

Abstract
Biological fluids contain a very high total concentration of macromolecules that leads to volume exclusion by one molecule to another. Theory and experiment have shown that this condition, termed macromolecular crowding, can have significant effects on molecular recognition. However, the influence of molecular crowding on recognition events involving virus particles, and their inhibition by antiviral compounds, is virtually unexplored. Among these processes, capsid self-assembly during viral morphogenesis and capsid-cell receptor recognition during virus entry into cells are receiving increasing attention as targets for the development of new antiviral drugs. In this study, we have analyzed the effect of macromolecular crowding on the inhibition of these two processes by peptides. Macromolecular crowding led to a significant reduction in the inhibitory activity of: 1), a capsid-binding peptide and a small capsid protein domain that interfere with assembly of the human immunodeficiency virus capsid, and 2), a RGD-containing peptide able to block the interaction between foot-and-mouth disease virus and receptor molecules on the host cell membrane (in this case, the effect was dependent on the conditions used). The results, discussed in the light of macromolecular crowding theory, are relevant for a quantitative understanding of molecular recognition processes during virus infection and its inhibition.
Introduction
The total concentration of macromolecules in intracellular compartments and extracellular fluids is extremely high (ranging from ∼80 mg/mL in blood plasma to ∼300 mg/mL in bacterial cells). Such conditions lead to excluded volume effects arising from the mutual impenetrability of soluble macromolecules, a situation known as macromolecular crowding (1–5). In a crowded solution, the reactivity of a given solute is determined by the number of solute molecules per unit of available (not total) volume. Thus, in a molecularly crowded medium, the thermodynamic activity (i.e., effective concentration) will be higher than in a dilute solution by a factor that corresponds to the activity coefficient of that solute.
Crowding is a nonspecific force that favors reactions leading to a reduction in the total excluded volume, such as the formation of soluble macromolecular complexes or insoluble aggregates, folding of proteins, and the binding of macromolecules to surface sites. Many of the effects predicted by macromolecular crowding theory have been observed experimentally using a large variety of systems involving proteins and/or other cellular components (e.g., (1–16)). The magnitude of the crowding effect is highly dependent on the sizes and shapes of crowding species and dilute macromolecular species. For example, macromolecular crowding in volume-occupied media will increase the thermodynamic activity of large and/or elongated macromolecules relative to that of small, compact solutes; this effect may lead to a significant enhancement of the binding of large and/or elongated ligands to a receptor relative to that of small, compact ligands (2).
During the life cycle of viruses, most reactions in which viral macromolecules or the viral particles themselves participate also occur in molecularly crowded environments, either intracellular or extracellular. To what extent macromolecular crowding could have an influence on the different specific recognition steps of the viral life cycle has remained, to our knowledge, a virtually unexplored question, despite its relevance for a better understanding of virus biology and the fight against viral disease. In a previous study, we used an in vitro system to show that addition of macromolecular crowding agents induced the efficient assembly of the human immunodeficiency virus type 1 (HIV-1) capsid in close to physiological conditions (10). In this study, our goal has been to experimentally analyze whether macromolecular crowding could have an influence on the inhibition by different compounds of essential steps of the viral cycle that involve virus particles.
Most licensed or candidate antiviral drugs are small molecules that act by binding a viral or cellular macromolecule or macromolecular complex, such as a virus capsid, and inhibiting its function (17–20). In particular, two very promising antiviral strategies that are receiving increasing attention are
-
1.
The inhibition of virus assembly.
-
2.
The inhibition of virus-host cell recognition or virus entry.
For example, several experimental inhibitors of HIV-1 capsid assembly have been described (21–25). In addition, two antiviral drugs that inhibit virus-cell recognition and entry have been recently licensed for chemotherapy against HIV-1 infection (20), and others are in different stages of development and/or trials. Some of these antiviral compounds are small peptides (e.g., (20,22–24,26–32).
Both virus self-assembly and virus-cell receptor recognition do occur in macromolecularly crowded environments in vivo. Because of their outstanding interest as new targets for antiviral therapy, in this work we have focused on these two crucial steps of the viral cycle. To this aim, we have used specific, in vitro simplified models of these viral processes that are well established in our laboratory.
As a model for the inhibition of virus capsid self-assembly, we chose HIV-1. During HIV morphogenesis, a molecular reorganization process occurs where the mature capsid is assembled from hundreds of copies of its monomeric component, the capsid protein CA (see recent reviews in (33,34)). The CA protein is composed of two domains. The smaller, C-terminal domain (CTD) is responsible for CA dimerization and, in its isolated form, is able to efficiently inhibit the in vitro assembly of the HIV-1 capsid (35,36). Other compounds such as small organic molecules (21), lipids (37), dendrimers (25), and peptides ((22,23) and Rebeca Bocanegra, José Luis Neira, and Mauricio G. Mateu, unpublished results) are also able to inhibit this process. A small peptide (CAI) that was identified by screening a combinatorial library of dodecapeptides binds the CTD and is able to impair the polymerization of CA into capsids by inducing a conformational distortion of CTD and/or sterically impairing self-association of CA (22,38,39).
A substantially modified, conformationally restricted peptide variant of CAI has been shown to penetrate susceptible cells and inhibit HIV-1 infection ex vivo (23). The inhibition of HIV-1 capsid assembly by CTD or CAI is a consequence of their binding to capsid-building blocks and growing capsid particles, thus interfering with CA-CA recognition (22,35,36). According to favored models (40), those building blocks include not only CA dimers, but also preassembled, higher-order oligomers of considerably larger size than CA, CTD, and CAI.
As a model for the inhibition of virus-host cell receptor recognition we chose foot-and-mouth disease virus (FMDV), the causative agent of one of the most economically important animal diseases worldwide (reviewed in (41,42)). Infection by FMDV involves the attachment of the virion to integrin receptors on the host cell membrane, through a RGD-containing protein loop (the G-H loop of protein VP1) on the virus capsid (reviewed in (43)). This loop is structurally free-standing and mobile, and participates in few or no contacts with other capsid regions (44). Accordingly, the VP1 G-H loop of FMDV can be mimicked by small RGD-containing peptides (43,45–53). In particular, synthetic peptides that reproduce the sequence of the central 15–20 amino acids of this viral loop are able to efficiently inhibit the infectivity of serotype C FMDV to BHK-21 cells, by competitively impairing the attachment of the virus to the cell receptors (49,51). The inhibition of FMDV infectivity by RGD peptides is due to a steric blockade of the recognition by the virion of integrin receptors on the surface of the host cells (43,47–49).
Using these two well-characterized in vitro or ex vivo systems, we have quantitatively compared the inhibitory activity of the above-mentioned molecules on either assembly of the HIV capsid or cell binding and infectivity of the FMDV virion, in the absence or presence of high concentrations of a macromolecular crowding agent. The conceptual framework and the implications for the inhibition of crucial steps of the viral cycle are discussed.
Materials and Methods
Virus and cell lines
FMDV C-S8c1 is a plaque-purified virus clone (54). The BHK-21c2 cell line used derives from a cell clone obtained by limiting dilution (55). Cells were cultivated in Dulbecco's modified Eagle's medium containing nonessential amino acids, gentamycin, and buthyl parahydroxybenzoate, and supplemented with 5% fetal bovine serum (56).
Synthetic peptides
Solid-phase synthesized and purified peptides with an amydated C-terminus were provided by Prof. D. Andreu (Universitat Pompeu Fabra, Barcelona, Spain). For the inhibition of HIV-1 capsid assembly, the CAI peptide (ITFEDLLDYYGP) (22) was used. An inactive peptide (KAFSPEVIPMFSALSEGAT), which represents a segment of the CTD sequence of CA from HIV-1, was used as a negative control.
For the inhibition of FMDV-cell receptor recognition, two overlapping synthetic peptides that represent the central amino-acid sequence of the G-H loop in VP1 of FMDV C-S8c1 were used: peptide A15 (VP1 residues 136–150: YTASARGDLAHLTTT) and peptide A19 (VP1 residues 138–156: ASARGDLAHLTTTHARHLP) (45,50). Peptide A15RGE, a variant A15 peptide with an inactivating, Asp-to-Glu substitution at the RGD motif (YTASARGELAHLTTT), was used as a negative control. The peptides were dissolved in complete phosphate-buffered saline (PBS). Peptide A19 coupled to keyhole limpet hemocyanin (A19-KLH) (46) was also used. The A19-KLH conjugate solution was dialyzed immediately before use to eliminate any free A19 peptide and other low-molecular-weight contaminants.
Purification of HIV-1 capsid protein CA and its C-terminal domain
CA of HIV-1 (strain BH10) and its CTD were purified, and their purity and concentration determined, essentially as described previously (10,57).
In vitro assembly of the HIV-1 capsid
Assembly reactions (10) were carried out in 50 mM sodium phosphate buffer pH = 7.4, 2.25 M NaCl and 46 μM CA in the absence or presence of 100 g/L Ficoll 70 or 50 g/L Dextran T40. A volume of CA solution at the appropriate concentration in 50 mM sodium phosphate buffer was introduced into a spectrophotometer cuvette (10 mm × 2 mm internal section), and the assembly reaction was triggered by adding a solution containing 4 M NaCl in the same buffer, with or without 179 g/L Ficoll 70 or 90 g/L Dextran T40 to get the final concentrations desired for each component in a final volume of 500 μL, followed by rapid mixing.
For inhibition assays, CA was mixed with the appropriate amount of CTD or CAI to get the desired inhibitor/CA molar ratio, and incubated for 30 min before triggering the reaction. The time-dependent increase in the optical density at 350 nm as a measure of the light scattered by the assembled particles was monitored at 25°C, using a model No. UV-1603 spectrophotometer (Shimadzu, Columbia, MD) with data points collected every 6 s. Traces of the variation in the turbidity were analyzed as described previously (10). The time-dependent increase in optical density fitted very well the empirical Hill function,
where OD is the optical density at incubation time t, ODf is the optical density at infinite time, t50 is the time at which the OD is equal to one-half the ODf, and n is a cooperativity parameter (9).
The polymerization rate kp in each tested condition was determined from the slope of the approximately linear portion of the curve. The ratio kpi/kp0 between the polymerization rate in the presence or absence of the inhibitor yielded the reduction in the polymerization rate at each concentration of inhibitor tested. The relative amount of capsid formed was estimated from the OD corresponding to the plateau of the turbidity curves (10,35,36). The ratio ODi/OD0 between the plateau OD in the presence or absence of the inhibitor yielded the reduction in the amount of capsid formed at each concentration of inhibitor tested.
Purification of the FMDV virion
FMD virions were purified as previously described (58) with some modifications. Briefly, BHK-21c2 cell monolayers in 12 B175 flasks were infected with FMDV C-S8c1 at a multiplicity of infection of 3. After complete cytopathic effect, the medium and remaining cells were recovered, and the cells were lysed by freezing and thawing, followed by the addition of 2% chloroform. The cell debris were removed by centrifugation, and the supernatant was loaded on a 20% sucrose cushion in TNE buffer (10 mM Tris-HCl pH = 7.5, 100 mM NaCl, 1 mM EDTA), and centrifuged at 25,000 rpm for 2.5 h at 4°C in a AH627 rotor (Sorvall, Newtown, CT). The pellet was thoroughly resuspended in ∼0.6 mL TNE and the suspension centrifuged through a 7.5–30% sucrose gradient in TNE at 37,000 rpm for 1 h at 4°C in a SW40 rotor (Beckman Coulter, Brea, CA). A quantity of 0.5 mL fractions was recovered, and those containing virions were pooled and dialyzed against incomplete PBS at 4°C. The purity of the preparation was assessed by SDS-Urea-PAGE.
FMDV inactivation
Purified FMDV virions were inactivated essentially as described previously (59–61). Briefly, 2.4 mL of a virus suspension in incomplete PBS were mixed with 1.2 mL of inactivating buffer (0.3 M Tris-HCl pH = 8.5, 1.5 M NH4Cl), filtered through a cellulose acetate membrane (0.45 μM pore; Pall Life Sciences, East Hills, NY), and incubated for 4 h at 37°C. Then, the suspension was dialyzed against incomplete PBS. Complete inactivation was assessed by titration in standard plaque assays.
Cell toxicity assays
A colorimetric method (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega, Fitchburg, WI) was used according to the instructions provided by the manufacturer. Briefly, near-confluent BHK-21 cell monolayers in 96-well plates were washed with incomplete PBS (Ca2+ and Mg2+-free), and incubated during 90 min at room temperature with increasing concentrations (0–200 g/L) of Ficoll 70. The cells were then washed with DMEM and incubated for 90 min at 37°C with MTS ([3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]), and the absorbance at 490 nm was determined in a microplate reader.
Inhibition of FMDV infectivity
The general outline of the assay was as described previously (51), but the conditions were appropriately varied. A quantity of 80% confluent BHK-21 cell monolayers in P60 petri dishes was washed with DMEM and incubated for 45 min at room temperature with a volume of 180 μL of either complete PBS, with or without Ficoll 70 (as a negative control), or with increasing concentrations (0.1–100 μM) of A15 or A19 peptides or A19-KLH conjugate diluted in complete PBS, with or without Ficoll 70. Except where stated otherwise, a final concentration of 100 g/L Ficoll 70 was used. Then, 20 μL of a virus suspension were added. The virus suspension contained 100 plaque-forming units of FMDV C-S8c1 (supernatant of infected cultures) in complete PBS at two physical particle/infectious particle ratios. In the experiments we used either 105 or 109 physical virus particles and ∼106 cells. If we assume a somewhat conservative figure of 103 αv-containing integrin receptor molecules able to bind FMDV per cell (62), the virus/receptor ratios used were 10−4 and 1.
The higher ratio was achieved by mixing the appropriate amounts of a suspension of infectious viral particles and a suspension of viral particles that had been completely inactivated as described above. In this way, the percent inhibition at different virus/receptor ratios could be obtained in the same type of experiment, and directly compared. The only relevant difference between the two conditions is the total number of viral particles used due to the different physical/infectious viral particle ratio; noninfectious and infectious viral particles show no differences in binding to the cell receptors. After the virus was added to the cell monolayers, these were incubated for 45 min at room temperature, washed with DMEM, covered with a semisolid agar overlay, and further incubated for 24 h at 37°C. The monolayers were fixed and stained, and the viral plaques counted. The inhibitory activity was compared by using the IC50 value, defined as the concentration of inhibitor needed to reduce the number of viral plaques to 50% of the initial value.
Results
Effects of macromolecular crowding on the inhibition of the in vitro assembly of the HIV-1 capsid by the capsid-binding CAI peptide and the CTD
The first model system used in this study involves a competition for binding to growing HIV-1 capsidlike particles between the capsid building blocks (CA dimers and other small oligomers (40)) and the inhibitor molecule, either the isolated CTD or the CAI peptide. The inhibitory activity of both molecules was compared in the absence or presence of a high concentration of an inert macromolecule, Ficoll 70.
The inhibitory effects on HIV-1 capsid assembly of the isolated CTD domain or CAI at different inhibitor/CA molar ratios are shown in Figs. 1 and 2. In the absence of crowding, addition of increasing amounts of free CTD (Fig. 1 a) or CAI (Fig. 1 c) led to decreasing CA polymerization rates (kp) and decreasing amounts of assembled capsids (indicated by the OD plateau in the polymerization curves). For example, at a CTD/CA molar ratio of 2, the kpi/kp0 ratio and the ODi/OD0 ratio were, respectively, ∼0.2 and 0.5 (a fivefold reduction of the polymerization rate and a twofold reduction in the total amount of capsid formed, relative to absence of inhibitor) (Fig. 1 a and Fig. 2). At a CAI/CA molar ratio as low as 0.5, both the kpi/kp0 ratio and the ODi/OD0 ratio were <0.1 (corresponding to 10-fold reduction in both polymerization rate and total amount of capsid formed, relative to absence of inhibitor) (Fig. 1 c and Fig. 2). Thus both the CTD and, even more so, the CAI peptide (which inhibits polymerization by a different mechanism than CTD), behaved in these experiments as effective inhibitors of HIV-1 capsid assembly in the absence of crowding. An inactive peptide used as a negative control had no inhibitory effect at all on CA polymerization (data not shown).
Figure 1.
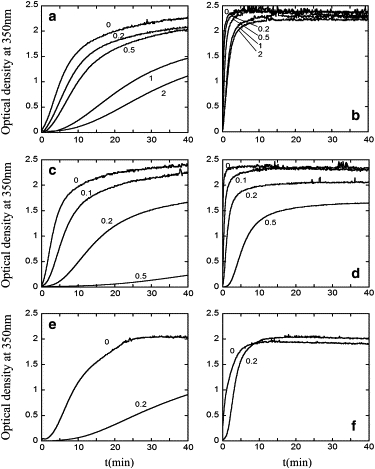
Effect of macromolecular crowding on the inhibition of the in vitro assembly of the HIV-1 capsid. Reaction kinetics of CA polymerization followed by measuring the optical density at 350 nm as a function of time after triggering the reaction are shown. (a and b) Inhibition by the CTD domain in the absence (a) or presence (b) of 100 g/L Ficoll 70 as an inert macromolecular crowding agent. (c and d) Inhibition by the CAI peptide in the absence (c) or presence (d) of 100 g/L Ficoll 70. (e and f) Inhibition by the CAI peptide in the absence (e) or presence (f) of 50 g/L Dextran T40. The CA (monomer) concentration used was 46 μM. In each panel, the inhibitor/CA molar ratios used (from 0 to 2) are indicated. In each panel, a representative experiment is shown. Average and standard deviation values from the processed results of several comparable experiments are shown in Fig. 2.
Figure 2.
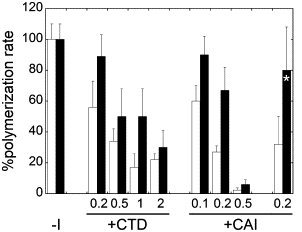
Inhibition of the CA polymerization rate by CTD and CAI. Each bar represents as a percentage the kpi/kp0 ratio, i.e., the ratio between the polymerization rate obtained in the presence of inhibitor relative to that obtained in its absence (bars labeled –I) for each inhibitor/CA molar ratio tested (from 0.1 to 0.5 for CAI or from 0.2 to 2 for CTD), either in the absence (open bars) or in the presence (solid bars) of 100 g/L Ficoll 70 (all solid bars but the one labeled with an asterisk) or 50 g/L Dextran T40 (solid bar labeled with an asterisk) as an inert macromolecular crowding agent. Each value represents the average of two, three, or four measurements obtained in independent experiments. The standard deviations are indicated. Experimental conditions were as described in the legend for Fig. 1.
Addition of 100 g/L Ficoll 70 as a macromolecular crowder had little effect on the total amount of capsid formed in the absence of inhibitor, but dramatically increased kp, both in the absence or presence of increasing amounts of CTD (Fig. 1 b) or CAI (Fig. 1 d). Remarkably, the inhibitory activities of CTD and CAI were substantially reduced in the presence of macromolecular crowding, relative to its absence. On average, in the presence of either inhibitor, and at different inhibitor/CA molar ratios, addition of the crowding agent roughly doubled the kpi/kp0 ratio (Fig. 2), and led also to a large increase in the ODi/OD0 ratio (Fig. 1). For example, at a CTD/CA molar ratio of 2, the ODi/OD0 ratio increased from ∼0.5 in the absence of crowder to ∼0.9 when the crowding agent was added, corresponding to a reduction of the inhibition by CTD from 50% to only 10%. Even more dramatically, at a CAI/CA molar ratio of 0.5, the ODi/OD0 ratio increased from ∼0.1 in the absence of crowder to ∼0.7 when the crowder was present, corresponding to a reduction of the inhibition by CAI from 90% to 30%.
Similar results were obtained when the inhibition of HIV-1 capsid assembly by CAI was analyzed in the presence of other inert crowding agents including dextran T40 (Fig. 1, e and f) or the protein bovine serum albumin (results not shown). For example, at a CAI/CA molar ratio of 0.2, the ODi/OD0 ratio increased from ∼0.5 in the absence of crowder to ∼1.0 when the crowder was present, corresponding to a reduction of the inhibition by CAI from 50% to close to 0%. As expected, the reduced inhibition under macromolecular crowding conditions was not dependent on the molecular nature of the crowding agent used.
To summarize this part of the study, we have used a high concentration of macromolecular crowding agents to mimic in vitro the macromolecular crowding conditions that prevail in the budding HIV-1 virion where the mature capsid is assembled in vivo (10,34). The results show that macromolecular crowding can lead to a significant reduction in the inhibitory activity of both the isolated CTD and the CTD-binding CAI peptide on the in vitro assembly of the HIV-1 capsid.
Effect of macromolecular crowding on the inhibition of FMDV-cell recognition and virus infectivity by RGD-containing peptides
Evaluation of the toxicity of the macromolecular crowding agent used
We first determined the maximum concentration of Ficoll 70 that could be used in the infectivity assays without a toxic effect on the target cells. No significant cytotoxic effect was found in repeated experiments, even at a concentration of 200 g/L Ficoll (Fig. 3). Thus, for the virus inhibition assays we generally chose a Ficoll concentration of 100 g/L, as this proved to be clearly nontoxic, and is enough to mimic the macromolecular crowding conditions exerted in physiological fluids.
Figure 3.
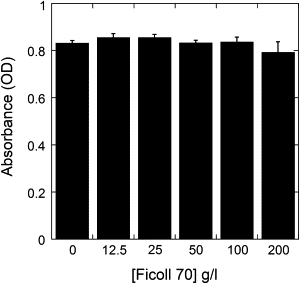
Evaluation of the cytotoxic effect of Ficoll 70 on cultured BHK21c2 cells. The values of absorbance, directly proportional to cell viability, are the average of six measurements. The standard deviations are indicated.
Evaluation of the inhibitory activity on FMDV infectivity of RGD-containing peptides
Next we determined the concentrations of the A15 and A19 peptides (either in free form or conjugated to a large protein) that were needed, in the absence of an external macromolecular crowding agent, to achieve efficient inhibition of cell attachment and infectivity of FMDV C-S8c1 (Fig. 4, a and b). The variant peptide A15RGE (with the RGD motif inactivated) was used as a negative control. Peptide A15RGE showed, as expected, no detectable inhibitory effect even at a concentration of 100 μM. In contrast, both A19 and A15 peptides showed a strong inhibitory activity, yielded a similar IC50 (∼1–2 μM), and reached close to 100% inhibition at a concentration of 100 μM (Fig. 4 a). The results using a multivalent A19-KLH conjugate showed that the peptide was about as active as a large protein conjugate (IC50 ∼1 μM peptide concentration; see Fig. 4 b) as it was as a free peptide (Fig. 4 a). Remarkably, as the peptide/KLH molar ratio in the conjugate is very high, this latter result indicates that multivalency of a receptor-binding motif per se may not necessarily lead to increased activity in the recognition of its receptor on the surface of host cells. From the above experiments, a range of 0.1 μM–100 μM peptide concentration (either free or coupled to KLH) was chosen for the subsequent assays.
Figure 4.
Inhibition of FMDV infectivity by synthetic peptides. (a and b) Inhibition in the absence of crowding agent by free peptides A19 (triangles), A15 (circles), or A15RGE (squares) (a) or by A19-KLH conjugate (b). The data points for A19, A15, A15RGE, and A19-KLH, respectively, represent the average of nine, four, four, or seven measurements obtained in three, two, two, or three independent experiments. The standard deviations are indicated. (c and d) Comparison of the inhibition of FMDV infectivity by free peptide A19 at two different virus/receptor ratios in the absence (open bars) or presence (solid bars) of Ficoll 70 as a crowding agent. The virus/receptor ratio in panel d is 104-fold higher than that in panel c. The values in panels c and d, respectively, represent the average of nine or four measurements obtained in three or two independent experiments. The standard deviations are indicated.
Evaluation of the effect of macromolecular crowding on the inhibition of FMDV infectivity by peptides
After the appropriate concentrations of crowding agent and RGD peptides had been determined, the effect of macromolecular crowding on the inhibition of FMDV infectivity by the A19 peptide was investigated. The ratio between the number of binding sites (FMDV receptors) in the membrane of infection-susceptible cells and the number of circulating viral particles may vary widely both in natural and laboratory infections. Thus, we decided to analyze the effects of macromolecular crowding at two very different virus particle/receptor ratios, estimated to be 10−4 and 1 (see Materials and Methods). The first ratio would leave most cell receptors unoccupied by the virions, while the second ratio would be closer to saturation of the viral receptors by virions. The number of binding sites (cell receptors) and other experimental conditions were kept invariant.
The results of the comparison are shown in Fig. 4, c and d. At the lower virus particle/receptor ratio tested, no significant differences in the inhibitory activity of the free A19 peptide in the absence or presence of 100 g/L Ficoll 70 were found (Fig. 4 c). The same lack of effect of macromolecular crowding conditions on the inhibitory activity was observed for a shorter RGD peptide, A15, or for a very large A19-KLH peptide conjugate (results not shown). In contrast, at the higher virus particle/receptor ratio tested, the inhibitory activity of the A19 peptide was significantly reduced in the presence of macromolecular crowding, relative to its absence (Fig. 4 d). The decrease was substantial only at low (up to 1 μM) peptide concentrations. However, in this situation as much as a 20-fold higher peptide concentration was needed to achieve in a crowded media (100 g/L Ficoll 70) the same inhibitory effect observed in a dilute solution.
To confirm that the reduced inhibitory activity of the peptide at the higher virus/receptor ratio tested is due to an effect of macromolecular crowding, we repeated the experiment, but this time using different concentrations of Ficoll 70 (0, 50, 100, and 200 g/L). As expected, an inverse dependence on Ficoll concentration of the inhibitory activity of the peptide was observed, both at low (1 μM) and higher (10 μM) peptide concentrations. For example, in the absence of Ficoll, a low concentration of peptide (1 μM) reduced virus infectivity by ∼25%. Addition of 50 g/L Ficoll had no significant effect. At 100 g/L Ficoll, the inhibition was reduced to only ∼5%; at 200 g/L Ficoll, the inhibitory activity of the peptide (at the same peptide concentration) was essentially abolished.
To summarize this second part of the study, we have used a high concentration of a macromolecular crowding agent to approximate in vitro the macromolecular crowding conditions that prevail in extracellular fluids where viruses bind their cellular receptors. The results show that macromolecular crowding, depending on the virus/cell receptor ratio, can lead to a significant reduction on the inhibitory activity of a RGD-containing peptide on FMD virus-host cell recognition and virus infectivity.
Discussion
The two models we have used to study the inhibition by small molecules of either capsid self-assembly or virus-cell recognition basically involve:
-
1.
A large target macromolecule or macromolecular assembly (T), either the growing HIV-1 capsid or the integrin receptor on the host cell membrane.
-
2.
A competition between an inhibitor peptide (I) of a relatively small size (molecular weight of ∼103–104), and larger or much larger macromolecules (M), either the CA building blocks that form by accretion capsidlike particles, or FMD virions (molecular weight 8 × 106).
-
3.
The conditional presence of an inert macromolecule (C). The presence of C at very high concentrations allowed us to achieve in vitro macromolecular crowding conditions that resemble those in the physiological fluids where assembly of the mature HIV-1 capsid (inside the budding virion) or FMDV-cell recognition (the extracellular fluid of certain tissues) occur in vivo.
Ficoll 70 was chosen as a model crowding agent because of its low viscosity, a size comparable to those of many proteins, and its low tendency to interact specifically with other solutes such as proteins (63). The Ficoll concentrations used in this study would approximately correspond to the volume occupancy estimated for extracellular conditions, or only somewhat lower than typical intracellular conditions. At these concentrations, this compound adequately promotes in vitro the crowded environment that in vivo originates mainly from a heterogeneous mixture of proteins.
The use of a single inert crowding agent to approximately mimic the volume occupancy due to complex mixtures of macromolecules in biological fluids allows the assignment of the observed effects to volume exclusion. This approach avoids the complications due to additional effects that could occur if complex macromolecular mixtures were used instead (see, for example, (64)). Because of the above reasons, Ficoll 70 is widely and extensively used by many researchers as a chemically defined, inert agent that leads to macromolecular crowding in simplified in vitro models.
The main result we obtained using either system (inhibition of capsid self-assembly or inhibition of virus-cell interactions) is that macromolecular crowding can significantly reduce the inhibitory effects of the antiviral peptides tested. Volume exclusion theory may provide a simplified conceptual framework and a general thermodynamic explanation for this experimental result: if the sizes of I (competitive inhibitor) and M (competed macromolecule) are similar, the inhibitory activity of I in a crowded solution would be similar to that in a nonphysiological, diluted (ideal) solution. In contrast, if I is clearly smaller than M, the inhibitory activity of I in crowded conditions would be significantly reduced, relative to that in a dilute solution, as theoretically justified by Minton (example I in (2)) and summarized next.
If M can bind T (target macromolecule) with an apparent equilibrium association constant KMT, and I can bind T with an apparent equilibrium association constant KIT, the relationships between KMT or KIT and the corresponding true equilibrium association constant KoMT or KoIT of the reaction are
where γM and γI, respectively, denote the thermodynamic (chemical) activity coefficient of M and I. Then, the preference ratio (P) of T for M, relative to I, will be
where P0 is the preference ratio of T for M relative to I, in an ideal solution (and thus, in the absence of crowding), where γM = γI = 1. When an inert background species C is present at high concentrations (acting as a macromolecular crowding agent), the value of the activity coefficient γx will depend, in part, on the size of the reactant X relative to the size of C (Ficoll 70 is larger than the inhibitors I used in our study). In addition, if I is smaller than M, as is the case in our two models, γM will increase with the concentration of C faster than γI because the available volume under these conditions is lower for M than for I. Thus, γM/γI will be higher than unity, and the value of P will be higher than P0.
In agreement with this theoretical prediction, it has been very recently shown that small chemical ligands that bind DNA G-quadruplexes, and thus inhibit telomerase activity, were significantly less effective under macromolecular crowding conditions (65). However, to our knowledge the prediction that macromolecular crowding may generally impair the inhibitory activity of small compounds (2), despite its biological relevance, had not been tested to date in any other cellular or viral system.
If the implicit assumptions and simplifications of the above theoretical analysis apply to the complex experimental models and under the conditions we have used, it could be expected that the preference ratio for association of the larger building blocks to the growing HIV-1 capsid, relative to the association of the smaller inhibitor peptides CAI and even CTD, would be higher in macromolecular crowding conditions than in a dilute solution. Likewise, the preference ratio for association of the large FMDV virions to the cell receptors, relative to the association of the smaller inhibitor peptide A19, would be higher in macromolecular crowding conditions than in a dilute solution. For both viral models, the antiviral activity of the peptide inhibitors is predicted to be reduced in the presence of macromolecular crowding, and this is precisely what we have experimentally observed in this study.
It must be noted, however, that the available theoretical analysis (2) does not seem to provide an explanation on why, in the case of the FMDV infection, the predicted effect of crowding was indeed observed, but apparently vanished at very low virus/receptor (V/R) ratios. We suggest that this may be due to an opposing, specific effect occurring in this complex system, but we are uncertain about its origin. Complexities of this system include:
-
1.
The virus, unlike the peptide, can recruit several receptor molecules; its internalization may depend on the number of receptors attached, which may be dependent in turn on the V/R ratio.
-
2.
FMDV is known to bind several different receptors with high affinity, but the same may not hold true for the peptide; multiple competing reactions could occur to different extents at different V/R ratios.
-
3.
Unproductive collision with receptors may conformationally alter the viral particles and its infectivity; the collision frequency will depend on the V/R ratio.
-
4.
The receptors were not in solution, but fixed to a cell membrane (see (66) for an example regarding predictions of macromolecular crowding theory on association reactions at cellular membranes).
Because of these or other reasons, some of the simplifying assumptions made by theory may have to be revised for this complex system. However, it is important to note that the theoretically predicted reduction in the inhibitory activity of the peptides occurred under some conditions in this system, and under any tested condition in the less complex viral capsid assembly system also investigated in this study.
It is also clear that the use of a single crowding agent provides only a first approximation to experimentally study some effects of high macromolecular concentrations. Biological environments are crowded but also heterogeneous media, and volume exclusion will not be the only factor affecting biochemical rates and equilibria. Further studies will be needed to evaluate whether the observed volume exclusion effects on virologically relevant macromolecular reactions are compounded by other effects derived from the presence of a complex mixture of macromolecules. These considerations underscore the need for more quantitative definitions of model systems and further theoretical developments to achieve a better understanding of association reactions in molecularly crowded environments.
The results obtained may also have practical implications for the initial testing of small molecules aimed at inhibiting virus assembly or virus-cell receptor recognition. The inhibitory activity of candidate antiviral drugs in vivo may be less than anticipated based on initial evaluation on test tube or cell culture, if these experiments are carried out in the absence of added macromolecular crowding agents. For example, in typical HIV-1 capsid assembly in vitro experiments, the total protein concentration is only a few mg/mL at most. In ex vivo experiments directed to assess the inhibition of virus-host cell recognition, the total protein concentration in the cell culture medium, even if supplemented with as much as 5–10% fetal bovine serum, will be ∼1 mg/mL or less (67).
These total macromolecular concentrations correspond to essentially diluted conditions, being approximately two-orders-of-magnitude lower than those leading to macromolecular crowding in vivo and to significant volume exclusion effects. The use of high concentrations of a macromolecular crowding agent in in vitro or ex vivo screening assays of candidate antiviral compounds may contribute to a better initial assessment of the inhibitory activity of these compounds at the total macromolecular concentrations they may encounter in vivo. This suggested approach could contribute to a better selection of candidate compounds for subsequent trials under more physiological conditions.
Conclusions
The experimental results obtained in this study demonstrate that macromolecular crowding can, at least under certain conditions, substantially impair the inhibition by small peptide molecules of capsid self-assembly or virus attachment to receptors on the membrane of host cells. These and other molecular recognition processes during the life cycle of viruses involve competition between ligands of widely different sizes and affinities, both outside and inside the cell. Because macromolecular crowding is widespread in both environments, volume exclusion effects may have to be considered for a more quantitative understanding of virus biology. In addition, most antiviral drugs in use for inhibiting the activity of viral proteins, or being developed for inhibiting virus assembly or virus-cell receptor recognition, are small organic compounds.
The results obtained with two very different virus model systems indicate that the inhibitory activity of antiviral drugs may be substantially tempered under macromolecular crowding conditions such as those present in the organism.
Acknowledgments
We gratefully acknowledge Prof. A. P. Minton for critical reading of the first version of the manuscript and suggestions, Prof. D. Andreu for the supply of peptides, and M. A. Fuertes for excellent technical assistance.
Work in M.G.M.'s laboratory is funded by grants from Fundación para la Investigación y Prevención del SIDA en España (No. 36557/06), the Spanish Ministerio de Ciencia e Innovación (No. BIO2009-10092), and Comunidad de Madrid (No. S-2009/MAT/1467), and by an institutional grant from Fundación Ramón Areces. Work in G.R.'s laboratory is funded by grant No. BIO2008-04478-C03-03 and No. S-BIO-0260/2006 from the Spanish Ministerio de Ciencia e Innovación and Comunidad de Madrid, respectively. V.R. is the recipient of a FPI predoctoral fellowship from the Spanish Ministerio de Ciencia e Innovación. M.G.M. is an associate member of the Instituto de Biocomputación y Física de los Sistemas Complejos, Zaragoza, Spain.
References
- 1.Minton A.P. The effect of volume occupancy upon the thermodynamic activity of proteins: some biochemical consequences. Mol. Cell. Biochem. 1983;55:119–140. doi: 10.1007/BF00673707. [DOI] [PubMed] [Google Scholar]
- 2.Minton A.P. Macromolecular crowding and molecular recognition. J. Mol. Recognit. 1993;6:211–214. doi: 10.1002/jmr.300060410. [DOI] [PubMed] [Google Scholar]
- 3.Minton A.P. Molecular crowding: analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 1998;295:127–149. doi: 10.1016/s0076-6879(98)95038-8. [DOI] [PubMed] [Google Scholar]
- 4.Minton A.P. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 2001;276:10577–10580. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- 5.Zhou H.-X., Rivas G., Minton A.P. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis R.J. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 2001;26:597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 7.Rivas G., Fernández J.A., Minton A.P. Direct observation of the enhancement of noncooperative protein self-assembly by macromolecular crowding: indefinite linear self-association of bacterial cell division protein FtsZ. Proc. Natl. Acad. Sci. USA. 2001;98:3150–3155. doi: 10.1073/pnas.051634398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis-Searles P.R., Saunders A.J., Pielak G.J. Interpreting the effects of small uncharged solutes on protein-folding equilibria. Annu. Rev. Biophys. Biomol. Struct. 2001;30:271–306. doi: 10.1146/annurev.biophys.30.1.271. [DOI] [PubMed] [Google Scholar]
- 9.Hatters D.M., Minton A.P., Howlett G.J. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J. Biol. Chem. 2002;277:7824–7830. doi: 10.1074/jbc.M110429200. [DOI] [PubMed] [Google Scholar]
- 10.del Alamo M., Rivas G., Mateu M.G. Effect of macromolecular crowding agents on human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol. 2005;79:14271–14281. doi: 10.1128/JVI.79.22.14271-14281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minton A.P. How can biochemical reactions within cells differ from those in test tubes? J. Cell Sci. 2006;119:2863–2869. doi: 10.1242/jcs.03063. [DOI] [PubMed] [Google Scholar]
- 12.Ellis R.J., Minton A.P. Protein aggregation in crowded environments. Biol. Chem. 2006;387:485–497. doi: 10.1515/BC.2006.064. [DOI] [PubMed] [Google Scholar]
- 13.Hu Z., Jiang J., Rajagopalan R. Effects of macromolecular crowding on biochemical reaction equilibria: a molecular thermodynamic perspective. Biophys. J. 2007;93:1464–1473. doi: 10.1529/biophysj.107.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batra J., Xu K., Zhou H.-X. Nonadditive effects of mixed crowding on protein stability. Proteins. 2009;77:133–138. doi: 10.1002/prot.22425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiao M., Li H.T., Liang Y. Attractive protein-polymer interactions markedly alter the effect of macromolecular crowding on protein association equilibria. Biophys. J. 2010;99:914–923. doi: 10.1016/j.bpj.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elcock A.H. Models of macromolecular crowding effects and the need for quantitative comparisons with experiment. Curr. Opin. Struct. Biol. 2010;20:196–206. doi: 10.1016/j.sbi.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Clercq E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Clercq E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004;30:115–133. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 19.Greene W.C., Debyser Z., Cihlar T. Novel targets for HIV therapy. Antiviral Res. 2008;80:251–265. doi: 10.1016/j.antiviral.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Melby T., Westby M. Inhibitors of viral entry. In: Kräusslich H.-G., Bartenschlager B., editors. Antiviral Strategies, Handbook of Experimental Pharmacology. Vol. 189. Springer-Verlag; Berlin, Germany: 2009. pp. 177–202. [DOI] [PubMed] [Google Scholar]
- 21.Tang C., Loeliger E., Summers M.F. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 2003;327:1013–1020. doi: 10.1016/s0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- 22.Sticht J., Humbert M., Kräusslich H.-G. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 2005;12:671–677. doi: 10.1038/nsmb964. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H., Zhao Q., Debnath A.K. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J. Mol. Biol. 2008;378:565–580. doi: 10.1016/j.jmb.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neira J.L. The capsid protein of human immunodeficiency virus: designing inhibitors of capsid assembly. FEBS J. 2009;276:6110–6117. doi: 10.1111/j.1742-4658.2009.07314.x. [DOI] [PubMed] [Google Scholar]
- 25.Doménech R., Abian O., Neira J.L. Dendrimers as potential inhibitors of the dimerization of the capsid protein of HIV-1. Biomacromolecules. 2010;11:2069–2078. doi: 10.1021/bm100432x. [DOI] [PubMed] [Google Scholar]
- 26.Yang Q.E., Stephen A.G., Sei S. Discovery of small-molecule human immunodeficiency virus type 1 entry inhibitors that target the gp120-binding domain of CD4. J. Virol. 2005;79:6122–6133. doi: 10.1128/JVI.79.10.6122-6133.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones J.C., Turpin E.A., Schultz-Cherry S. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J. Virol. 2006;80:11960–11967. doi: 10.1128/JVI.01678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porotto M., Carta P., Moscona A. Molecular determinants of antiviral potency of paramyxovirus entry inhibitors. J. Virol. 2007;81:10567–10574. doi: 10.1128/JVI.01181-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akkarawongsa R., Potocky T.B., Brandt C.R. Inhibition of herpes simplex virus type 1 infection by cationic β-peptides. Antimicrob. Agents Chemother. 2008;52:2120–2129. doi: 10.1128/AAC.01424-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall P.R., Hjelle B., Larson R.S. Multivalent presentation of antihantavirus peptides on nanoparticles enhances infection blockade. Antimicrob. Agents Chemother. 2008;52:2079–2088. doi: 10.1128/AAC.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsubara T., Sumi M., Sato T. Inhibition of influenza virus infections by sialylgalactose-binding peptides selected from a phage library. J. Med. Chem. 2009;52:4247–4256. doi: 10.1021/jm801570y. [DOI] [PubMed] [Google Scholar]
- 32.Vitiello M., Galdiero M., Galdiero M. Inhibition of viral-induced membrane fusion by peptides. Protein Pept. Lett. 2009;16:786–793. doi: 10.2174/092986609788681742. [DOI] [PubMed] [Google Scholar]
- 33.Ganser-Pornillos B.K., Yeager M., Sundquist W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008;18:203–217. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mateu M.G. The capsid protein of human immunodeficiency virus: intersubunit interactions during virus assembly. FEBS J. 2009;276:6098–6109. doi: 10.1111/j.1742-4658.2009.07313.x. [DOI] [PubMed] [Google Scholar]
- 35.Lanman J., Sexton J., Prevelige P.E., Jr. Kinetic analysis of the role of intersubunit interactions in human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol. 2002;76:6900–6908. doi: 10.1128/JVI.76.14.6900-6908.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.del Alamo M., Mateu M.G. Electrostatic repulsion, compensatory mutations, and long-range non-additive effects at the dimerization interface of the HIV capsid protein. J. Mol. Biol. 2005;345:893–906. doi: 10.1016/j.jmb.2004.10.086. [DOI] [PubMed] [Google Scholar]
- 37.Barrera F.N., del Álamo M., Neira J.L. Envelope lipids regulate the in vitro assembly of the HIV-1 capsid. Biophys. J. 2008;94:L08–L10. doi: 10.1529/biophysj.107.118083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ternois F., Sticht J., Rey F.A. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat. Struct. Mol. Biol. 2005;12:678–682. doi: 10.1038/nsmb967. [DOI] [PubMed] [Google Scholar]
- 39.Bartonova V., Igonet S., Kraüsslich H.-G. Residues in the HIV-1 capsid assembly inhibitor binding site are essential for maintaining the assembly-competent quaternary structure of the capsid protein. J. Biol. Chem. 2008;283:32024–32033. doi: 10.1074/jbc.M804230200. [DOI] [PubMed] [Google Scholar]
- 40.Barklis E., Alfadhli A., López C.S. Characterization of the in vitro HIV-1 capsid assembly pathway. J. Mol. Biol. 2009;387:376–389. doi: 10.1016/j.jmb.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahy B.W.J., editor. Foot-and-Mouth Disease Virus. Current Topics in Microbiology and Immunology Series. Vol. 288. Springer Verlag; Berlin, Germany: 2005. [DOI] [PubMed] [Google Scholar]
- 42.Sobrino F., Domingo E., editors. Foot and Mouth Disease: Current Perspectives. Horizon Bioscience; Norfolk, UK: 2004. [Google Scholar]
- 43.Baxt B., Rieder E. Molecular aspects of foot-and-mouth disease virus virulence and host range: role of host cell receptors and viral factors. In: Sobrino F., Domingo E., editors. Foot and Mouth Disease: Current Perspectives. Horizon Bioscience; Norfolk, UK: 2004. pp. 145–172. [Google Scholar]
- 44.Hewat E.A., Verdaguer N., Stuart D.I. Structure of the complex of a Fab fragment of a neutralizing antibody with foot-and-mouth disease virus: positioning of a highly mobile antigenic loop. EMBO J. 1997;16:1492–1500. doi: 10.1093/emboj/16.7.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowlands D.J., Clarke B.E., Lerner R.A. Chemical basis of antigenic variation in foot-and-mouth disease virus. Nature. 1983;306:694–697. doi: 10.1038/306694a0. [DOI] [PubMed] [Google Scholar]
- 46.Mateu M.G., Martínez M.A., Domingo E. Implications of a quasispecies genome structure: effect of frequent, naturally occurring amino acid substitutions on the antigenicity of foot-and-mouth disease virus. Proc. Natl. Acad. Sci. USA. 1989;86:5883–5887. doi: 10.1073/pnas.86.15.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fox G., Parry N.R., Brown F. The cell attachment site on foot-and-mouth disease virus includes the amino acid sequence RGD (arginine-glycine-aspartic acid) J. Gen. Virol. 1989;70:625–637. doi: 10.1099/0022-1317-70-3-625. [DOI] [PubMed] [Google Scholar]
- 48.Baxt B., Becker Y. The effect of peptides containing the arginine-glycine-aspartic acid sequence on the adsorption of foot-and-mouth disease virus to tissue culture cells. Virus Genes. 1990;4:73–83. doi: 10.1007/BF00308567. [DOI] [PubMed] [Google Scholar]
- 49.Hernández J., Valero M.L., Mateu M.G. Antibody and host cell recognition of foot-and-mouth disease virus (serotype C) cleaved at the Arg-Gly-Asp (RGD) motif: a structural interpretation. J. Gen. Virol. 1996;77:257–264. doi: 10.1099/0022-1317-77-2-257. [DOI] [PubMed] [Google Scholar]
- 50.Verdaguer N., Mateu M.G., Fita I. Structure of the major antigenic loop of foot-and-mouth disease virus complexed with a neutralizing antibody: direct involvement of the Arg-Gly-Asp motif in the interaction. EMBO J. 1995;14:1690–1696. doi: 10.1002/j.1460-2075.1995.tb07158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mateu M.G., Valero M.L., Domingo E. Systematic replacement of amino acid residues within an Arg-Gly-Asp-containing loop of foot-and-mouth disease virus and effect on cell recognition. J. Biol. Chem. 1996;271:12814–12819. doi: 10.1074/jbc.271.22.12814. [DOI] [PubMed] [Google Scholar]
- 52.Mateu M.G., Verdaguer N. Functional and structural aspects of the interaction of foot-and-mouth disease virus with antibodies. In: Sobrino F., Domingo E., editors. Foot and Mouth Disease: Current Perspectives. Horizon Bioscience; Norfolk, UK: 2004. pp. 223–260. [Google Scholar]
- 53.Rowlands D. Foot-and-mouth disease virus peptide vaccines. In: Sobrino F., Domingo E., editors. Foot and Mouth Disease: Current Perspectives. Horizon Bioscience; Norfolk, UK: 2004. pp. 335–354. [Google Scholar]
- 54.Sobrino F., Dávila M., Domingo E. Multiple genetic variants arise in the course of replication of foot-and-mouth disease virus in cell culture. Virology. 1983;128:310–318. doi: 10.1016/0042-6822(83)90258-1. [DOI] [PubMed] [Google Scholar]
- 55.de la Torre J.C., Martínez-Salas E., Domingo E. Coevolution of cells and viruses in a persistent infection of foot-and-mouth disease virus in cell culture. J. Virol. 1988;62:2050–2058. doi: 10.1128/jvi.62.6.2050-2058.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Domingo E., Dávila M., Ortín J. Nucleotide sequence heterogeneity of the RNA from a natural population of foot-and-mouth-disease virus. Gene. 1980;11:333–346. doi: 10.1016/0378-1119(80)90073-6. [DOI] [PubMed] [Google Scholar]
- 57.Mateu M.G. Conformational stability of dimeric and monomeric forms of the C-terminal domain of human immunodeficiency virus-1 capsid protein. J. Mol. Biol. 2002;318:519–531. doi: 10.1016/S0022-2836(02)00091-8. [DOI] [PubMed] [Google Scholar]
- 58.Díez J., Dávila M., Domingo E. Unique amino acid substitutions in the capsid proteins of foot-and-mouth disease virus from a persistent infection in cell culture. J. Virol. 1990;64:5519–5528. doi: 10.1128/jvi.64.11.5519-5528.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amadori M., Barei S., Panina G.F. Safety and efficacy of foot-and-mouth disease vaccines containing endonuclease-inactivated virions. Vaccine. 1987;5:219–222. doi: 10.1016/0264-410x(87)90104-6. [DOI] [PubMed] [Google Scholar]
- 60.Patil P.K., Suryanarayana V., Natarajan C. Integrity of GH-loop of foot-and-mouth disease virus during virus inactivation: detection by epitope specific antibodies. Vaccine. 2002;20:1163–1168. doi: 10.1016/s0264-410x(01)00431-5. [DOI] [PubMed] [Google Scholar]
- 61.Scodeller E.A., Lebendiker M.A., Vasquez C. Inactivation of foot-and-mouth disease virus vaccine strains by activation of virus-associated endonuclease. J. Gen. Virol. 1984;65:1567–1573. doi: 10.1099/0022-1317-65-9-1567. [DOI] [PubMed] [Google Scholar]
- 62.Benedetto S., Pulito R., Hamm J. Quantification of the expression level of integrin receptor αVβ3 in cell lines and MR imaging with antibody-coated iron oxide particles. Magn. Reson. Med. 2006;56:711–716. doi: 10.1002/mrm.21023. [DOI] [PubMed] [Google Scholar]
- 63.Fodeke A.A., Minton A.P. Quantitative characterization of polymer-polymer, protein-protein, and polymer-protein interaction via tracer sedimentation equilibrium. J. Phys. Chem. B. 2010;114:10876–10880. doi: 10.1021/jp104342f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ando T., Scolnick J. Crowding and hydrodynamic interactions likely dominate in vivo macromolecular motion. Proc. Natl. Acad. Sci. USA. 2010;107:18457–18462. doi: 10.1073/pnas.1011354107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Z., Zheng K.W., Tan Z. Reduced or diminished stabilization of the telomere G-quadruplex and inhibition of telomerase by small chemical ligands under molecular crowding condition. J. Am. Chem. Soc. 2009;131:10430–10438. doi: 10.1021/ja9010749. [DOI] [PubMed] [Google Scholar]
- 66.Kim J.-S., Yethiraj A. Crowding effects on association reactions at membranes. Biophys. J. 2010;98:951–958. doi: 10.1016/j.bpj.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Rizzo E., Vaz C.A.C., Yano A.F. Study on the growth promoting capacity of calf and fetal bovine serum for animal cells “in vitro”. II. Electrophoretic study and survey on the antiproteolytic activity of pools of calf and fetal bovine serum. Rev. Inst. Med. Trop. Sao Paulo. 1984;26:97–104. doi: 10.1590/s0036-46651984000200005. [DOI] [PubMed] [Google Scholar]