

Abstract
The structure and dynamics of soluble misfolded aggregates are poorly understood, despite their importance in protein science and disease. Water-soluble self-associated species that do not become insoluble over time are invaluable tools for high-resolution conformational studies aimed at dissecting the determinants of self-association. Here, we characterize the soluble model aggregate apomyoglobin119 (apoMb119), generated upon truncating the residues corresponding to the C-terminal helix of sperm whale apomyoglobin. The secondary structure and backbone dynamics of apoMb119, determined by multidimensional NMR at pH 6.0, reveal the presence of an N-terminal slow-tumbling core and a highly disordered flexible C-terminus displaying residual helicity and large-amplitude backbone motions on the picosecond-to-nanosecond timescale. The backbone of the apoMb119 aggregate assumes progressively increased mobility as residues get further removed from the nonpolar core and closer to the more hydrophilic C-terminal end. This structural motif establishes a useful paradigm for the topology of soluble misfolded protein aggregates in aqueous solution in the absence of stabilizing additives. The partially helical and flexible C-terminus of apoMb119's aggregate is in interesting contrast with the amyloid-related globulomers, which display dangling ends rich in β-strand. Finally, we investigate how a molecular chaperone, the substrate-binding domain of DnaK, interferes with apoMb119's aggregation.
Introduction
The formation of high-order soluble or insoluble polypeptide aggregates is generally undesired in vivo and often correlates with the insurgence of toxicity and(or) pathogenic phenotypes (1–4). Soluble small (1–10 nm) peptide and protein aggregates generated in vivo under aberrant conditions are particularly interesting, due to the recent discovery of their links with a variety of neurodegenerative and brain diseases (4–7). Indeed, such small soluble self-associated species are currently believed to be more closely connected with neural misfunction than the more widely studied large amyloid fibrils (8–11). Soluble aggregates can be either on-path or off-path to fibril formation (12), and the origin of their pathogenicity has been related to their ability to interfere with the normal cell metabolism (2,4,12–14) and cause neuronal damage (15–21).
The study of small soluble aggregates can be more challenging than the analysis of fibrillar adducts. While the latter species are generally kinetically and thermodynamically stable, and therefore easier to analyze for prolonged time, soluble aggregates are often transient in nature (and frequently also heterogeneous in size and morphology), therefore harder to tackle experimentally. Hence, high-resolution studies on such species are scarce to date.
Soluble protein aggregates bearing different morphological and structural characteristics have been reported so far for foldable protein sequences, under specific solution conditions (22–29). However, these aggregates are often heterogeneous and only transiently soluble. With time, they tend to convert to large, fibril-like insoluble species, thus defying high-resolution structural analysis. Two recent high-resolution NMR studies focused on the structure of globular oligomers (1–5 nm in diameter) formed by amyloid-β (Aβ) (30,31). Bocian et al. (32) showed that such species have a β-strand-like structure and a different topology from that of amyloid fibrils. One additional investigation focused on the self-association kinetics of amorphous insulin oligomers by high-resolution NMR. Nonfibrillar soluble globular Aβ oligomers were even shown to display in vitro neurotoxicity, and were suggested to be relevant toxic species in Alzheimer's disease (33). These soluble aggregates, often denoted as globulomers, are formed by the Aβ(1–42) or other peptides in the presence of small amounts of detergent or fatty acids (33).
To date, there are only very few reports (34–36) providing high-resolution information on both the secondary structure and backbone dynamics of soluble aggregates in the absence of stabilizing detergents. Characterization of both the dynamics and conformation of soluble aggregates is important to better understand their solution behavior and potential pathogenicity.
Here, we focus on the secondary structure and backbone dynamics of a model soluble nonfibrillar protein aggregate by high-resolution NMR in the absence of any detergents or cosolvents. Apomyoglobin (apoMb; see Fig. 1) from sperm whale is an excellent example of a globular monomeric protein that undergoes aggregation under particular conditions. This protein is well characterized and has been extensively employed as a model α-helical fold in a large variety of biophysical studies. While full-length native apoMb lacks any elements of β-strand secondary structure, C-terminal truncation results in a loss of the ability to fold, and in oligomerization accompanied by β-strand formation (37). Additionally, even full-length apoMb has been reported to display a tendency to aggregate under both nonnative (22) and nativelike (26) conditions. The apomyoglobin amino-acid sequence is capable of forming species of various association states bearing either nativelike or nonnative secondary structure, depending upon environmental conditions and the presence or absence of its C-terminal residues (26).
Figure 1.
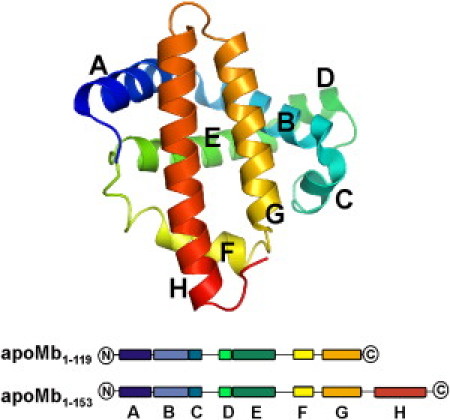
Three dimensional structure of apoMb. Atomic coordinates were derived from PDB file 1VXF, corresponding to the high-resolution crystal structure of carbonmonoxy-myoglobin (70). The image was created with the software PyMOL (DeLano Scientific, Palo Alto, CA). Below, a cartoon representation of the N-terminal fragment apoMb119 and full-length apoMb are given. The helices are color-coded and labeled from A to H (N- to C-terminus).
The 1-119 fragment of apoMb studied in this work (apoMb119, Fig. 1) lacks only the C-terminal helix, and forms water-soluble aggregates of low oligomeric state (37) with up to 18% nonnative β-strand secondary structure. Preliminary NMR characterization of ApoMb119 done in the absence of any specific resonance assignments focused on the general effect of molecular chaperones on the fragment's solubility (39). The low NMR chemical shift dispersion in the proton dimension of the apoMb119 1H, 15N-HSQC indicates that the residues corresponding to the detectable resonances have a globally unfolded conformation in solution. ApoMb119 resonance intensities increase significantly upon addition of an Hsp70 chaperone analog, supporting the fact that apoMb119 interacts with the chaperone (39).
The soluble apoMb119 aggregates are not fibrillogenic and do not display any significant thioflavin T binding at approximately neutral pH (37). Upon increasing the pH or further chain truncation, however, extended structures characterized by intense thioflavin T fluorescence are formed (37).
The high-resolution analysis of the secondary structure and backbone dynamics of the soluble apoMb119 aggregate presented here shows that the NMR-detectable C-terminal portion of the chain is highly dynamic (picosecond-to-nanosecond timescale) with some residual helicity. On the other hand, the hydrophobic N-terminal region does not give rise to any detectable NMR signal, suggesting that the apoMb119 aggregate has a slow-tumbling core, possibly undergoing some internal conformational exchange on the microsecond-to-millisecond timescale. In summary, this study provides a high-resolution glimpse at the conformation and backbone motions of the model soluble aggregate apoMb119 in solution.
Materials and Methods
Expression and purification of apoMb119 and DnaK-β
Uniformly 15N-labeled apoMb119 was overexpressed in Escherichia coli in M9 minimal medium containing 15N-NH4Cl and purified by reverse phase high-performance liquid chromatography as described (37,39).
Cell growth and purification of the unlabeled DnaK-β chaperone (the substrate-binding domain of DnaK) was carried out as described (39). DnaK-β was refolded by slow dropwise addition of the purified chaperone into an excess of 10 mM sodium acetate adjusted to pH 6.0 under gentle stirring.
NMR sample preparation and chaperone-binding titration
ApoMb119 NMR samples at pH 6.0 were prepared from concentrated stock solutions of 15N-labeled apoMb119 at pH 2.5, which were diluted to 300 μM in 20 mM sodium acetate, 5 mM KCl, and 5% D2O. The pH of the resulting solution was adjusted to 6.0 with 0.1 M KOH. The samples containing apoMb119 and DnaK-β at pH 6.0 were prepared as above, except that the final solution also contained stoichiometric amounts of DnaK-β and the final concentration of both apoMb119 and DnaK-β was 100 μM. Such low concentration ensured that DnaK-β, which is not highly soluble, did not undergo any undesired aggregation before the addition of substrate. After mixing all components, the solution pH was adjusted to 6.0. All NMR samples were equilibrated overnight at 4°C before data acquisition.
The chaperone-binding titration was carried out as described in the Supporting Material.
NMR spectroscopy: general procedures for data collection and processing
NMR experiments were performed at 4.0°C on an INOVA 600 MHz NMR spectrometer on a 1H{13C, 15N} cold probe equipped with a z-axis gradient (Varian, Cary, NC). Translational diffusion measurement experiments were carried out with a room temperature probe and relaxation experiments were performed on a cold probe. Temperature calibration was done using neat methanol as described (40). Spectral widths in the 1H and 15N dimensions, respectively, were set to 8000 and 1780 Hz for apoMb119 in the absence of DnaK-β and to 6600 and 1500 Hz in the presence of DnaK-β. Relaxation delays were set to 1.2 and 1.5 s in the absence and presence of DnaK-β, respectively. The NMRPipe (41) and NMRView (42) software packages were used for NMR data processing. Resonance intensity assessments were based on previously collected (39) sensitivity-enhanced 1H,15N-CPMG-HSQC (43) intensity distribution data reported on unassigned resonances.
Here, we report the intensity of the subset of assigned amide proton backbone resonances that are well resolved by 1H,15N-CPMG-HSQC as a function of residue number. Due to the very low concentrations demanded by the scarce solubility of apoMb119 and DnaK-β, TROSY-type experiments (44–48) were not performed. Based on our prior experience on similar (non-2H-enriched) systems, the expected gain in spectral resolution would likely not outbalance the signal loss resulting from multiple component selection. Data collection at low temperature (4°C) was required to prevent macroscopic protein aggregation. Procedures for backbone resonance assignments are described in the Supporting Material.
Secondary chemical shifts
Secondary chemical shifts (SCSs) of the assigned apoMb119 Cα nuclei were calculated by subtracting residue-specific random coil chemical shift reference values, obtained from Ac-GGXVGG-NH2 peptides in 1 M urea at 25°C at pH 5.0, from experimentally determined chemical shifts, as described in Wishart et al. (49). Reference random coil chemical shifts were corrected for the proximity of proline (49).
NMR relaxation measurements
15N T1, T2, and 1H-15N steady-state nuclear Overhauser effect (NOE) relaxation data were collected using pulse sequences described by Farrow et al. (50). T1 delays of 10, 100, 200, 300, 400, 550, 700, and 900 ms and T2 delays of 10, 30, 50, 70, 90, 110, 130, and 150 ms were used for apoMb119 samples, both in the absence and presence of DnaK-β. A 3 s saturation delay was applied in all 1H-15N steady-state NOE experiments. Data were recorded as 256(t1) × 2048(t2) total points for all experiments. The free induction decay was then apodized with an unshifted Gaussian function and zero-filled to 512(t1) × 4096(t2) total points. Maximum peak heights were determined with the NMRView software (42).
The T1, T2 values were fit to a two-parameter single exponential expression using the CURVEFIT program (http://www.palmer.hs.columbia.edu) developed by Arthur G. Palmer, III. The 1H-15N steady-state NOE values were determined via the NMRView software package (42). T1 and T2 errors for the apoMb119 samples were determined with the Jackknife simulation routine (51) using the average values from three (T1 measurements) and two (T2 measurements) repeats for each delay. In the case of the apoMb119-DnaK-β sample, the poor signal/noise resulting from the low concentration prevented the collection of multiple T1 and T2 data sets for apoMb119. Steady-state NOE data for this sample were collected but, due to the poor signal/noise, the results are not reported here.
Reduced spectral density analysis
15N R1 and R2 relaxation rates and steady-state 1H-15N heteronuclear NOEs are mainly affected by dipole-dipole interactions and chemical shift anisotropy according to (52)
| (1) |
| (2) |
and
| (3) |
where c and d are defined as
and J(ω) is the spectral density at frequency ω; μ0 is vacuum permeability; h is Planck's constant; γH is the 1H gyromagnetic ratio; γN is the 15N gyromagnetic ratio; ωH is 1H Larmor frequency; ωN is 15N Larmor frequency; rNH is the NH bond length; σ‖ and σ⊥ are the parallel and perpendicular components of the axially symmetric 15N chemical shift tensor, respectively; and (σ‖ − σ⊥) = −160 ppm (53). Reduced spectral density analysis (54–56) was performed according to Farrow et al. (54). Briefly, this approach assumes that the high-frequency spectral density terms that contribute to the relaxation process are of nearly equal magnitude. Therefore, they can be replaced by an equivalent spectral density J(0.870 ωH), such that
Equations 1–3 can be rearranged to yield the spectral densities J(0.870 ωH), J(ωN), and J(0), as
| (4) |
| (5) |
and
| (6) |
Numerical values for the above reduced spectral densities were derived by replacing the proper values of c, d, and the experimentally determined T1, T2 and steady-state NOE values in Eqs. 4–6.
Results and Discussion
ApoMb119 is a soluble aggregate
The native gel band for the apoMb119 soluble aggregate at pH 6.0 (see Fig. S1 in the Supporting Material) suggests the presence of one main species. The broad profile of this band and the published size exclusion chromatography data (37) are compatible with the presence of conformational heterogeneity within the main populated state.
Consistent with prior computational predictions (57) and NMR translational diffusion experiments under conditions similar to those of this work (39), apoMb119 is a soluble aggregate in aqueous buffer. A translational diffusion constant (D) of 3.65 × 10−11 m2 s−1 was measured (39), consistent with the presence of an apparent self-associated 8-mer. The self-association state was estimated via the Stokes-Einstein equation and a well-known relation connecting hydrodynamic radius and apparent molecular weight, upon assuming a soluble aggregate with overall spherical shape. Shape deviations from spherical in a monomer could also account for the small observed D (58). Several lines of evidence, however, suggest that this is not the case.
First, the D of apoMb119 at pH 6.0 is even smaller than the D observed for monomeric pH-2.5 apoMb119 (D ∼ 4.0 × 10−11 m2 s−1) (39), which is a globally unfolded random coil expected to display slow translational diffusion. Therefore, apoMb119 at pH 6.0 could not be composed uniquely by disordered monomers.
Second, if apoMb119 at pH 6.0 were a rigid elongated monomer (e.g., a prolate ellipsoid, which could account for the small D), its size exclusion and native gel profiles would be much sharper. Indeed, both size exclusion chromatography (37) and native gel electrophoresis (see Fig. S1) display a very broad peak/band for apoMb119 at pH 6.0, inconsistent with the presence of a rigid monomer. Therefore, we conclude that apoMb119 is a soluble aggregate under our experimental conditions.
General NMR features and secondary structure of apoMb119
The 1H,15N-HSQC spectrum of 15N-labeled apoMb119 is shown in Fig. 2. Only a fraction of the expected resonances is detectable. The assignments of the detectable resonances are labeled on the spectrum. Interestingly, all of the detectable resonances correspond to the C-terminal portion of apoMb119. The narrow chemical shift dispersion of the backbone amide protons indicates that the detectable portion of apoMb119 (i.e., the second half of the sequence) is highly disordered.
Figure 2.
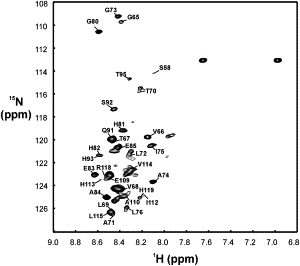
1H,15N-HSQC spectrum of 15N,13C-labeled apoMb119 (100 μM), in 20 mM sodium acetate, 5 mM KCl, and 5% D2O, at pH 6.0. The known spectral assignments (69) are indicated on the side of the respective resonances. In addition, the H64, K98, I111, H116, and S117 resonances were also assigned but are not labeled on the spectrum due to their low intensity.
The Cα secondary chemical shifts (SCSs) of apoMb119 are displayed in Fig. 3. Cα SCSs are good indicators of backbone secondary structure (59,60). Positive deviations from random coil values indicate α-helical secondary structure. The observed SCS values are much smaller than those typically observed for full helices in native proteins, including full-length apoMb (61). Residual helicity is observed in regions corresponding to the native E and G helices. Surprisingly, there is also helicity in the EF loop region, which is completely absent in the monomeric native full-length apomyoglobin. Such residual secondary structure is rapidly fluctuating and highly dynamic, as detailed in the next section.
Figure 3.
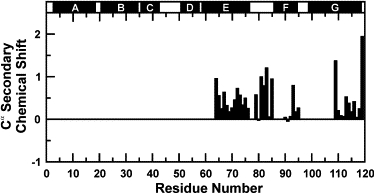
Cα NMR secondary chemical shifts for the detectable resonance of the apoMb119 soluble aggregate. Positive deviations from random coil values indicate α-helical secondary structure. (Solid bars above the plot) Sequence regions corresponding to native α-helices in full-length apoMb.
Backbone dynamics of apoMb119 by solution NMR
Valuable insights on the soluble aggregate of apoMb119 are provided by 15N backbone relaxation data. As shown by the native gel and translational diffusion experiments, apoMb119 behaves as a self-associated soluble species, under our experimental conditions. Fig. 4 shows the 15N relaxation rates R1, R2 and the steady-state 1H-15N NOE of apoMb119 at pH 6.0 in solution.
Figure 4.
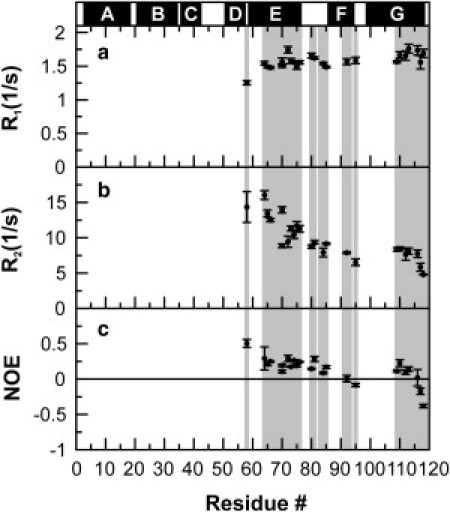
15N (a) R1 and (b) R2 relaxation rates, and (c) 1H-15N heteronuclear NOEs for apoMb119 at pH 6.0 and 4°C. (Shaded areas) Sequence regions corresponding to the assigned backbone residues. (Solid bars above the plot) Sequence regions corresponding to the native α-helices of full-length apomyoglobin.
All of the above three relaxation parameters are influenced by motions in the picosecond-to-nanosecond timescale, but each of them to a different extent. R1 depends mostly on picosecond-to-nanosecond motions. R2 is more sensitive to slower motions in the nanosecond range and is also influenced by slow dynamics and(or) chemical exchange taking place on the microsecond-to-millisecond timescale. Steady-state NOE is particularly sensitive to fast motions taking place on the picosecond-to-lower-nanosecond timescale.
A comparison among 1H,15N-HSQC and 1H,15N-CPMG-HSQC data has previously shown that conformational exchange on the microsecond-to-millisecond timescale is unlikely to significantly contribute to the broad lineshapes observed for most of the visible resonances (39), though intermediate-exchange processes slower than ∼1 ms cannot be technically ruled out. This study established that the intensity ratio of sensitivity-enhanced 1H,15N CPMG-HSQC (43), and HSQC (62) intensities (ICPMG-HSQC/IHSQC) equals 1 for most apoMb119 residues. H64 and V66 are an exception, as further discussed below.
The backbone 15N-R1, 15N-R2, and the 1H-15N-steady-state NOEs for the detectable well-resolved apoMb119 resonances are reported in Fig. 4.
The most salient properties of the graphs are 1), the generally flat profile of the R1 plot, 2), the large dynamic range of R2 values, and 3), the pronounced slope-down profiles of both R2 and steady-state NOEs.
The third property generally suggests an increasing mobility on the picosecond-to-nanosecond timescale as the chain approaches the C-terminus. A more rigorous description of the different regimes of motion is provided by the reduced spectral density analysis discussed below.
A major spectral feature of self-associated apoMb119 is that the resonances of N-terminal residues are broadened beyond detection. Hence, the relaxation parameters for the N-terminal residues cannot be reported. The severe line-broadening of the N-terminal residues is due to either very slow tumbling rates or to the presence of conformational exchange on the microsecond-to-millisecond timescale. Back-of-the-envelope calculations, assuming 15N relaxation to be dominated by dipole-dipole and chemical shift anisotropy mechanisms, show that the expected linewidths for an approximately spherical 8-mer at the temperature of our experiments would yield detectable resonances. This estimate suggests that intermediate chemical exchange may contribute, at least in part, to the observed undetectability of the N-terminal resonances.
As shown by the NMR relaxation profiles, self-associated apoMb119 is a complex species displaying plenty of site-specific local motions with widely different features. Therefore, it cannot generally be assumed to tumble isotropically. As a consequence, apoMb119 cannot be effectively described by traditional model-free analysis involving a single global rotational correlation time and either one (63) or two (64) effective correlation times for local motions.
Dynamically complex molecules sharing many of the above NMR features are often encountered in unfolded states and natively unfolded or partially folded proteins. The NMR relaxation of these species is typically treated by a variety of modified model-free (65,66) or alternative (54–56,67) approaches, which require limited or no assumptions on the structural features of the molecule of interest. We chose to analyze our data by the reduced spectral density approach introduced in 1995 by Farrow et al. (54). The residue-specific spectral density is proportional to the probability of motion at a given frequency (52). Therefore, knowledge on spectral densities provides a direct assessment of the rates of local motions of backbone amide bonds, allowing a prompt estimate of the dynamics timescales and their residue-specific variations.
The results are shown in Fig. 5. J(0) provides information on motions on the nanosecond timescale and slower microsecond-to-millisecond chemical exchange processes. J(0.870 ωH) reports on the presence of fast motions on the subnanosecond-to-picosecond timescale. J(ωN) results from significant contributions due to fast picosecond-to-nanosecond motions.
Figure 5.
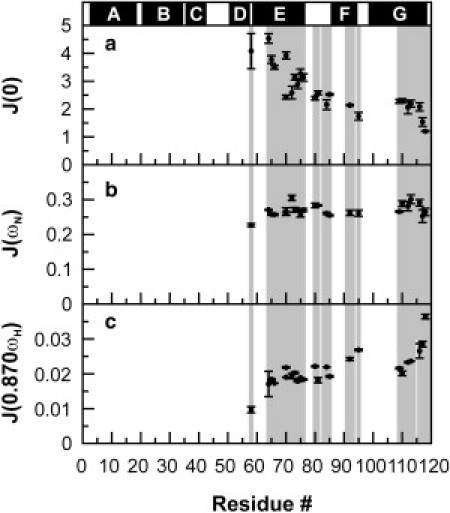
(a) J(0), (b) J(ωN), and (c) J(0.870 ωH) spectral densities (nanoseconds/radians) for apoMb119 at pH 6.0 and 4°C, calculated as described in the Materials and Methods. (Shaded areas) Chain regions corresponding to the assigned backbone residues. (Solid bars above the plot) Position of the native α-helices in full-length apoMb.
The apoMb119 J(0.870 ωH) values increase significantly from the intermediate portion of the fragment to the C-terminal end, indicating a progressively increasing population of high-frequency (picosecond) motions from the central region to the very C-terminal portion of the chain. Conversely, J(0) decreases from the intermediate portion of the fragment to the C-terminal end, consistent with smaller contributions of lower frequency (nanosecond) motions by the C-terminal part of the chain. While millisecond chemical exchange cannot be entirely ruled out, it is unlikely the main contributor to either R2 or J(0) due to the fact that ICPMG-HSQC/IHSQC = 1 for most of the detectable backbone resonances (see also discussion above). However, the H64 and V66 resonances have a higher 1H,15N-CMPG-HSQC intensity than the corresponding HSQC experiment (ICPMG-HSQC/IHSQC = 1.1–1.2), suggesting that millisecond-type slow conformational processes are present for this portion of the backbone, which is quite close to the compact self-associated core.
Interestingly, the NMR resonances for the region corresponding to the first portion of the native G helix, which is highly nonpolar (Fig. 6 a), could not be assigned. These residues are likely exchange-broadened due to some intramolecular conformational rearrangements taking place on the intermediate NMR chemical shift timescale (microsecond-to-millisecond). The broadening of these resonances is less likely to arise from nonlocal effects resulting from exchange with the self-associated N-terminal core, given that we do not detect any of the expected variations in J(0.870 ωH) and J(0) for the amino acids close to the missing resonances in the G helix region.
Figure 6.
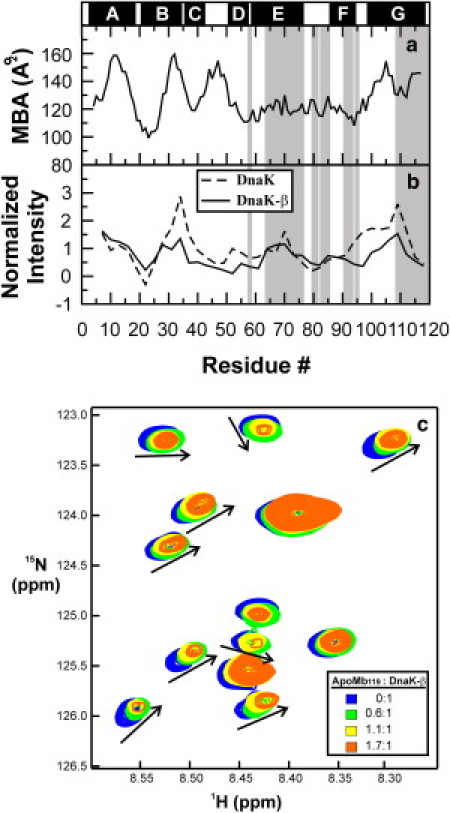
(a) Mean buried area (MBA) of the apoMb119 sequence calculated according to Rose et al. (71). High scores correspond to nonpolar regions of the chain. (b) Normalized experimental binding affinities of full-length DnaK (dashed line) and DnaK-β (solid line) for the apoMb119 sequence (72). (Shaded areas) Chain regions corresponding to the assigned backbone residues. (Solid bars above the plot) Position of the native α-helices in full-length apoMb. (c) 1H,15N-HSQC spectrum of 15N-DnaK-β chaperone in the absence and presence of variable amounts of unlabeled apoMb119, in 10 mM sodium acetate and 5% D2O, at pH 6.0. Spectra acquired at different stoichiometric ratios are color-coded according to the scheme provided in the legend. Specific resonance shifts observed upon addition of increasing amounts of apoMb119 (indicated by arrows to guide the eye).
Nonpolar nature of apoMb119 and local binding sites for DnaK-β
ApoMb119 comprises the majority of the nonpolar residues of the parent 153-residue full-length apomyoglobin and it is overall rather hydrophobic, as assessed by the plot of the apoMb119 mean buried area (MBA) distribution across sequence, shown in Fig. 6 a. The N-terminal region of the chain is particularly nonpolar, with three major hydrophobic regions, suggesting that this portion of the chain is particularly prone to self-association.
ApoMb119 exhibits a tendency to bind molecular chaperones. Fig. 6 b shows that the local affinities (68) for both the full-length DnaK chaperone and the substrate-binding domain of DnaK, denoted as DnaK-β (39), are similar. A comparison of panels a and b of Fig. 6 also shows that the main predicted DnaK-β local binding sites correspond to the major nonpolar regions of the apoMb119 sequence. These sites include the chaperone affinity sites along the apoMb119 A and B helices, located in the N-terminal half of the chain, and the local chaperone affinity site along the residues corresponding to the apoMb119 G helix.
Binding of apoMb119 to DnaK-β
Upon titrating 15N-DnaK-β with unlabeled apoMb119, a selected number of chaperone resonances are shifted as shown in Fig. 6 c, demonstrating the presence of specific interactions between the chaperone and apoMb119. On the other hand, the detectable 1H,15N-HSQC NMR resonances of apoMb119 undergo essentially no chemical shift changes when stoichiometric amounts of DnaK-β are added to the medium (Fig. 2 and Fig. S3). The inability to detect changes in the apoMb119 spectrum shows that the residues responsible for interactions with the chaperone are in the N-terminal region, whose resonances are undetectable by NMR.
A previous report (39) indicated that the average intensity of the detectable NMR resonances increases by >100% (for samples with identical substrate concentration) after addition of stoichiometric amounts of chaperone to apoMb119. At that time, no assignments were available. Given that chemical shift assignments are now known (69), the relative changes in the well-resolved resonance intensities upon addition of chaperone can be plotted at the residue-specific level, as shown in Fig. 7. An overall increase in the detectable intensities is observed, suggesting either a decrease in the size of the aggregate (ruled out by the relaxation and translational diffusion data, see below) or an enhancement in the concentration of the NMR-detectable aggregate. The secondary structure of the residues corresponding to the detectable NMR resonances is weakly helical and entirely similar both in the absence (Fig. 3) and presence (69) of chaperone.
Figure 7.
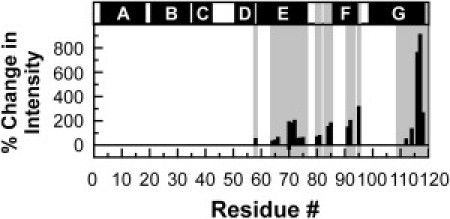
Plot illustrating the percent change in apoMb1191H,15N-HSQC resonance intensity upon addition of stoichiometric amounts of DnaK-β as a function of residue number, defined as [(IapoMb119-DnaK-β − IapoMb119)/IapoMb119]. (Shaded areas) Sequence regions corresponding to the assigned backbone residues. (Solid bars above the plot) Regions corresponding to the native α-helices of full-length apoMb.
The enhancement in resonance intensities (Fig. 7) coupled with the absence of chemical shift changes in the C-terminal portion of the chain (Fig. 2 and Fig. S3), the faster R2 relaxation rates of the C-terminal residues (see Fig. S5), and the slightly smaller translational diffusion coefficient (see the Supporting Material) can only be reconciled if the presence of the chaperone enables the production of a larger population of soluble aggregate of similar structure but slightly larger than the aggregate present in the absence of chaperone. The experiments in the presence of chaperone are performed by mixing a low-pH protein stock of unfolded monomeric apoMb119 with an excess of buffer containing the DnaK-β chaperone. Therefore, after the two solutions are mixed, the chaperone may interact with apoMb119 transiently, before it aggregates, and skew the distribution of aggregates toward a larger population of NMR-detectable aggregates. A similar scenario was previously observed for the effect of the DnaK-β chaperone on full-length apoMb, apoMb153 (39).
A structural model for the ApoMb119 soluble aggregate
The combination of 1), translational diffusion experiments, 2), native gel analysis, 3), the macroscopic aggregation observed at high concentrations, and 4), the high MBA values detected for the N-terminal region (Fig. 6 a) show that N-terminal region of the apoMb119 ensemble is self-associated and comprises a slow tumbling core.
Given that the detectable resonances bear only residual helical structure, and that prior far-UV circular dichroism data showed the presence of 30% helix and 18% β-strand for apoMb119 under similar solution conditions (37), it is likely that the slow-tumbling core contains some β-strand secondary structure. Such structure cannot be explicitly quantified here, as resonances corresponding to N-terminal region of the polypeptide chains are undetectable by NMR.
The self-associated apoMb119 shows an unusual distribution of native and nonnative secondary structure and topology, which is in interesting contrast with the globulomer structure displayed by other more amyloidogenic sequences such as Aβ (30). In the latter case, a compact core with dangling ends was detected, similar to the apoMb119 aggregate. However, the Aβ globulomer displayed only β-strand secondary structure.
The nearly exclusive absence of any exchange contributions to R2 in the microsecond-to-millisecond timescale for the visible resonances (39) is consistent with a model according to which the N-terminal portion of apoMb119 forms a self-associated hydrophobic core with fraying ends and a highly dynamic (picosecond-to-nanosecond timescale) C-terminal portion, bearing some residual α-helical structure. Some of the residual helicity is nonnative, e.g., in the EF loop region, which is a turn in native apomyoglobin but has distinct helical character in apoMb119. A simple model illustrating the proposed conformation and dynamics of the apoMb119 soluble aggregate, consistent with the above data, is provided in Fig. 8.
Figure 8.
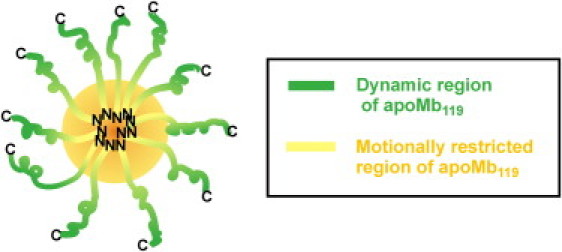
Cartoon illustrating the proposed model for the conformational state of apoMb119 oligomers in solution. (Green and yellow regions) Motionally free and dynamically restricted portion of apoMb119, respectively. (Orange circle) Core of the slow-tumbling apoMb119 soluble aggregate. The residual helicity of the highly dynamic C-terminal region is qualitatively illustrated. N and C denote apoMb119's N- and C-termini, respectively.
Conclusions
This study underscores the widely heterogeneous conformation and dynamics of a soluble protein aggregate. ApoMb119 has a nonpolar moderately dynamic N-terminal core with nonnative β-strands, and a more polar, highly dynamic C-terminal region with residual helical secondary structure. The conformation of apoMb119 is in interesting contrast with that of amyloidogenic globulomers, which display exclusively β-strand secondary structure.
Acknowledgments
We thank Carolina Vega and Marco Tonelli for technical assistance, and Ashok Sekhar and Daria Fedyukina for a critical reading of the manuscript.
This work was supported by National Institutes of Health (NIH) grant No. GM068538 and by the Vilas Associates Award to S.C. This study made use of the National NMR Facility at Madison, which is supported by NIH grants No. P41RR02301 (Biomedical Research Training Program/National Center for Research Resources) and No. P41GM66326 (National Institute of General Medical Sciences).
Footnotes
Senapathy Rajagopalan's present address is The Methodist Hospital Research Institute, Genomic Medicine, 6565 Fannin St., F8-060, Houston, TX 77030 and UTMD Anderson Cancer Center, 1515 Holcombe Blvd., Unit 1000, Houston, TX 77030.
Supporting Material
References
- 1.Jellinger K.A. Basic mechanisms of neurodegeneration: a critical update. J. Cell. Mol. Med. 2010;14:457–487. doi: 10.1111/j.1582-4934.2010.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uversky V.N. Mysterious oligomerization of the amyloidogenic proteins. FEBS J. 2010;277:2940–2953. doi: 10.1111/j.1742-4658.2010.07721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramirez-Alvarado M. Principles of protein misfolding. In Progress in Molecular Biology and Translational Science. Molecular Biology of Protein Folding, Part B. 2008;84:115–160. doi: 10.1016/S0079-6603(08)00404-2. [DOI] [PubMed] [Google Scholar]
- 4.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 5.Dember L.M. Amyloidosis-associated kidney disease. J. Am. Soc. Nephrol. 2006;17:3458–3471. doi: 10.1681/ASN.2006050460. [DOI] [PubMed] [Google Scholar]
- 6.Sakono M., Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 7.Kitamura A., Kubota H. Amyloid oligomers: dynamics and toxicity in the cytosol and nucleus. FEBS J. 2010;277:1369–1379. doi: 10.1111/j.1742-4658.2010.07570.x. [DOI] [PubMed] [Google Scholar]
- 8.Goto Y., Yagi H., Ban T. Structure, formation and propagation of amyloid fibrils. Curr. Pharm. Des. 2008;14:3205–3218. doi: 10.2174/138161208786404146. [DOI] [PubMed] [Google Scholar]
- 9.Tycko R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Q. Rev. Biophys. 2006;39:1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 10.Nelson R., Eisenberg D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006;16:260–265. doi: 10.1016/j.sbi.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Irie K., Murakami K., Shirasawa T. Structure of β-amyloid fibrils and its relevance to their neurotoxicity: implications for the pathogenesis of Alzheimer's disease. J. Biosci. Bioeng. 2005;99:437–447. doi: 10.1263/jbb.99.437. [DOI] [PubMed] [Google Scholar]
- 12.Roychaudhuri R., Yang M., Teplow D.B. Amyloidβ-protein assembly and Alzheimer disease. J. Biol. Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teplow D.B., Lazo N.D., Stanley H.E. Elucidating amyloid β-protein folding and assembly: a multidisciplinary approach. Acc. Chem. Res. 2006;39:635–645. doi: 10.1021/ar050063s. [DOI] [PubMed] [Google Scholar]
- 14.Lazo N.D., Grant M.A., Teplow D.B. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005;14:1581–1596. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lesné S., Koh M.T., Ashe K.H. A specific amyloid β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 16.Townsend M., Shankar G.M., Selkoe D.J. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: a potent role for trimers. J. Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S.M., Hong S.Y., Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh D.M., Selkoe D.J. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Lambert M.P., Barlow A.K., Klein W.L. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsia A.Y., Masliah E., Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc. Natl. Acad. Sci. USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roher A.E., Chaney M.O., Emmerling M.R. Morphology and toxicity of Aβ-(1-42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J. Biol. Chem. 1996;271:20631–20635. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- 22.Fändrich M., Fletcher M.A., Dobson C.M. Amyloid fibrils from muscle myoglobin. Nature. 2001;410:165–166. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- 23.Pallarès I., Vendrell J., Ventura S. Amyloid fibril formation by a partially structured intermediate state of α-chymotrypsin. J. Mol. Biol. 2004;342:321–331. doi: 10.1016/j.jmb.2004.06.089. [DOI] [PubMed] [Google Scholar]
- 24.Frare E., Polverino de Laureto P., Fontana A. A highly amyloidogenic region of hen lysozyme. J. Mol. Biol. 2004;340:1153–1165. doi: 10.1016/j.jmb.2004.05.056. [DOI] [PubMed] [Google Scholar]
- 25.Wiseman R.L., Powers E.T., Kelly J.W. Partitioning conformational intermediates between competing refolding and aggregation pathways: insights into transthyretin amyloid disease. Biochemistry. 2005;44:16612–16623. doi: 10.1021/bi0511484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chow C., Kurt N., Cavagnero S. Structural characterization of apomyoglobin self-associated species in aqueous buffer and urea solution. Biophys. J. 2006;90:298–309. doi: 10.1529/biophysj.105.070227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calamai M., Chiti F., Dobson C.M. Amyloid fibril formation can proceed from different conformations of a partially unfolded protein. Biophys. J. 2005;89:4201–4210. doi: 10.1529/biophysj.105.068726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmad B., Winkelmann J., Chiti F. Searching for conditions to form stable protein oligomers with amyloid-like characteristics: the unexplored basic pH. Biochim. Biophys. Acta. 2010;1804:223–234. doi: 10.1016/j.bbapap.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Khare S.D., Dokholyan N.V. Molecular mechanisms of polypeptide aggregation in human diseases. Curr. Protein Pept. Sci. 2007;8:573–579. doi: 10.2174/138920307783018703. [DOI] [PubMed] [Google Scholar]
- 30.Yu L.P., Edalji R., Olejniczak E.T. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 31.Gellermann G.P., Byrnes H., Barghorn S. Aβ-globulomers are formed independently of the fibril pathway. Neurobiol. Dis. 2008;30:212–220. doi: 10.1016/j.nbd.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 32.Bocian W., Sitkowski J., Kozerski L. Direct insight into insulin aggregation by 2D NMR complemented by PFGSE NMR. Proteins. 2008;71:1057–1065. doi: 10.1002/prot.21969. [DOI] [PubMed] [Google Scholar]
- 33.Barghorn S., Nimmrich V., Hillen H. Globular amyloid β-peptide oligomer—a homogenous and stable neuropathological protein in Alzheimer's disease. J. Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- 34.Chugh J., Chatterjee A., Hosur R.V. Structural characterization of the large soluble oligomers of the GTPase effector domain of dynamin. FEBS J. 2006;273:388–397. doi: 10.1111/j.1742-4658.2005.05072.x. [DOI] [PubMed] [Google Scholar]
- 35.Chugh J., Sharma S., Hosur R.V. NMR insights into a megaDalton-size protein self-assembly. Protein Sci. 2008;17:1319–1325. doi: 10.1110/ps.035840.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Juneja J., Bhavesh N.S., Hosur R.V. NMR identification and characterization of the flexible regions in the 160 kDa molten globule-like aggregate of barstar at low pH. Biochemistry. 2002;41:9885–9899. doi: 10.1021/bi026034w. [DOI] [PubMed] [Google Scholar]
- 37.Chow C.C., Chow C., Cavagnero S. Chain length dependence of apomyoglobin folding: structural evolution from misfolded sheets to native helices. Biochemistry. 2003;42:7090–7099. doi: 10.1021/bi0273056. [DOI] [PubMed] [Google Scholar]
- 38.Reference deleted in proof.
- 39.Kurt N., Rajagopalan S., Cavagnero S. Effect of Hsp70 chaperone on the folding and misfolding of polypeptides modeling an elongating protein chain. J. Mol. Biol. 2006;355:809–820. doi: 10.1016/j.jmb.2005.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Geet A.L. Calibration of methanol nuclear magnetic resonance thermometer at low temperature. Anal. Chem. 1970;42:679–680. [Google Scholar]
- 41.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 42.Johnson B.A., Blevins R.A. NMRView—a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 43.Mulder F.A.A., Spronk C., Boelens R. Improved HSQC experiments for the observation of exchange broadened signals. J. Biomol. NMR. 1996;8:223–228. doi: 10.1007/BF00211169. [DOI] [PubMed] [Google Scholar]
- 44.Pervushin K., Riek R., Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tzakos A.G., Grace C.R.R., Riek R. NMR techniques for very large proteins and RNAs in solution. Annu. Rev. Biophys. Biomol. Struct. 2006;35:319–342. doi: 10.1146/annurev.biophys.35.040405.102034. [DOI] [PubMed] [Google Scholar]
- 46.Wider G., Wüthrich K. NMR spectroscopy of large molecules and multimolecular assemblies in solution. Curr. Opin. Struct. Biol. 1999;9:594–601. doi: 10.1016/s0959-440x(99)00011-1. [DOI] [PubMed] [Google Scholar]
- 47.Wider G. NMR techniques used with very large biological macromolecules in solution. In Methods in Enzymology. Nuclear Magnetic Resonance of Biological Macromolecules, Part C. 2005;394:382–398. doi: 10.1016/S0076-6879(05)94015-9. [DOI] [PubMed] [Google Scholar]
- 48.Fernández C., Wider G. TROSY in NMR studies of the structure and function of large biological macromolecules. Curr. Opin. Struct. Biol. 2003;13:570–580. doi: 10.1016/j.sbi.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 49.Wishart D.S., Bigam C.G., Sykes B.D. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 50.Farrow N.A., Muhandiram R., Kay L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 51.Mosteller F., Tukey J.W. Addison-Wesley; Boston, MA: 1977. Data Analysis and Regression: a Second Course in Statistics. [Google Scholar]
- 52.Abragam A. Clarendon Press; Oxford, UK: 1961. The Principles of Nuclear Magnetism. pp.599. [Google Scholar]
- 53.Hiyama Y., Niu C.H., Torchia D.A. Determination of 15N chemical-shift tensor via 15N-2H dipolar coupling in boc-glycylglycyl[15N]glycine benzyl ester. J. Am. Chem. Soc. 1988;110:2378–2383. [Google Scholar]
- 54.Farrow N.A., Zhang O.W., Kay L.E. Spectral density function mapping using 15N relaxation data exclusively. J. Biomol. NMR. 1995;6:153–162. doi: 10.1007/BF00211779. [DOI] [PubMed] [Google Scholar]
- 55.Peng J.W., Wagner G. Mapping of spectral density functions using heteronuclear NMR relaxation measurements. J. Magn. Reson. 1992;98:308–332. [Google Scholar]
- 56.Peng J.W., Wagner G. Investigation of protein motions via relaxation measurements. Methods Enzymol. 1994;239:563–596. doi: 10.1016/s0076-6879(94)39022-3. [DOI] [PubMed] [Google Scholar]
- 57.Kurt N., Cavagnero S. The burial of solvent-accessible surface area is a predictor of polypeptide folding and misfolding as a function of chain elongation. J. Am. Chem. Soc. 2005;127:15690–15691. doi: 10.1021/ja0560682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cantor C.R., Schimmel P.R. Freeman; New York: 1980. Biophysical Chemistry. [Google Scholar]
- 59.Wishart D.S., Sykes B.D. Chemical shifts as a tool for structure determination. In Methods in Enzymology. Nuclear Magnetic Resonance, Part C. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 60.Spera S., Bax A. Empirical correlation between protein backbone conformation and C-α and C-β C-13 nuclear-magnetic-resonance chemical-shifts. J. Am. Chem. Soc. 1991;113:5490–5492. [Google Scholar]
- 61.Eliezer D., Wright P.E. Is apomyoglobin a molten globule? Structural characterization by NMR. J. Mol. Biol. 1996;263:531–538. doi: 10.1006/jmbi.1996.0596. [DOI] [PubMed] [Google Scholar]
- 62.Kay L.E., Keifer P., Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 1992;114:10663–10665. [Google Scholar]
- 63.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- 64.Clore G.M., Szabo A., Gronenborn A.M. Deviations from the simple 2-parameter model-free approach to the interpretation of N-15 nuclear magnetic-relaxation of proteins. J. Am. Chem. Soc. 1990;112:4989–4991. [Google Scholar]
- 65.Modig K., Poulsen F.M. Model-independent interpretation of NMR relaxation data for unfolded proteins: the acid-denatured state of ACBP. J. Biomol. NMR. 2008;42:163–177. doi: 10.1007/s10858-008-9280-0. [DOI] [PubMed] [Google Scholar]
- 66.Buevich A.V., Baum J. Dynamics of unfolded proteins: Incorporation of distributions of correlation times in the model free analysis of NMR relaxation data. J. Am. Chem. Soc. 1999;121:8671–8672. [Google Scholar]
- 67.Lefevre J.F., Dayie K.T., Wagner G. Internal mobility in the partially folded DNA binding and dimerization domains of GAL4: NMR analysis of the N-H spectral density functions. Biochemistry. 1996;35:2674–2686. doi: 10.1021/bi9526802. [DOI] [PubMed] [Google Scholar]
- 68.Rüdiger S., Germeroth L., Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997;16:1501–1507. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Z., Kurt N., Cavagnero S. Secondary structure mapping of DnaK-bound protein fragments: chain helicity and local helix unwinding at the binding site. Biochemistry. 2006;45:12325–12333. doi: 10.1021/bi0612263. [DOI] [PubMed] [Google Scholar]
- 70.Kuriyan J., Wilz S., Petsko G.A. X-ray structure and refinement of carbon-monoxy (Fe II)-myoglobin at 1.5 Å resolution. J. Mol. Biol. 1986;192:133–154. doi: 10.1016/0022-2836(86)90470-5. [DOI] [PubMed] [Google Scholar]
- 71.Rose G.D., Geselowitz A.R., Zehfus M.H. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 72.Vega C.A., Kurt N., Cavagnero S. Binding specificity of an α-helical protein sequence to a full-length Hsp70 chaperone and its minimal substrate-binding domain. Biochemistry. 2006;45:13835–13846. doi: 10.1021/bi061432a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.