(0) having no unshared electrons. However, Hg is a class B soft metal with a strong tendency to coordinate or complex with soft bases such as thiols (21, 32). We found that this complexation is occurring not under oxidizing conditions but under strongly reducing conditions, in which Hg(II) is formed by ligand-induced oxidative complexation due to the strong tendency of Hg to react with reduced sulfur or thiols. However, we cannot rule out potential reactions between oxidized thiolate species such as disulfide (–S–S–) that may exist in reduced DOM. In this case, Hg(0) becomes oxidized and subsequently complexed with the intermediate thiolate products, as expressed in reactions 3 and 4,
where R–S–S–R′ represents DOM with oxidized sulfur or disulfides. Both sets of reactions (reactions 1–2 and 3–4) can thus take place and lead to ligand-induced oxidative complexation under reducing conditions. However, it would be difficult to distinguish the two mechanisms due to the formation of the same products. The key point here is that the oxidation state of the sulfur in DOM, which can either be oxidized or reduced, affects the ability of the overall DOM macromolecule to participate in charge-transfer and complexation reactions.
Reactions between Hg(0) and HA were further examined to determine their affinity and stability. At a fixed HA concentration of 5 mg/L, the amount of Hg retained by the HA increased linearly with increasing Hg(0) up to ~20 nM at pH 7 (Fig. 3A). Purgeable Hg(0) was essentially undetectable with the addition of 20 nM Hg(0), suggesting that all of the Hg was removed or stabilized by the HA. However, when excess Hg(0) was added, purgeable Hg(0) concentration increased linearly and was recovered almost completely, indicating that the HA had reached its capacity for binding and that Hg(0) could no longer react and be stabilized by the HA. The amount of Hg retained by HA shows a plateau when plotted against purgeable Hg(0) in solution (Fig. 3B). The maximum amount of Hg retained by the HA was ~3.5 μmol/g (or 0.7 mg/g) HA at pH 7. This binding capacity should be regarded as a conservative estimate because it accounts only for the high-affinity interaction (the initial slope) between Hg(0) and thiols on the HA. Fig. 3B also shows that this capacity decreased to ~1.6 μmol/g at pH 3.5 (Fig. S2), suggesting that lower pH decreases interaction between Hg(0) and HA. This observation is consistent with the view that the reaction involves deprotonation following bond cleavage of thiol (–SH) groups on HA (reaction 2), leading to the formation of HA–Hg complexes. Such complexes are so strong that even 10 mM Zn2+ (added as a competitive ion) had only limited effect on the complexation (<15%) (Fig. S3), consistent with the ability of Hg(II) to bind with thiols, with stability constants orders of magnitude greater than those of Zn2+ (45).
Fig. 3.

Interaction of dissolved gaseous Hg(0) with reduced soil HA. (A) Hg retained by HA (5 mg/L) at pH 7 and purgeable Hg(0) at varying initial Hg(0) concentrations. (B) Determination of binding capacity and affinity between Hg and reduced HA. The estimated binding affinity from the initial slope (dashed line) was >106 mL/g. Estimate strong binding sites are ~3.5 μmol/g HA at pH 7 and ~1.6 μmol/g HA at pH 3.5, respectively, assuming the formation of 1:1 Hg:thiol complexes. Controls were performed under the same conditions in water (asterisks) and in the culture solution (▽) prepared identically to that of the reduced HA solution.
Using the determined binding capacity at pH 7, we estimate a reactive thiol content of ~7 μmol/g of the reduced HA, assuming the formation of 1:2 Hg:thiol complexes, or ~3.5 μmol/g if 1:1 Hg:thiol complexes are formed. Previous studies established that the formation of Hg(II)–DOM complexes depends on the Hg(II):thiol ratio in the DOM (20, 21). Analyses using extended X-ray absorption fine structure spectroscopy (EXAFS) suggest that Hg(II) forms 1:2 complexes at relatively low Hg:thiol ratios (0.1–0.15) (21). Once these strong binding sites are saturated, weak binding groups such as amine and carboxyls become dominant (18, 20, 21). This pattern of Hg complexation is well illustrated in Fig. 3B, in which the amount of Hg retained by HA shows a sharp initial increase followed by a plateau as Hg(0) concentrations increase at both pH 3.5 and 7. The partitioning coefficient, estimated from the initial slope, exceeds 106 mL/g. At higher Hg(0):HA ratios, however, the slope decreases by five orders of magnitude to ~25 mL/g, indicating weaker interactions between Hg and the amine and/or carboxyls in the HA. We infer that, under these conditions, excess Hg(0) could no longer react with the HA and was thus purged from solution. This result is supported by the ease with which Hg(0) is purged from solutions containing weakly binding ligands such as acetate, citrate, and small amine–thiol ligands (14, 44).
We therefore summarize below our reasoning concerning the ligand-induced oxidative complexation of Hg(0) on the basis of the following lines of evidence: (i) Hg(0) in solution decreases as reduced HA increases; (ii) addition of reducing agents (SnCl2 and ascorbic acid) does not alter this trend, and loss of Hg(0) continues; (iii) Hg(0) is not lost due to volatilization, as indicated by the recovery of Hg after reaction with reduced HA; (iv) when oxidized HA is used, 80% of the Hg(0) remains in solution (Fig. 2); (v) reduced HA can reduce Hg(II), but complexation outcompetes reduction at increasing HA concentrations (Fig. 1); and (vi) reactions between Hg(0) and reduced HA are very strong, not hydrophobic, and show a complexation capacity similar to that of Hg(II)–DOM (Fig. 3). These observations indicate that both thiolate and disulfide functional groups in the reduced DOM facilitate the interaction with Hg(0) shown in reactions 1–4. This proposed reaction between reduced DOM and Hg(0) is also consistent with the observations that sorption of Hg(0) in soils and lake sediments increases with soil organic matter (28, 50). Reduced DOM thus sequesters Hg(0), but as Hg(II), not as Hg(0) itself.
Environmental Implications.
This study demonstrates that, in anoxic sediments and water, reduced DOM is not only capable of converting Hg(II) to Hg(0) but also capable of reacting with Hg(0) to form Hg–DOM complexes via ligand-induced oxidative complexation. These results shed light on the dual role of DOM in Hg reduction and complexation in anoxic environments where both bacterial methylation and DOM reduction occur (4, 5, 10, 38). Hg concentrations higher than those generally encountered in natural environments were used in this study to determine the reaction mechanisms and to demonstrate saturation of the binding sites. However, higher Hg concentrations, up to 500 nM, can be observed in contaminated environments (14, 45, 52, 53). At these sites, Hg(0) is present due to its prior industrial use and/or production. Microbial (25, 26, 33) and photochemical reduction (16, 17, 27–31) can also produce Hg(0) in natural sediments and water. Our study suggests that the fate of the Hg(0) is influenced by its interaction with DOM. Even in pristine environments, Hg(0) has been observed to be stabilized in sediments but released following biodegradation of DOM at the sediment–water interface (28). These observations would be accounted for by DOM-induced oxidative complexation of Hg(0) found in this study. We thus conclude that DOM at relatively low levels [low DOM:Hg(II) ratios] facilitates the reduction of Hg(II) under anoxic conditions. The Hg(0) produced by this direct reduction can be released through the water column to the atmosphere, thereby decreasing Hg availability for microbial methylation. In contrast, relatively high levels of DOM increase retention of Hg through complexation particularly under reducing conditions. Subsequent changes in redox conditions and/or in microbial activity can result in changes of the redox state of DOM and thus Hg species transformation and its availability for methylation.
This study also highlights the importance of the largely overlooked interaction between Hg(0) and biologically reduced DOM that exists widely in anoxic sediments and water (38–41). Over the past few decades, considerable attention has been paid to complexation between Hg(II) and DOM (11, 18–21). However, most of these studies have isolated and experimented on DOM under oxic conditions, which are known to cause the oxidation of redox-sensitive groups such as thiols in DOM. It may not therefore be possible to generalize results obtained under these conditions to reactions in anoxic environments where both Hg(II) and Hg(0) species may coexist with reduced DOM. Future studies of these reactions and processes could provide mechanistic insights into the factors controlling Hg species transformation, geochemical cycling, and especially toxic methylmercury production.
Materials and Methods
Reduction of mercuric Hg(II) ions by HA was determined by reacting Hg(II) [10 nM as Hg(NO3)2] with Elliott soil HA at concentrations ranging from 0 to 40 mg/L. The Elliott soil HA was obtained from the International Humic Substances Society (IHSS). It was used either without modification or after biological reduction as follows. The reduced HA was prepared by dissolving HA at 2,000 mg/L in deoxygenated water, adjusting its pH (~7), and then incubating with washed cells of Shewanella putrefaciens CN32 (~109 cells/mL) anaerobically, using established procedures with ethanol as an electron donor (38–41). Following reduction, the HA was filtered (0.2-μm filter) to remove cells, stored in the anaerobic chamber, and used to prepare fresh HA working solutions for each batch of the experiment. Control culture solutions, without the addition of HA, were prepared identically to examine the potential influence of bacterial metabolites and media components on the experiments. The HA samples were then prepared in a series of 20-mL amber vials, followed by the addition of Hg(II) from the working solution, which was made up in Milli-Q water directly from a 1,000-mg/L standard (Ricca Chemical Company, preserved in 3% HNO3). The final volume of each sample was 5 mL, and all samples were wrapped with aluminum foil or kept dark during equilibration. After equilibrating for 4 h, the reduced and purgeable gaseous Hg(0) in solution was determined directly by purging the solution with ultra-high-purity nitrogen and analyzing the vapor-phase Hg(0) (Lumex 915+) (14, 44). Samples with relatively low Hg(0) concentrations were analyzed by purging and trapping the vapor-phase Hg(0) onto a gold-coated sand trap, followed by thermal desorption under a flow of argon and detection by cold-vapor atomic fluorescence spectroscopy (CVAFS) (Brooks Rand Model III fluorescence spectrophotometer) (14). A purging time of 15 min was found to be sufficient to purge gaseous Hg(0) out of solution, as indicated by a nondetectable Hg(0) signal and a steady baseline. For mass-balance analysis, the complexed or nonpurgeable Hg in solution was determined following oxidation of the HA or Hg–HA complexes by bromine chloride (BrCl) for a minimum of 24 h (14). After oxidation, the total Hg in solution was reduced with SnCl2 and determined as described above. Duplicate samples were prepared for Hg(II) interactions in the presence or absence of HA with an estimated experimental error within ±14% or better. Parallel experiments were also performed using the same HA that was not biologically reduced but prepared by directly dissolving the HA under ambient conditions (referred to as the oxidized HA) to compare their reduction potential under nonreducing conditions.
Reactions between HA and dissolved Hg(0) were studied first by equilibrating the HA solutions (0–40 mg/L) in 20-mL amber vials with Hg(0) (1.2 nM) in an anoxic glove chamber. The Hg(0) stock solution was prepared following techniques reported by Whalin and Mason (51). Briefly, a droplet of elemental Hg(0) (~20 μL) was pipetted into a piece of small silicon tubing with one end capped with a Teflon plug. Following the transfer, the other end of the tubing was also plugged before the tube was immersed in water in a 40-mL glass vial. The gaseous Hg(0) was allowed to equilibrate with water for at least 48 h and kept in an anoxic glove chamber until use. The Hg(0) concentration in the stock was determined for every experimental batch and varied between ~200 and 300 nM, depending on the equilibration time. The working solution was then prepared from this stock after appropriate dilutions. Similarly, all samples were prepared in duplicate, and the final volume was made up to 5 mL. Following equilibration, the free, purgeable Hg(0) concentration in solution was determined directly by purging and analysis, described earlier. Additional series of experiments were performed in the presence of 100 nM SnCl2 or ascorbic acid in the HA solution to ensure strongly reducing conditions before the addition of Hg(0). For the determination of the reactions and partitioning of Hg(0) in HA, experiments were performed by equilibrating a fixed concentration (5 mg/L) of the HA solution with concentrations of Hg(0) ranging from 0 to 65 nM. Similarly, for mass-balance analysis, selected samples were analyzed for total Hg content following oxidation by BrCl (14) to determine the complexed or nonpurgeable Hg in solution.
Supplementary Material
Acknowledgments
We thank X. Yin for technical support and two anonymous reviewers for helpful comments and suggestions. This research was supported by the Office of Biological and Environmental Research, Office of Science, US Department of Energy, as part of the Mercury Science Focus Area Program at Oak Ridge National Laboratory. The Oak Ridge National Laboratory is managed by UT-Battelle LLC for the Department of Energy under Contract DE-AC05-00OR22725.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1008747108/-/DCSupplemental.
References
- 1.Morel FMM, Kraepiel AML, Amyot M. The chemical cycle and bioaccumulation of mercury. Annu Rev Ecol Syst. 1998;29:543–566. [Google Scholar]
- 2.Harris HH, Pickering IJ, George GN. The chemical form of mercury in fish. Science. 2003;301:1203. doi: 10.1126/science.1085941. [DOI] [PubMed] [Google Scholar]
- 3.Harris RC, et al. Whole-ecosystem study shows rapid fish-mercury response to changes in mercury deposition. Proc Natl Acad Sci USA. 2007;104:16586–16591. doi: 10.1073/pnas.0704186104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benoit JM, Gilmour CC, Heyes A, Mason RP, Miller CL. Geochemical and biological controls over methylmercury production and degradation in aquatic ecosystems. Biogeochem Environ Imp Trace Elem. 2003;835:262–297. [Google Scholar]
- 5.Schaefer JK, Morel FMM. High methylation rates of mercury bound to cysteine by Geobacter sulfurreducens. Nat Geosci. 2009;2:123–126. [Google Scholar]
- 6.Compeau GC, Bartha R. Sulfate-reducing bacteria: Principal methylators of mercury in anoxic estuarine sediment. Appl Environ Microbiol. 1985;50:498–502. doi: 10.1128/aem.50.2.498-502.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerin EJ, et al. Mercury methylation by dissimilatory iron-reducing bacteria. Appl Environ Microbiol. 2006;72:7919–7921. doi: 10.1128/AEM.01602-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilmour CC, Henry EA, Mitchell R. Sulfate stimulation of mercury methylation in fresh-water sediments. Environ Sci Technol. 1992;26:2281–2287. [Google Scholar]
- 9.Barkay T, Gillman M, Turner RR. Effects of dissolved organic carbon and salinity on bioavailability of mercury. Appl Environ Microbiol. 1997;63:4267–4271. doi: 10.1128/aem.63.11.4267-4271.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barkay T, Wagner-Döbler I. Microbial transformations of mercury: Potentials, challenges, and achievements in controlling mercury toxicity in the environment. Adv Appl Microbiol. 2005;57:1–52. doi: 10.1016/S0065-2164(05)57001-1. [DOI] [PubMed] [Google Scholar]
- 11.Skyllberg U. Competition among thiols and inorganic sulfides and polysulfides for Hg and MeHg in wetland soils and sediments under suboxic conditions: Illumination of controversies and implications for MeHg net production. J Geophys Res Biogeosci. 2008;113:G00C03. [Google Scholar]
- 12.Mason RP, Kim EH, Cornwell J, Heyes D. An examination of the factors influencing the flux of mercury, methylmercury and other constituents from estuarine sediment. Mar Chem. 2006;102:96–110. [Google Scholar]
- 13.Miller CL, Mason RP, Gilmour CC, Heyes A. Influence of dissolved organic matter on the complexation of mercury under sulfidic conditions. Environ Toxicol Chem. 2007;26:624–633. doi: 10.1897/06-375r.1. [DOI] [PubMed] [Google Scholar]
- 14.Miller CL, Southworth G, Brooks SC, Liang L, Gu B. Kinetic controls on the complexation between mercury and dissolved organic matter in a contaminated environment. Environ Sci Technol. 2009;43:8548–8553. doi: 10.1021/es901891t. [DOI] [PubMed] [Google Scholar]
- 15.Xiao ZF, Stromberg D, Lindqvist O. Influence of humic substances on photolysis of divalent mercury in aqueous-solution. Water Air Soil Pollut. 1995;80:789–798. [Google Scholar]
- 16.Amyot M, Mierle G, Lean D, McQueen DJ. Effect of solar radiation on the formation of dissolved gaseous mercury in temperate lakes. Geochim Cosmochim Acta. 1997;61:975–987. [Google Scholar]
- 17.O'Driscoll NJ, Lean DRS, Loseto LL, Carignan R, Siciliano SD. Effect of dissolved organic carbon on the photoproduction of dissolved gaseous mercury in lakes: Potential impacts of forestry. Environ Sci Technol. 2004;38:2664–2672. doi: 10.1021/es034702a. [DOI] [PubMed] [Google Scholar]
- 18.Haitzer M, Aiken GR, Ryan JN. Binding of mercury(II) to dissolved organic matter: The role of the mercury-to-DOM concentration ratio. Environ Sci Technol. 2002;36:3564–3570. doi: 10.1021/es025699i. [DOI] [PubMed] [Google Scholar]
- 19.Xia K, et al. X-ray absorption spectroscopic evidence for the complexation of Hg(II) by reduced sulfur in soil humic substances. Environ Sci Technol. 1999;33:257–261. [Google Scholar]
- 20.Hesterberg D, Chou JW, Hutchison KJ, Sayers DE. Bonding of Hg(II) to reduced organic sulfur in humic acid as affected by S/Hg ratio. Environ Sci Technol. 2001;35:2741–2745. doi: 10.1021/es001960o. [DOI] [PubMed] [Google Scholar]
- 21.Skyllberg U, Bloom PR, Qian J, Lin CM, Bleam WF. Complexation of mercury(II) in soil organic matter: EXAFS evidence for linear two-coordination with reduced sulfur groups. Environ Sci Technol. 2006;40:4174–4180. doi: 10.1021/es0600577. [DOI] [PubMed] [Google Scholar]
- 22.Hintelmann H, Keppel-Jones K, Evans RD. Constants of mercury methylation and demethylation rates in sediments and comparison of tracer and ambient mercury availability. Environ Toxicol Chem. 2000;19:2204–2211. [Google Scholar]
- 23.Miskimmin BM, Rudd JWM, Kelly CA. Influence of dissolved organic-carbon, pH, and microbial respiration rates on mercury methylation and demethylation in lake water. Can J Fish Aquat Sci. 1992;49:17–22. [Google Scholar]
- 24.Golding GR, et al. Evidence for facilitated uptake of Hg(II) by Vibrio anguillarum and Escherichia coli under anaerobic and aerobic conditions. Limnol Oceanogr. 2002;47:967–975. [Google Scholar]
- 25.Barkay T, Liebert C, Gillman M. Environmental significance of the potential for mer(Tn21)-mediated reduction of Hg2+ to Hg0 in natural waters. Appl Environ Microbiol. 1989;55:1196–1202. doi: 10.1128/aem.55.5.1196-1202.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaefer JK, Letowski J, Barkay T. mer-mediated resistance and volatilization of Hg(II) under anaerobic conditions. Geomicrobiol J. 2002;19:87–102. [Google Scholar]
- 27.Poulain AJ, et al. Biological and photochemical production of dissolved gaseous mercury in a boreal lake. Limnol Oceanogr. 2004;49:2265–2275. [Google Scholar]
- 28.Bouffard A, Amyot M. Importance of elemental mercury in lake sediments. Chemosphere. 2009;74:1098–1103. doi: 10.1016/j.chemosphere.2008.10.045. [DOI] [PubMed] [Google Scholar]
- 29.Rolfhus KR, Fitzgerald WF. The evasion and spatial/temporal distribution of mercury species in Long Island Sound, CT-NY. Geochim Cosmochim Acta. 2001;65:407–418. [Google Scholar]
- 30.Costa M, Liss PS. Photoreduction of mercury in sea water and its possible implications for Hg0 air-sea fluxes. Mar Chem. 1999;68:87–95. [Google Scholar]
- 31.Lalonde JD, Amyot M, Doyon MR, Auclair JC. Photo-induced Hg(II) reduction in snow from the remote and temperate Experimental Lakes Area (Ontario, Canada) J Geophys Res. 2003;108:4200. [Google Scholar]
- 32.Schuster E. The behavior of mercury in the soil with special emphasis on complexation and adsorption processes—a review of the literature. Water Air Soil Pollut. 1991;56:667–680. [Google Scholar]
- 33.Wiatrowski HA, Ward PM, Barkay T. Novel reduction of mercury (II) by mercury-sensitive dissimilatory metal reducing bacteria. Environ Sci Technol. 2006;40:6690–6696. doi: 10.1021/es061046g. [DOI] [PubMed] [Google Scholar]
- 34.Alberts JJ, Schindler JE, Miller RW, Nutter DE., Jr Elemental mercury evolution mediated by humic acid. Science. 1974;184:895–897. doi: 10.1126/science.184.4139.895. [DOI] [PubMed] [Google Scholar]
- 35.Allard B, Arsenie I. Abiotic reduction of mercury by humic substances in aquatic system—an important process for the mercury cycle. Water Air Soil Pollut. 1991;56:457–464. [Google Scholar]
- 36.Rocha JC, et al. Reduction of mercury(II) by tropical river humic substances (Rio Negro)—A possible process of the mercury cycle in Brazil. Talanta. 2000;53:551–559. doi: 10.1016/s0039-9140(00)00532-4. [DOI] [PubMed] [Google Scholar]
- 37.Mauclair C, Layshock J, Carpi A. Quantifying the effect of humic matter on the emission of mercury from artificial soil surfaces. Appl Geochem. 2008;23:594–601. [Google Scholar]
- 38.Lovley DR. Humic substances as electron acceptors for microbial respiration. Nature. 1996;382:445–448. [Google Scholar]
- 39.Royer RA, et al. Enhancement of hematite bioreduction by natural organic matter. Environ Sci Technol. 2002;36:2897–2904. doi: 10.1021/es015735y. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Gu B, Royer RA, Burgos WD. The roles of natural organic matter in chemical and microbial reduction of ferric iron. Sci Total Environ. 2003;307:167–178. doi: 10.1016/S0048-9697(02)00538-7. [DOI] [PubMed] [Google Scholar]
- 41.Gu B, Chen J. Enhanced microbial reduction of Cr(VI) and U(VI) by different natural organic matter fractions. Geochim Cosmochim Acta. 2003;67:3575–3582. [Google Scholar]
- 42.Marsili E, et al. Shewanella secretes flavins that mediate extracellular electron transfer. Proc Natl Acad Sci USA. 2008;105:3968–3973. doi: 10.1073/pnas.0710525105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Canstein H, Ogawa J, Shimizu S, Lloyd JR. Secretion of flavins by Shewanella species and their role in extracellular electron transfer. Appl Environ Microbiol. 2008;74:615–623. doi: 10.1128/AEM.01387-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamborg CH, Tseng CM, Fitzgerald WF, Balcom PH, Hammerschmidt CR. Determination of the mercury complexation characteristics of dissolved organic matter in natural waters with “reducible Hg” titrations. Environ Sci Technol. 2003;37:3316–3322. doi: 10.1021/es0264394. [DOI] [PubMed] [Google Scholar]
- 45.Dong WM, Liang L, Brooks SC, Southworth G, Gu B. Roles of dissolved organic matter in the speciation of mercury and methylmercury in a contaminated ecosystem in Oak Ridge, Tennessee. Environ Chem. 2009;7:94–102. [Google Scholar]
- 46.Scott DT, McKnight DM, Blunt-Harris EL, Kolesar SE, Lovley DR. Quinone moieties act as electron acceptors in the reduction of humic substances by humic-reducing microorganisms. Environ Sci Technol. 1998;32:2984–2989. [Google Scholar]
- 47.Arias ZG, Alvarez JLM, Fonseca JML. Electrochemical characterization of the self-assembled monolayer of 6-thioguanine on the mercury electrode. Electroanalysis. 2004;16:1044–1050. [Google Scholar]
- 48.Lee JY, Ju YH, Keener TC, Varma RS. Development of cost-effective noncarbon sorbents for Hg(0) removal from coal-fired power plants. Environ Sci Technol. 2006;40:2714–2720. doi: 10.1021/es051951l. [DOI] [PubMed] [Google Scholar]
- 49.Makkuni A, Varma RS, Sikdar SK, Bhattacharyya D. Vapor phase mercury sorption by organic sulfide modified bimetallic iron-copper nanoparticle aggregates. Ind Eng Chem Res. 2007;46:1305–1315. [Google Scholar]
- 50.Fang SC. Studies on the sorption of elemental mercury vapor by soils. Arch Environ Contam Toxicol. 1981;10:193–201. doi: 10.1007/BF01055621. [DOI] [PubMed] [Google Scholar]
- 51.Whalin LM, Mason RP. A new method for the investigation of mercury redox chemistry in natural waters utilizing deflatable Teflon (R) bags and additions of isotopically labeled mercury. Anal Chim Acta. 2006;558:211–221. [Google Scholar]
- 52.Azzaria LM, Aftabi A. Stepwise thermal-analysis technique for estimating mercury phases in soils and sediments. Water Air Soil Pollut. 1991;56:203–217. [Google Scholar]
- 53.Biester H, Nehrke G. Quantification of mercury in soils and sediments—Acid digestion versus pyrolysis. Fresenius J Anal Chem. 1997;358:446–452. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.