

Abstract
Homozygous mutations in the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene cause hereditary inclusion body myopathy type 2 (HIBM2). We describe two unrelated American patients with novel GNE mutations. While one patient followed a typical disease course for HIBM2 with an onset at age 25 and rimmed vacuole pathology on muscle biopsy, the second patient had several features atypical for HIBM2. This patient's onset was at age 55, included distal weakness, quadriceps sparing and respiratory insufficiency. His muscle biopsy showed prominent necrosis without rimmed vacuoles. This study expands the phenotype and illustrates the clinical spectrum of HIBM2 identified in a U.S. based neuromuscular clinic.
Keywords: IBM, GNE, distal myopathy, rimmed vacuoles
Introduction
Hereditary inclusion body myopathy type 2 (HIBM2) is due to homozygous mutations in the bifunctional enzyme UDP-GlcNAc 2-Epimerase/ManNAc Kinase gene (GNE) [1]. GNE is an essential enzyme necessary for sialic acid synthesis. Whether HIBM2 associated GNE mutations cause a deficiency in sialic acid synthesis remains unclear [2, 3]. GNE mutations were initially identified and later found in two clinically similar autosomal recessively inherited neuromuscular disorders [1, 4]. These two disorders are quadriceps sparing inclusion body myopathy in patients of Iranian Jewish descent and distal myopathy with rimmed vacuoles or Nonaka myopathy in Japanese patients. HIBM2 patients have adult onset progressive distal weakness that typically spares the quadriceps muscle [1]. Biopsy of affected muscle has characteristic rimmed vacuoles and tubulofilamentous inclusions [1]. To date greater than 60 unique missense mutations in the GNE gene have been identified in HIBM2 patients [5]. In the current report, we describe two patients with HIBM2 identified in our clinic. These patients were identified based upon their pattern of distal weakness with relative sparing of the quadriceps. Sequencing of the GNE gene identified four missense mutations, three of them being novel. Interestingly, the patients are clinically distinct from each other in that one followed a more traditional clinical course for HIBM2 whereas the second patient had several unique features including late onset at 55 years of age, respiratory weakness and a biopsy with prominent necrotic features. To our knowledge this would be latest presentation for a patient with defined GNE mutations. This study highlights the phenotypic spectrum of HIBM2 patients that can be identified in a U.S. based neuromuscular clinic. Our study suggests that patients with distal weakness regardless of family history, muscle biopsy features or age of onset should be tested for GNE mutations in order to make the diagnosis of HIBM2.
Case Report
Patient 1
A 54 year-old Caucasian woman initially presented at the age of 25 with progressive bilateral foot dorsiflexion weakness of two years duration. In retrospect she had been told by others that she “walked funny” for the past several years An early exam at the age of 25 demonstrated bilateral foot drop with a high steppage gait, 3/5 weakness in neck extensors and bilateral winging of the scapula. This pattern of weakness progressed gradually. She began using a cane and a walker around the age of 35 and was wheelchair-dependent by the age of 45. At 54, she is now totally dependent in activities of daily living. Family history is notable for a distant maternal cousin with a presumed diagnosis of Duchenne Muscular Dystrophy. No other family members have muscle weakness.
On examination she had no ocular, facial, or bulbar weakness. She had severe weakness (3/5) of neck extensors and moderate (4/5) weakness of neck flexors. There was bilateral scapular winging. She had very severe weakness (1-2/5) in the proximal and distal muscles of all limbs except the quadriceps which were relatively spared at 5/5 bilaterally. Reflexes were diffusely absent and sensory examination was normal. Patient was unable to stand or walk.
Patient 2
A 65 year-old Caucasian man presented with bilateral leg weakness. He developed a gradually progressive bilateral foot drop 10 years prior to presentation. A few years later, he noted difficulty in climbing steps and rising from a chair. These difficulties were associated with muscle wasting in the lower legs and thighs. He also complained of numbness in his feet and on the posterior aspects of his thighs. Notably, he played basketball in college and later golf without any remarkable weakness until the age of 55. He had a post-ischemic dilated cardiomyopathy and atrial fibrillation secondary to a myocardial infarction at the age of 47. His CK was initially noted to be elevated after starting ezetimibe for high cholesterol. A repeat CK demonstrated a persistent elevation after ezetimibe was discontinued. He had never taken any statin medications and takes colesevelam for his hyperlipidemia. His father died at the age of 82 from heart disease and mother died at the age of 62 from ovarian cancer. There is no history of neuromuscular disease in the family.
On exam his vital capacity was 53% and FEV1 was 35% of predicted. Cranial nerves were intact except for mild ptosis on the left. Symmetric and diffuse atrophy was noted in both lower legs. He had limitation of ankle dorsiflexion at 90°. Weakness was symmetric in the following muscle groups, foot dorsiflexion (3+/5), hamstrings (4/5), hip flexors (4+/5), finger and wrist extensors (4/5). Strength in other muscle groups was normal, most notably in the bilateral quadriceps (5/5). Pinprick, temperature and vibration were diminished in the feet and distal legs. Tendon reflexes were +2, symmetric in the arms and knees but absent at the ankles.
Laboratory tests, electrophysiology and ultrasound
Patient 1 had a CK of 371 IU/L (nl 30-200) and patient 2 had a CK of 1610 IU/L and aldolase of 18.2 (nl 0-8.0) at the time of presentation. Other lab tests were reported as normal. Patient 1 had normal nerve conduction studies whereas patient 2 had normal nerve conduction studies with the exception of absent posterior tibial H-reflexes bilaterally which may be compatible with an early neuropathy. Needle electromyography of both patients showed distal worse than proximal myopathic potentials with the presence of fibrillations and positive sharp waves in some of the muscles examined. Ultrasound of the quadriceps in patient 1 revealed increased echogenicity in the rectus femoris, vastus medius, lateralis and intermedius (Figure 1). In contrast, a similar study performed in patient 2 identified only mild atrophy of the rectus femoris as compared to normal control quadriceps muscle (Figure 1).
Figure 1.
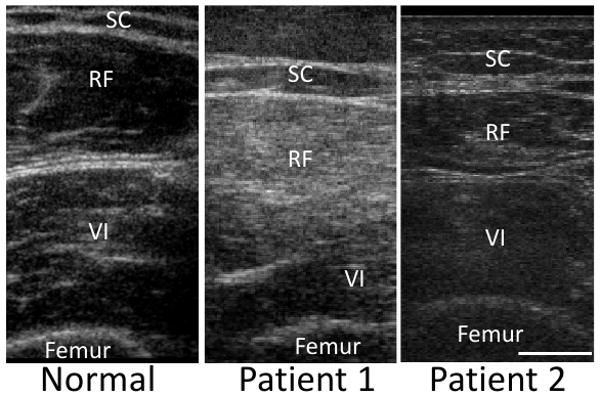
Ultrasonic imaging of quadriceps muscle from normal control or one of two HIBM2 patients. In patient 1, echogenicity is increased in the rectus femoris (RF) and vastus intermedius (VI) compared to the subcutaneous fat (SC). In contrast, only mild atrophy of the rectus femoris is seen in patient 2. Scale is 1 cm.
Muscle biopsy
Patient 1 had an open muscle biopsy performed on her right deltoid. It showed variation in fiber size and rimmed vacuoles in many small angular fibers. These vacuoles were appreciated on hematoxylin and eosin, modified gomori trichrome (Figure 2) and congo red stains. Vacuoles did not react with acid phosphatase or non-specific esterase. They were most commonly in type II fibers. There was no evidence of inflammation on alkaline phosphatase or by secondary immune stains (anti-MHCI, anti-C5b9, anti-CD4). Occasional fibers had the accumulation of the autophagosome marker MAP1-LC3 (Figure 2) and most fibers had a global increase in lysosome associated membrane protein-2 (Lamp2) staining. Neither MAP1-LC3 or Lamp2 were associated with vacuolar structures.
Figure 2.
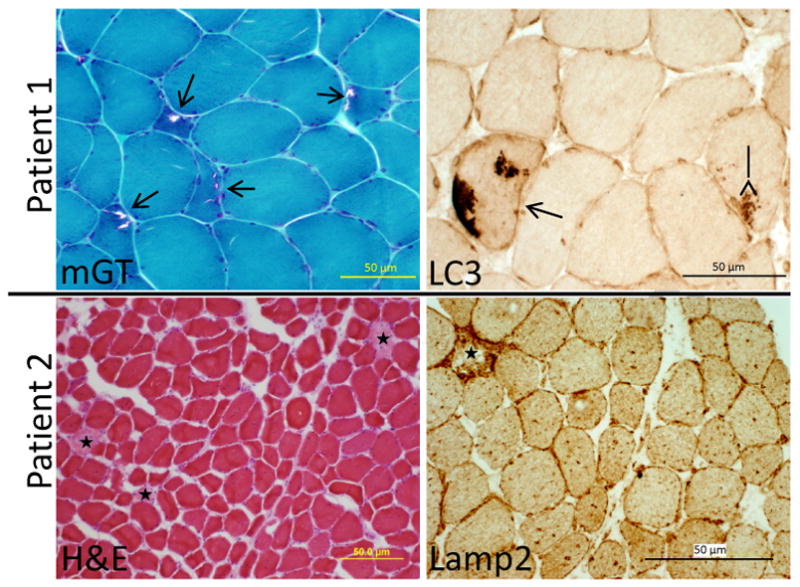
Muscle histochemistry with modified gomori trichrome (mGT) or hematoxylin and eosin (H&E) and immunohistochemistry with anti-LC3 or anti-Lamp2 of patient 1 (top) and patient 2 (bottom). Open arrows denote rimmed vacuoles, closed arrows mark LC3 positive inclusions and stars highlight necrotic fibers.
Patient 2 had an open biopsy of his left quadriceps. It demonstrated scattered necrotic and regenerating fibers with numerous internal nuclei (Figure 2). There were no vacuoles appreciated on any of the muscle stains. Necrosis and an absence of associated inflammation was visualized on hematoxylin and eosin, acid phosphatase stains and secondary immunostaining with anti-C5B9. MAP1-LC3 was again seen in scattered fibers but more striking was the accumulation of large Lamp2 positive vesicles in all fibers including non-necrotic ones (Figure 2).
Genetic Analysis
The coding sequence of the GNEgene was amplified and sequenced as previously described [1]. Patient 1 is heterozygous for two mutations: c.76G>C and c.1892C>T, producing two respective missense mutations, p.A26P which is novel, and p.A631V in the kinase domain which has been previously reported. Patient 2 is heterozygous for two novel mutations: c.998A>G producing an p.H333R missense mutation and c.1083T>G producing a p.Y361X nonsense mutation.
Discussion
The current report presents two unrelated patients with HIBM2 identified in a U.S. based specialty neuromuscular clinic. Patient 1 followed a classic presentation for HIBM2. She developed distal weakness in her early 20s that progressed to wheelchair dependence after 20 years. Now nearly 30 years after her initial symptoms she is profoundly weak in all muscle groups with the exception of her quadriceps which, although abnormal on ultrasound, still has normal (5/5) strength. Her muscle biopsy demonstrated classic histochemical features for HIBM2 with small angular fibers and rimmed vacuoles.
In contrast, patient 2 presented with several atypical features for HIBM2. He had marked respiratory weakness as measured by bedside spirometry. Unlike other inclusion body myopathies, the diaphragm and intercostals muscle are typically spared in patients with GNE mutations [4, 6]. He developed weakness in his 6th decade, the oldest onset of weakness reported in a patient with HIBM2. Most manifesting patients with homozygous GNE mutations develop symptoms by the end of the 3rd decade. Argov et al reported 5 non-manifesting patients with homozygous GNE mutations the oldest of which was 68 [6]. It is not known if this patient went on to develop symptoms of HIBM2. Unlike patient 1 and other patients with HIBM2, patient 2 had only minimal abnormalities on ultrasound of the rectus femoris [7]. His muscle biopsy was also atypical for HIBM2 and revealed scattered necrosis with no obvious vacuoles. A “necrotizing myopathy” has been previously described in another a patient with homozygous GNE mutations although a subsequent biopsy did find rimmed vacuoles [8]. Interestingly both our patient and this previously reported patient had necrosis evident on a quadriceps biopsy. The quadriceps muscle is typically unaffected in HIBM2. Proper selection of muscle biopsy site may avoid misdiagnosis and repeat biopsies in patients with HIBM2 [9]. Patient 2's sensory findings and their relation to GNE mutations is unclear. Many patients with HIBM2 report parethesias or extremities that feel cold [6], however; our patient had subject sensory complaints and nerve conduction changes that could be consistent with an axonal polyneuropathy.
Our study identified three novel GNE mutations adding to the growing list of HIBM2 mutations. Patient 1 is a compound heterozygote with a novel A26P and A631V mutation. A missense mutation at residue 631 has been previously reported in multiple patients and families [1]. The novel A26P mutation lies next to a previously reported P27S mutation [10]. Patient 2 is a compound heterozygote for two novel mutations, H333R and Y361X. These mutations lie within exon 6 and 7 respectively. Interestingly a previously reported family with homozygous mutations in exon 7 (L379H) identified rimmed vacuoles in only 2/5 family members [11] and another report of a single patient with homozygous V367I mutations in GNE found necrosis, inflammation and rimmed vacuoles [12]. Perhaps patients homozygous for mutations within this region have less classic HIBM2 pathology.
The vacuoles in patients with HIBM2 are thought to be autophagic in nature. This is evidenced by detailed pathologic studies of HIBM2 patient biopsies which found that hydrolytic enzyme stains such as acid phosphatase are present around and within sarcoplasmic vacuoles [13]. Other studies have identified an increase in Lamp2 around vacuoles and within the sarcoplasm of muscle fibers from HIBM2 patients [14]. The accumulation of the autophagic marker LC3 and its localization to vacuolar structures is seen in sporadic inclusion body myositis [15, 16] and inclusion body myopathy associated with paget's disease of the bone and fronto-temporal dementia [17]. We found an increase in acid phosphatase staining as well as LC3 and Lamp2 positive immunoreactivity in both our HIBM2 patients. This staining was not exclusively localized to vacuoles but instead was found throughout the sarcoplasm of scattered fibers. This data supports a defect in the autophagosome-lysosomal system as a pathogenic mechanism in HIBM2 and further suggests that a dysregulation in autophagy may be more widespread than vacuolated fibers.
We identified these two patients based upon their phenotypic presentation of distal weakness with relative sparing of quadriceps strength. Despite this preservation of strength, the quadriceps of patients with HIBM2 can demonstrate pathology on ultrasound [7]. Although our two, sequence confirmed, HIBM2 patients had differing presentations, one feature unifies them: a distal myopathy with relative sparing of the quadriceps strength. Patients with this distinctive pattern of weakness should be evaluated for GNE mutations regardless of family history, time of onset, radiologic evidence of quadriceps pathology, or absence of vacuoles on muscle pathology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Eisenberg I, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29(1):83–7. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 2.Huizing M, et al. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab. 2004;81(3):196–202. doi: 10.1016/j.ymgme.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 3.Salama I, et al. No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNE mutation. Biochem Biophys Res Commun. 2005;328(1):221–6. doi: 10.1016/j.bbrc.2004.12.157. [DOI] [PubMed] [Google Scholar]
- 4.Nishino I, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59(11):1689–93. doi: 10.1212/01.wnl.0000041631.28557.c6. [DOI] [PubMed] [Google Scholar]
- 5.Saechao C, et al. Novel GNE mutations in hereditary inclusion body myopathy patients of non-Middle Eastern descent. Genet Test Mol Biomarkers. 14(2):157–62. doi: 10.1089/gtmb.2009.0157. [DOI] [PubMed] [Google Scholar]
- 6.Argov Z, et al. Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology. 2003;60(9):1519–23. doi: 10.1212/01.wnl.0000061617.71839.42. [DOI] [PubMed] [Google Scholar]
- 7.Adler RS, et al. Muscle sonography in six patients with hereditary inclusion body myopathy. Skeletal Radiol. 2008;37(1):43–8. doi: 10.1007/s00256-007-0367-6. [DOI] [PubMed] [Google Scholar]
- 8.Motozaki Y, et al. Hereditary inclusion body myopathy with a novel mutation in the GNE gene associated with proximal leg weakness and necrotizing myopathy. Eur J Neurol. 2007;14(9):e14–5. doi: 10.1111/j.1468-1331.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- 9.Vasconcelos OM, Raju R, Dalakas MC. GNE mutations in an American family with quadriceps-sparing IBM and lack of mutations in s-IBM. Neurology. 2002;59(11):1776–9. doi: 10.1212/01.wnl.0000039780.13681.ad. [DOI] [PubMed] [Google Scholar]
- 10.Broccolini A, et al. Novel GNE mutations in Italian families with autosomal recessive hereditary inclusion-body myopathy. Hum Mutat. 2004;23(6):632. doi: 10.1002/humu.9252. [DOI] [PubMed] [Google Scholar]
- 11.Amouri R, et al. Allelic heterogeneity of GNE gene mutation in two Tunisian families with autosomal recessive inclusion body myopathy. Neuromuscul Disord. 2005;15(5):361–3. doi: 10.1016/j.nmd.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Krause S, et al. A novel homozygous missense mutation in the GNE gene of a patient with quadriceps-sparing hereditary inclusion body myopathy associated with muscle inflammation. Neuromuscul Disord. 2003;13(10):830–4. doi: 10.1016/s0960-8966(03)00140-8. [DOI] [PubMed] [Google Scholar]
- 13.Sugie K, et al. Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J Neuropathol Exp Neurol. 2005;64(6):513–22. doi: 10.1093/jnen/64.6.513. [DOI] [PubMed] [Google Scholar]
- 14.Tsuruta Y, et al. Expression of the lysosome-associated membrane proteins in myopathies with rimmed vacuoles. Acta Neuropathol. 2001;101(6):579–84. doi: 10.1007/s004010000329. [DOI] [PubMed] [Google Scholar]
- 15.Temiz P, Weihl CC, Pestronk A. Inflammatory myopathies with mitochondrial pathology and protein aggregates. J Neurol Sci. 2009;278(1-2):25–9. doi: 10.1016/j.jns.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Lunemann JD, et al. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol. 2007;61(5):476–83. doi: 10.1002/ana.21115. [DOI] [PubMed] [Google Scholar]
- 17.Ju JS, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187(6):875–88. doi: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]