

Abstract
The constraint-based reconstruction and analysis (COBRA) framework has been widely used to study steady-state flux solutions in genome-scale metabolic networks. One shortcoming of current COBRA methods is the possible violation of the loop law in the computed steady-state flux solutions. The loop law is analogous to Kirchhoff's second law for electric circuits, and states that at steady state there can be no net flux around a closed network cycle. Although the consequences of the loop law have been known for years, it has been computationally difficult to work with. Therefore, the resulting loop-law constraints have been overlooked. Here, we present a general mixed integer programming approach called loopless COBRA (ll-COBRA), which can be used to eliminate all steady-state flux solutions that are incompatible with the loop law. We apply this approach to improve flux predictions on three common COBRA methods: flux balance analysis, flux variability analysis, and Monte Carlo sampling of the flux space. Moreover, we demonstrate that the imposition of loop-law constraints with ll-COBRA improves the consistency of simulation results with experimental data. This method provides an additional constraint for many COBRA methods, enabling the acquisition of more realistic simulation results.
Introduction
A primary aim of researchers in the field of systems biology is to understand the properties of large-scale biochemical networks through the construction and use of predictive in silico models. One common approach is the constraint-based reconstruction and analysis (COBRA) framework (1–4). Genome-scale metabolic models are built in a bottom-up fashion from various sources of biological knowledge, such as genome annotations, metabolic databases, and published biochemical information (5–7). This quality-controlled reconstruction process results in validated mathematical models that can make predictions about reaction fluxes inside a cell. These predictions have a wide variety of applications (8–10). Because these models are generally underdetermined, steady-state flux solutions are calculated by imposing constraints on the system and optimizing an objective function (2,11–13). Popular constraints include the steady-state assumption, reaction reversibility, and bounds on reaction capacity. The various methods developed under this framework have been described elsewhere (2,4–6,14).
COBRA models are defined primarily by their stoichiometric matrix (S) and reaction lower (lb) and upper (ub) bounds. The stoichiometric matrix encodes information about reactions (columns) and metabolites (rows), such that each entry, Si,j, is the signed stoichiometric coefficient of metabolite i in reaction j. The stoichiometric matrix relates the reaction flux (v) to the change in metabolite concentrations (x):
At steady state, it is assumed that concentrations do not change. Thus, the equation reduces to . Upper and lower bounds can be placed on each reaction flux. Many reactions are considered irreversible (vi > 0), whereas others, such as uptake and secretion reactions, can be set to experimentally measured values (vi = vexp). If no information is available, arbitrarily large bounds are set (e.g., −10,000 < vi < 10,000). Together, the flux bounds and the steady-state equation define a bounded space of possible flux states.
The constraint-based method known as flux balance analysis (FBA) is commonly used to compute the likely state of the network by optimizing a metabolic objective (15,16). Common objectives include biomass production (12), ATP production (17), and biomass production per unit input (13).
Many COBRA methods, including FBA, ignore the imposition of the loop law (18). The loop law is analogous to Kirchhoff's second law for electrical circuits, in that it states that the thermodynamic driving forces around a metabolic loop must add up to zero. As such, there cannot be a net flux around a closed cycle in a network at steady state. Methods for detecting loops have been developed (19); however, these methods are too computationally intensive and not flexible enough to be included within optimization computations (20).
An alternative approach for addressing the loop law is to include additional thermodynamic information. Such an approach relies on the relation ΔGr = ΔG0 + RT ln Q, where Q is a ratio of metabolic concentrations and ΔGr is the Gibbs energy of a reaction. ΔGr directly relates to the sign of the flux through the associated reaction (i.e., if ΔGr > 0, then vnet < 0 and vice versa). This imposes additional constraints on reaction directionality, as well as on feasible concentrations. This approach has been used to compute potential regulatory sites (21), determine reaction directionality (22), and compute feasible concentration ranges (23). A slightly different formalism with decoupled forward and reverse reactions has been used to modify FBA (24,25). All of these methods require a priori knowledge of the standard free-energy change of reactions (ΔGr) or the standard energies of formation (ΔGf) of all metabolites in the network. These values can be found in databases (e.g., the NIST Chemical Kinetics Database (26)) or estimated computationally with methods such as group contribution theory (27,28). However, the lack of accuracy and coverage in some cases can pose challenges.
Here, we present a simpler method to incorporate the loop-law constraints into many current COBRA methods. This method does not require additional inputs or data (e.g., metabolite concentrations and ΔGf), and turns any linear programming (LP), quadratic programming (QP), or mixed integer problem (MIP) into a modified MIP problem. The solution to the modified MIP problem solves the initial problem, with an additional constraint. This constraint does not allow the inclusion of network fluxes that contain loops. We demonstrate this technique on three popular COBRA methods: FBA, flux variability analysis (FVA), and Monte Carlo sampling, thereby producing loopless versions of each method (ll-FBA, ll-FVA, and ll-sampling, respectively). Given the extensive application of COBRA methods, this simple method for imposing the loop-law constraint will likely find widespread use.
Materials and Methods
The loopless condition
First, consider the simpler problem of determining whether a given flux solution, v, contains a loop. For v to satisfy the loop law, the reaction energies around any cycle must add to zero. This condition can be written concisely as vT × G = 0, where G is a vector of energies for each reaction. Extreme pathway (29) and elementary mode analysis (30) can be used to identify all cycles. However, these methods have shown that the number of loops (type III pathways) grows rapidly with the network size, and that enumerating all loops is not possible for medium- to large-scale networks (31). Fortunately, it is not necessary to enumerate all loops. As shown in Fig. 1, all loops lie within the internal network, Sint. Any steady-state pathway in Sint is a loop, and all such paths can be expressed as a linear combination of the null basis of Sint (3). All loops can be expressed in the form v = Nint × αi, where Nint = null(Sint), and αi are weights. If it can be shown that if NTint × G = 0, then vT G = 0 for all loops v.
Figure 1.
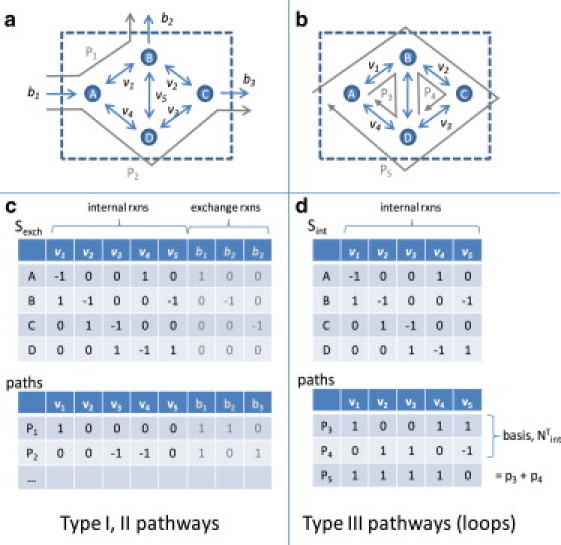
Loops in metabolic networks. (a and c) A small network illustrates pathways with and without loops, in which the network contains five internal reactions and three exchanges. (b and d) The internal part of the network. Steady-state pathways are superimposed (P1-P5). EPA (29) indicates that Type I and II pathways use exchange reactions. A partial subset of these pathways is listed in c. Type III pathways do not contain exchange reactions and form a set of loops. In this example there are three type III pathways (d). P3 and P4 form the basis of the internal null space (Nint), and P5 can be written as a linear combination of the other two type III pathways.
The loopless condition forms an LP problem. A vector of continuous variables (Gi) indicates the driving force of each reaction. This quantity can be thought of as being analogous to the ΔGr of each reaction, in that sign(G) = sign(ΔGr), although numerically they may be quite different. A loop is purely defined by the sign (direction) of the flux distribution (19). Therefore, if Nint × Gi = 0, no loop is present.
To verify that a flux distribution does not contain loops, a solution to Nint × G = 0 is found with the following constraints:
In practice, it is necessary to restrict Gi to be strictly positive or strictly negative to avoid the degenerate solution Gi = 0 for all i. This requirement is another reason why Gi may not be interpreted directly as ΔGr-values. The following correction restricts Gi to [−1000,−1] or [1,1000], and Gi may never be exactly zero:
If a solution exists, then v contains no loops. Otherwise, v contains a loop. Unlike most LP problems, the objective (max cT × G) is of no concern because only the feasibility is relevant.
Adding the loop-law constraints to COBRA problems: ll-FBA
The linear loop-law constraints described above can be added to almost any COBRA LP, mixed integer linear programming (MILP), QP, or mixed integer quadratic programming (MIQP) problem, as long as this problem contains a variable, vi, for each of the internal fluxes in the model. The only necessary addition is the condition that ensures sign(v) = −sign(G). This is achieved by adding a binary indicator variable (ai) for each internal reaction.
The full set of constraints can be expressed as follows:
This is converted to the following MILP problem:
As before, to avoid degenerate solutions, Gi is not allowed to be zero.
These constraints may be added to almost any LP COBRA method. For example, the full formulation for loopless FBA (ll-FBA) is as follows:
subject to
where Skj is the stoichiometric matrix; j iterates over all reactions; i iterates over internal reactions; lbj and ubj are the lower and upper bounds, respectively, for each reaction; and cj are the coefficients of optimization. See the Supporting Material for additional performance enhancements that can be added to speed up the computation.
Models
Several COBRA models were used to validate the ll-COBRA methods. All models (except the toy network) have been published elsewhere (see Table 1). Models were exported from the BiGG knowledgebase (32) as SBML files and subsequently imported in the COBRA toolbox, using default parameters. Only the Staphylococcus aureus model was modified. This model contains several potential biomass objective functions, so only the default objective, biomass_SA_8a, was retained. In addition, this model had unrealistic uptake rates of carbon substrates, so these rates were reduced to 10 mmol glucose gDW−1 h−1 (similar to uptake rates in other prokaryotic models). These changes do not affect the occurrence of loops, but aided in the comparison of loop fluxes before and after the imposition of loop law constraints.
Table 1.
Five models of increasing size used in this work
Loopless FVA
Loopless FVA (ll-FVA) was performed with the use of the ll-FBA method described above, followed by the sequential maximization and minimization of each reaction in the model. This computation was performed both with and without the loop-law constraints. Reactions in which the range (max vi − min vi) differed by >10−6 mmol gDW−1 h−1 between FVA and ll-FVA were classified as loop reactions, affected by the loop-law constraints.
Sampling of the steady-state solution space
Monte Carlo sampling was used to generate a set of flux distributions that uniformly sample the space of all feasible fluxes (possibly including loops). The method is based on the artificially centered hit and run algorithm with slight modifications (33,34). Initially, a set of 2000 nonuniform pseudo-random points, called warm-up points, was generated. In a series of iterations, we randomly moved each point while keeping it within the feasible flux space. We accomplished this by 1), choosing a random direction; 2), computing the limits on how far a point could travel in that direction (positive or negative); and 3), choosing a new point randomly along that line. After numerous iterations, the set of points was mixed and approached a uniform sample of the solution space. We used a mixed fraction, as described previously (34), to determine when the solution space was uniformly sampled. For this work, 2000 points were generated and mixed for 1 h, at which time the space was determined to be uniformly sampled with a mixed fraction of 0.495.
Removing loops: ll sampling
Sampling the loopless solution space can be implemented as a postprocessing step in Monte Carlo sampling. Once a set of flux distributions has been generated, the loops can be removed. To do this, we find a loopless flux, wi, that is nearest to a given flux distribution, vi:
subject to:
where |wi − vi| is the distance to be minimized. If the Euclidian norm (2-norm) is used as the distance metric, this becomes an MIQP problem in which (wj − vj)2 is minimized. The Manhattan norm (1-norm) may be implemented as an MILP problem by introducing a helper variable v+, such that |v − w| = v+.
subject to:
Computer configuration and availability
Computations were performed in the MATLAB (The MathWorks, Natick, MA) version 2009b environment with the COBRA toolbox (35) and SBML toolbox version 3.0 (36). Linear and mixed integer linear programming were performed with the TOMLAB/CPLEX package version 7.4 (Tomlab Research, Pullman, WA). All computations were run on a Dell Studio XPS workstation (core i7 920 processor, 12GB ram, Windows 2003 R2, 64 bit). The MATLAB parallel toolbox was used for both the ll-FVA and ll-sampling applications to take advantage of its multicore architecture.
The methods presented here can be added to any implementation of FBA or similar methods. However, to facilitate their use, the code used for the computations in this publication are available in the COBRA toolbox 2.0 (35), in which the optimizeCbModel (FBA) and fluxVariability functions have an optional flag to exclude loops. Monte Carlo sampling is called through the nearest loopless flux function, which returns the nearest loopless flux distribution using the MILP or MIQP methods. Internally, these three methods call an addLoopLawConstraints function, which adds loop-law constraints to any COBRA LP, MILP, QP, or MIQP problem and turns it into an ll-COBRA problem.
Microarray analysis
Previously published (37) Affymetrix E. coli antisense genome arrays were obtained and subjected to GC robust multiarray average (gcrma) normalization using the bioconductor package in R. This data set includes 213 expression profiles, representing ∼70 variations in experimental conditions and genetic perturbation of E. coli K-12 MG1655. We used these data to assess loop flux before and after imposition of the loop-law constraints, and to compare these fluxes with gene expression levels. To that end, we first identified the genes associated with loop and nonloop reactions. We then selected 2500 random pairs, containing one loop- and one nonloop-associated gene, and computed the ratios between the expression levels for each pair. In like manner, we randomly selected pairs of loop and nonloop reactions, and computed the ratios of the maximum fluxes through these reactions before and after adding the loop law constraints.
Results
FBA with a toy network
A toy network is used to illustrate FBA in Fig. 2, a and b. The S matrix defines the topology of this network mathematically. Upper and lower bounds are imposed on the reaction rates. In this case, the internal reactions (v1, v2, and v3) are reversible and bounded in the range [−10, 10]. Metabolites A and C can be exchanged with the environment at lower rates [0,1]. This setup is commonly used because uptake and secretion rates may be constrained to experimentally measured values, whereas internal reaction bounds are often unknown. Maximizing reaction v3 by conventional FBA yields the flux distribution shown in Fig. 2 c. This solution is not unique. However, all of the solutions have a flux of 10 through v3 and at least 9 units through reactions v1, v2, and v3 forming a loop around A→B→C→A. This scenario violates the second law of thermodynamics because there can be no chemical driving force for all three reactions at the same time. However, the ll-FBA solution that maximizes flux through v3 effectively removes the loop by allowing only 1 unit of flux through v3 while deactivating v2 and v1 (Fig. 2 d).
Figure 2.
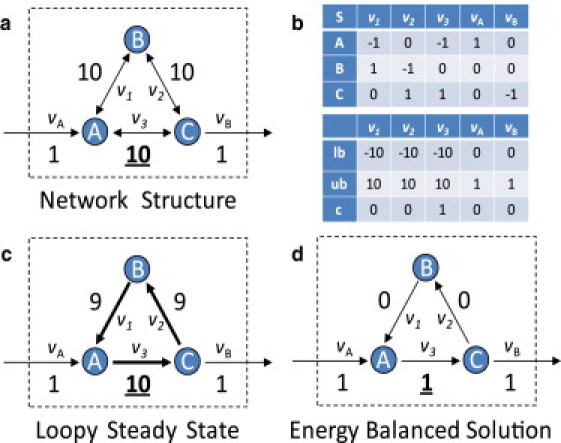
Toy network with loops. A five-reaction toy network is used to illustrate the effects of the loop law. (a) The structure of the network and the reaction bounds are represented graphically. Reactions v1, v2, and v3 form a loop. (b) The stoichiometric matrix, lower bounds and upper bounds (S, lb, and ub, respectively) mathematically describe the network. The objective coefficient, c, indicates which reaction should be maximized (in this case, reaction v3). (c) Classical FBA returns a solution that contains a loop and an objective value of 10. (d) By eliminating the loop, we obtain a lower, thermodynamically consistent objective of 1 unit of flux, with flux passing through reaction v3 and not around the center loop.
FVA
The above analysis provides a simple example of how the loop-law constraints eliminate thermodynamically infeasible loops from FBA simulations. Of importance, imposing loop-law constraints has a similar effect on genome-scale metabolic networks. FVA is a common technique for evaluating the scope of metabolic states a network can achieve (Fig. 3 a). In FVA, each reaction is sequentially minimized and maximized, thereby providing the range of feasible steady-state fluxes for each reaction. Here, six models of increasing size were tested with FVA (Table 1). The scale of these models ranges from the toy network (five reactions) to the human metabolic network (3742 reactions). The results of the six networks are shown in Table 2. Although each network has a different number of loop reactions, the fraction of reactions that participate in the loops is roughly 0.4–4% (except for the toy model).
Figure 3.
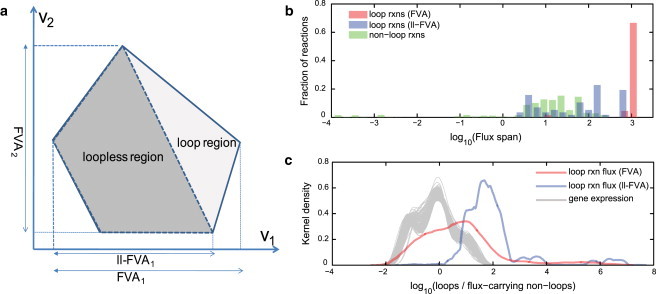
FVA computes the range of each reaction in a network. (a) An example of the loop and loopless regions in the flux solution space shows that removing loops (ll-FVA) reduces the range of reaction 1 but not that of reaction 2. In all cases, ll-FVA is as or more constraining than classical FVA. (b) After the se of ll-FVA on the iAF1260 model of E. coli metabolism, the flux spans for most loop reactions are comparable to the ranges of nonloop reactions. (c) The use of ll-FVA on the iAF1260 model also brings the ratio of loop to nonloop reaction flux to a level that is more consistent with the loop to nonloop gene expression ratios, as determined from 213 different microarray experiments (each gray line represents gene expression level ratios from one microarray) . Thus, ll-FVA reduces the range of feasible reaction flux to values that are more consistent with nonloop flux and gene expression.
Table 2.
Comparison of FVA and ll-FVA, performed on models spanning a wide range of sizes
| Model | Number of reactions | FVA time (s) | ll-FVA time (s) | Time per iteration (s) | Number of changed reactions | % of model changed | Number of deactivated reactions | Changed reactions |
|---|---|---|---|---|---|---|---|---|
| Toy network | 5 | 0.08 | 0.19 | 0.04 | 3 | 60.00% | 0 | v1 v2 v3 |
| Core E. coli | 95 | 1.1 | 7.35∗ | 0.08 | 2 | 2.11% | 0 | FRD7 SUCDi |
| H. pylori iIT341 | 554 | 7.8 | 123 | 0.22 | 22 | 3.97% | 6 | 4HGLSD ACKr AHSERL2 H2CO3D H2CO3D2 HCO3E HPROa HPROx HSERTA HSK METB1r NAt3_1 PHCD PHCHGS PROt2r PROt4r PTAr SHSL1r SHSL2r SHSL4r THRD_L THRS |
| S. aureus iSB619 | 730 | 12.1 | 604 | 0.83 | 6 | 0.82% | 1 | FLDO FMNRx GLUt2 GLUt2r NADTRHD NAt3 |
| E. coli iAF1260 | 2382 | 58.8 | 18222∗∗ | 7.6 | 68 | 2.85% | 7 | ABUTt2pp ACACT1r ACCOAL ACKr ACS ACt2rpp ACt4pp ADK1 ADK3 ADNt2pp ADNt2rpp ALATA_L CA2t3pp CAt6pp CRNDt2rpp CRNt2rpp CRNt8pp CYTDt2pp CYTDt2rpp GLBRAN2 GLCP GLCP2 GLCS1 GLCtex GLCtexi GLDBRAN2 GLGC GLUABUTt7pp GLUt2rpp GLUt4pp GLYCLTt2rpp GLYCLTt4pp HPYRI HPYRRx ICHORS ICHORSi INDOLEt2pp INDOLEt2rpp INSt2pp INSt2rpp KAT1 NAt3pp NDPK1 PPAKr PPCSCT PPKr PPM PROt2rpp PROt4pp PRPPS PTA2 PTAr R15BPK R1PK SERt2rpp SERt4pp SUCOAS THMDt2pp THMDt2rpp THRt2rpp THRt4pp TRSARr URAt2pp URAt2rpp URIt2pp URIt2rpp VALTA VPAMT |
| Human Recon 1 | 3742 | 199.95 | 111163 | 29.7 | 34 | 0.91% | 10 | 10FTHFtm 25HVITD2t 25HVITD2tm 4HGLSDm ACCOAtn ACHtn CHAT CHATn CHOLtn COAtn CYTK1n CYTK2n DCK1n DCK2n GALT H2CO3D2m H2CO3Dm HCO3Em NADHtpu NADtpu NDPK1n NDPK2n NDPK3n NDPK5n NDPK7n NDPK8n NNATr P5CRx PHCDm PHCHGSm PPDOx UMPKn VITD3t VITD3tm |
Standard deviation of 0.3 s across four different growth conditions and two different objective functions.
Standard deviation of 6358 s across four different growth conditions and two different objective functions.
Flux through loops in constraint-based models is limited only by the reaction directionality and the arbitrary upper limits in flux imposed by the user. Thus, the predicted maximum flux values through loop reactions are expected to be significantly higher than fluxes through nonloop reactions. If the loopless methods effectively remove loops, then the maximum flux values for loop reactions should be comparable to nonloop reactions. To demonstrate this, we used ll-FVA to compute the range of feasible flux values for all reactions. Using ll-FVA on the E. coli iAF1260 model, all loop reaction flux ranges decrease, and most decrease to levels within the range of nonloop reactions (Fig. 3 b). This reduction of all loop reaction flux spans occurs in the other models tested here (see Table 2 and Fig. S1). In addition, the larger models exhibit cases in which a few reactions are completely deactivated, suggesting that these reactions can only carry a flux in the growth conditions tested here if they participate in a loop. Thus, ll-FVA reduces the flux span for all loop reactions and provides flux ranges that are more consistent with the ranges seen in nonloop reactions.
ll-FVA and expression levels of loop reactions
Although ll-FVA effectively reduces the range of feasible flux values for all loop reactions, it remains to be shown that this is consistent with biological reality. Previous studies reported a moderate correlation between model-predicted flux and gene expression levels (13,38,39). However, when comparisons are made between flux and gene expression, loop reactions are regularly removed from the analysis. Thus, if ll-FVA effectively improves the prediction of loop-reaction fluxes, one would expect the loop reaction fluxes to be more consistent with the gene expression levels. Here, we tested this by comparing the ratios between loop and nonloop reaction fluxes with the ratios between gene expression levels for loop and nonloop genes. First, were removed all reactions that could not carry flux in the given growth conditions from the E. coli i AF1260 model. Second, we selected 2500 randomly chosen pairs of gene-associated loop and nonloop reactions, and used FVA to compute the maximum flux for the reactions. The ratio of loop reaction flux to nonloop flux was subsequently computed. Third, we identified genes associated with loop and nonloop reactions. We then selected 2500 random pairs of loop and nonloop genes, and computed the ratios of their expression values using 213 different microarrays (37).
The ratios of loop reaction fluxes to nonloop fluxes are beyond the ranges of the loop to nonloop gene expression ratios computed from the expression data. However, when we repeated this process using ll-FVA, we found that the loop/nonloop ratios were more similar to the distribution of loop/nonloop gene expression ratios (Fig. 3 c). Thus, for most reactions, ll-FVA effectively reduces the range of feasible flux values to ranges that are more consistent with gene expression differences between sets of loop- and nonloop-associated genes.
Performance considerations
ll-FVA reduces flux spans of loop reactions to levels that are more consistent with nonloop reactions and with real gene expression levels. However, ll-FVA is more computationally intensive than FVA. MILP is known to be NP complete, and the running time is highly problem- and solver-specific. Average running times for the Tomlab/CPLEX solver are shown in Table 2. For a medium-sized problem such as the H. pylori model, the addition of the loop-law constraints increases the computation time by a factor of 12. A different solver, GLPK, was unable to solve this problem after several hours. Thus, the solver choice and network size significantly affect the running time of ll-FVA. However, variations in model growth media and objective function choice only have a small effect on running time and thus do not significantly affect the feasibility of ll-FVA calculations (see Table 2 and Supporting Material).
Monte Carlo sampling of networks
Monte Carlo sampling of the steady-state solution space allows one to study the set of feasible flux distributions that a network is capable of supporting. For this study, we chose the H. pylori iIT431 model, and sampled the model using a modified ACHR method. We then postprocessed the 2000 uniformly sampled points to remove the loops using either the MILP or MIQP method as shown in Fig. 4 a (see Materials and Methods). All 2000 points contain loop fluxes (Fig. 4 b). The MILP and MIQP methods show different properties in removing looping reactions. On average, the MILP method altered 22.6 reactions in the flux distribution, which is very close to the number of reactions with altered FVA range (22 reactions). The MIQP method on average shifted 252.6 reactions by 0.01 units or more. This difference results because the MIQP method is able to find nearer loopless flux distributions by adding small modifications to nonloop reactions. However, these modifications are orders of magnitude smaller than the changes in the loop reaction fluxes (Fig. S2). Thus, for both methods, loop reactions are primarily changed.
Figure 4.
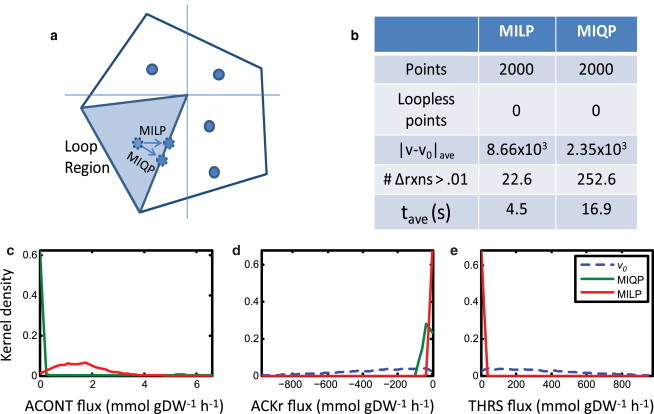
Monte Carlo sampling. A random Monte Carlo sample of 2000 points was generated for the H. pylori model (50). (a) Points are initially sampled from the complete steady-state solution space. Points within the infeasible region (shaded) are moved to the nearest feasible loopless point using either a Euclidian distance (MIQP) or a Manhattan distance (MILP). (b) Differences between the two samples are quantified, including the average distance moved (|v-v0|) and the average number of reactions that change their flux values as the point is corrected (#Δrxns > 0.01). (c) Histograms show the distributions of sample flux values for three reactions (ACONT, ACKr, and THRS) of initial points (v0), and MILP/MIQP loopless points.
These samples allow visualization of the solution space within which the cell operates. By plotting the samples on a histogram, one can obtain the projection across the solution space for any reaction. This projection shows the distribution of feasible flux values for each reaction, given the model topology and constraints. The projections of three reactions are shown in Fig. 4 c. The MILP loop removal does not shift points for nonloop reactions, such as ACONT. However, when loops are removed by MIQP, small modifications are introduced in these nonloop reactions because the modifications lead to slightly closer loopless flux distributions. However, for loop reactions, both methods shift the bulk of points toward fluxes of lower magnitude (e.g., ACKr and THRS).
Performance considerations
Removing loops in the iIT431 model with 554 reactions requires ∼4.5 s of postprocessing per point using the MILP method. The MIQP method requires ∼16.9 s. It is possible to sample larger networks. The iJR904 E. coli model with 1075 reactions is the largest successfully sampled model (results not shown). However, the larger E. coli model, iAF1260, and the human reconstruction could not be sampled as described in the Materials and Methods section, due to scaling issues. The MILP/MIQP solver was not able to find the nearest solution in a reasonable time.
Discussion
We have presented an enhancement of COBRA methods that incorporates the constraints associated with the loop law by disallowing steady-state flux solutions containing closed loops. We applied this method to FBA, FVA, and Monte Carlo sampling to demonstrate how it refines the solutions to various COBRA methods. In these cases, we showed that the traditional method, which allows loops, predicted flux distributions that are known to be infeasible, whereas ll-COBRA provided better solutions. The success of ll-COBRA in this respect has three broader implications for the modeling community, as discussed below.
First, our results demonstrate that for certain applications, detailed quantitative parameterization of thermodynamic constants can be bypassed through the use of more global governing constraints. The COBRA framework has developed rapidly in part because the associated models do not demand the rigorous parameterization required by kinetic models. Thus, many COBRA methods can be used even on large FBA models, since model predictions are subject to more global physicochemical constraints such as mass balance and the delineation of possible chemical transformations in the cell. The successes and, in part, the limitations of these models have motivated the development of hybrid methods that integrate additional elements of thermodynamics with the FBA approach. The different modeling approaches that can be used to address thermodynamics are summarized in Table 3.
Table 3.
Assumptions, required parameters, and computational complexity of different metabolic modeling formalisms
| Formalism | FBA | ll-FBA | FBA + thermodynamics | Kinetics |
| Assumptions and constraints | Steady state, well mixed | Steady state, well mixed, loop law | Steady state, well mixed, ΔG = ΔG0 + RT ln(Q) | Well mixed, mass action∗∗ |
| Parameters | Enzyme capacities, uptake rates | Enzyme capacities, uptake rates | Enzyme capacities, uptake rates, metabolite concentrations, ΔG0 | Kinetic parameters, metabolite concentrations, initial conditions, mechanisms∗∗ |
| Computation type | Linear | Mixed integer | Nonlinear | Nonlinear |
| Computational difficulty | Easy | Medium | Hard∗ | Hard |
| Example studies | (16) | This study | (21–23) |
Depends on the implementation (can be MILP, convex optimization, or generalized NLP).
One has to either assume mass action kinetics or know the mechanism of reaction.
Many methods that incorporate thermodynamics rely on a priori knowledge of the ΔGf for the metabolites in the model (21,23). These quantities can be obtained from the literature or estimated computationally (27). In theory, only these parameters and metabolite concentrations are needed to compute the reaction directionality. An advantage of this formalism is that no flux distributions with loops are allowed. However, these methods are hindered by the requirement for accurate ΔGf-values. When these values are not available, many of the thermodynamic constraints can no longer be evaluated. Moreover, as shown in Table 3, these methods significantly vary in their descriptive power, computational difficulty, and number of required parameters.
The loop-law method proposed here incorporates thermodynamic constraints while also addressing concerns about computational difficulty and missing thermodynamic constants. Its primary advantage over classical FBA is that it excludes loop-containing fluxes without requiring additional parameters. The additional constraints come free as a direct consequence of imposing the second law of thermodynamics on the model. It also has an advantage over other hybrid approaches in that it reduces the need for rigorous parameterization by including a ΔGf for each metabolite. Also, as an MIP, it is computationally more tractable. Thus, detailed parameterization can be minimized and the models can be more easily and rapidly scaled up.
Second, this approach affects model construction. The ll-COBRA formulation provides an alternative to the laborious and potentially incorrect manual removal of loops in the reconstruction process. The process of reconstructing COBRA models has been described in detail elsewhere (5–7). When reconstructing a metabolic network, one must manually evaluate each reaction and assess the reaction directionality. An important step in network reconstruction is to verify that the model produces accurate results when optimizing for physiological functions such as ATP or biomass production. Unfortunately, if the models are not adequately constrained, unbounded internal reaction rates can result from loops. To remedy these unrealistic and problematic loops, one can change the directionality of a reaction, thereby eliminating the loop. Although this approach solves the problem of unbounded solutions, it may inadvertently introduce an artificial constraint on the system. For example, studies have indicated (40,41) that most reactions in the iJR904 model of E. coli metabolism (42) are estimated to be near equilibrium. Thus, in this model, few reactions witness additional constraints from ll-FVA, because iJR904 has been constructed to avoid loops. This is only a concern because all reactions are inherently reversible, and marking them as irreversible may restrict the accuracy of the model under extreme conditions. Therefore, the use of ll-COBRA instead of classical COBRA methods may lead to better models in the future because it will not be necessary to add many of these artificial reversibility constraints to the models. Condition-specific loop elimination can instead be done with ll-COBRA.
Third, the ll-COBRA formulism is broadly applicable to COBRA methods. In this study we have provided examples of how the loop-law constraints can be added, and how the loop law affects COBRA solutions. Many other COBRA methods may benefit from this addition. The only requirements are that 1), the method must involve an LP, MILP, or QP problem; and 2), the method must include a variable for each internal reaction. Example candidate methods include minimization of metabolic adjustment (43), regulatory on/off minimization (44), gene deletion analysis (45), regulatory FBA (46), flux coupling analysis (47), Parsimonious enzyme usage FBA (38), and geometric FBA (48). By integrating the loop law constraints with these established methods, loop reactions can be corrected. Thus, beyond the five genome-scale models used in this study (49-53), additional insight may be obtained from additional metabolic models of prokaryotic function (8,9,54), host-pathogen responses (34), off-target drug effects (55), and multicellular tissues (56), using a host of additional COBRA methods.
Conclusions
Solutions to the classical FBA problem formulation regularly violate thermodynamic constraints. Although other thermodynamic methods can provide more insight into network functions than the ll-COBRA methods, the ll-COBRA method presented here successfully removes thermodynamically infeasible loops, is more scalable, and does not require additional parameters for implementation in a COBRA model. The code used to implement the ll-COBRA methods here is freely available in COBRA toolbox 2.0 (35).
Acknowledgments
We thank Ronan Fleming for many insightful discussions on the topic of thermodynamics.
This work was supported in part by the National Institutes of Health (grants R01 GM068837 and R01 GM57089) and a Plant Systems Biology training grant from the National Science Foundation Integrative Graduate Education and Research Traineeship Program (DGE-0504645).
Supporting Material
References
- 1.Durot M., Bourguignon P.Y., Schachter V. Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol. Rev. 2009;33:164–190. doi: 10.1111/j.1574-6976.2008.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Price N.D., Reed J.L., Palsson B.O. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol. 2004;2:886–897. doi: 10.1038/nrmicro1023. [DOI] [PubMed] [Google Scholar]
- 3.Palsson B. Cambridge University Press; Cambridge/New York: 2006. Systems biology: properties of reconstructed networks. [Google Scholar]
- 4.Lewis N.E., Jamshidi N., Palsson B.Ø. Metabolic systems biology: a constraint-based approach. In: Meyers R.A., editor. Encyclopedia of Complexity and Systems Science. Springer; New York: 2009. p. 5535. [Google Scholar]
- 5.Reed J.L., Famili I., Palsson B.O. Towards multidimensional genome annotation. Nat. Rev. Genet. 2006;7:130–141. doi: 10.1038/nrg1769. [DOI] [PubMed] [Google Scholar]
- 6.Feist A.M., Herrgård M.J., Palsson B.Ø. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 2009;7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiele I., Palsson B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010;5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feist A.M., Palsson B.O. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat. Biotechnol. 2008;26:659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberhardt M.A., Palsson B.O., Papin J.A. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 2009;5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palsson B. Metabolic systems biology. FEBS Lett. 2009;583:3900–3904. doi: 10.1016/j.febslet.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varma A., Boesch B.W., Palsson B.O. Biochemical production capabilities of Escherichia coli. Biotechnol. Bioeng. 1993;42:59–73. doi: 10.1002/bit.260420109. [DOI] [PubMed] [Google Scholar]
- 12.Feist A.M., Palsson B.O. The biomass objective function. Curr. Opin. Microbiol. 2010;13:344–349. doi: 10.1016/j.mib.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuetz R., Kuepfer L., Sauer U. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 2007;3:119. doi: 10.1038/msb4100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gianchandani E.P., Chavali A.K., Papin J.A. The application of flux balance analysis in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010;2:372–382. doi: 10.1002/wsbm.60. [DOI] [PubMed] [Google Scholar]
- 15.Varma A., Palsson B.O. Metabolic flux balancing: basic concepts, scientific and practical use. Nat. Biotechnol. 1994;12:994–998. [Google Scholar]
- 16.Orth J.D., Thiele I., Palsson B.O. What is flux balance analysis? Nat. Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pramanik J., Keasling J.D. Stoichiometric model of Escherichia coli metabolism: incorporation of growth-rate dependent biomass composition and mechanistic energy requirements. Biotechnol. Bioeng. 1997;56:398–421. doi: 10.1002/(SICI)1097-0290(19971120)56:4<398::AID-BIT6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18.Price N.D., Famili I., Palsson B.Ø. Extreme pathways and Kirchhoff's second law. Biophys. J. 2002;83:2879–2882. doi: 10.1016/S0006-3495(02)75297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beard D.A., Babson E., Qian H. Thermodynamic constraints for biochemical networks. J. Theor. Biol. 2004;228:327–333. doi: 10.1016/j.jtbi.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Price N.D., Thiele I., Palsson B.O. Candidate states of Helicobacter pylori's genome-scale metabolic network upon application of “loop law” thermodynamic constraints. Biophys. J. 2006;90:3919–3928. doi: 10.1529/biophysj.105.072645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kummel A., Panke S., Heinemann M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100074. 2006.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kümmel A., Panke S., Heinemann M. Systematic assignment of thermodynamic constraints in metabolic network models. BMC Bioinformatics. 2006;7:512. doi: 10.1186/1471-2105-7-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry C.S., Broadbelt L.J., Hatzimanikatis V. Thermodynamics-based metabolic flux analysis. Biophys. J. 2007;92:1792–1805. doi: 10.1529/biophysj.106.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian H., Beard D.A., Liang S.D. Stoichiometric network theory for nonequilibrium biochemical systems. Eur. J. Biochem. 2003;270:415–421. doi: 10.1046/j.1432-1033.2003.03357.x. [DOI] [PubMed] [Google Scholar]
- 25.Fleming R.M., Thiele I., Nasheuer H.P. Integrated stoichiometric, thermodynamic and kinetic modelling of steady state metabolism. J. Theor. Biol. 2010;264:683–692. doi: 10.1016/j.jtbi.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.National Institute of Standards and Technology. Chemical Kinetics Database on the Web. http://kinetics.nist.gov/.
- 27.Mavrovouniotis M.L. Estimation of standard Gibbs energy changes of biotransformations. J. Biol. Chem. 1991;266:14440–14445. [PubMed] [Google Scholar]
- 28.Alberty R.A. Calculation of standard transformed Gibbs energies and standard transformed enthalpies of biochemical reactants. Arch. Biochem. Biophys. 1998;353:116–130. doi: 10.1006/abbi.1998.0638. [DOI] [PubMed] [Google Scholar]
- 29.Schilling C.H., Letscher D., Palsson B.O. Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. J. Theor. Biol. 2000;203:229–248. doi: 10.1006/jtbi.2000.1073. [DOI] [PubMed] [Google Scholar]
- 30.Schuster S., Hilgetag C. On elementary flux modes in biochemical reaction systems at steady state. J. Biol. Syst. 1994;2:165–182. [Google Scholar]
- 31.Yeung M., Thiele I., Palsson B.O. Estimation of the number of extreme pathways for metabolic networks. BMC Bioinformatics. 2007;8:363. doi: 10.1186/1471-2105-8-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schellenberger J., Park J.O., Palsson B.Ø. BiGG: a biochemical genetic and genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics. 2010;11:213. doi: 10.1186/1471-2105-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thiele I., Price N.D., Palsson B.Ø. Candidate metabolic network states in human mitochondria. Impact of diabetes, ischemia, and diet. J. Biol. Chem. 2005;280:11683–11695. doi: 10.1074/jbc.M409072200. [DOI] [PubMed] [Google Scholar]
- 34.Bordbar A., Lewis N.E., Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol. Syst. Biol. 2010;6:422. doi: 10.1038/msb.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schellenberger J., Que R., Palsson B.O. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 2011 doi: 10.1038/nprot.2011.308. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keating S.M., Bornstein B.J., Hucka M. SBMLToolbox: an SBML toolbox for MATLAB users. Bioinformatics. 2006;22:1275–1277. doi: 10.1093/bioinformatics/btl111. [DOI] [PubMed] [Google Scholar]
- 37.Lewis N.E., Cho B.K., Palsson B.O. Gene expression profiling and the use of genome-scale in silico models of Escherichia coli for analysis: providing context for content. J. Bacteriol. 2009;191:3437–3444. doi: 10.1128/JB.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis N.E., Hixson K.K., Palsson B.Ø. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010;6:390. doi: 10.1038/msb.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bordel S., Agren R., Nielsen J. Sampling the solution space in genome-scale metabolic networks reveals transcriptional regulation in key enzymes. PLOS Comput. Biol. 2010;6:e1000859. doi: 10.1371/journal.pcbi.1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henry C.S., Jankowski M.D., Hatzimanikatis V. Genome-scale thermodynamic analysis of Escherichia coli metabolism. Biophys. J. 2006;90:1453–1461. doi: 10.1529/biophysj.105.071720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fleming R.M., Thiele I., Nasheuer H.P. Quantitative assignment of reaction directionality in constraint-based models of metabolism: application to Escherichia coli. Biophys. Chem. 2009;145:47–56. doi: 10.1016/j.bpc.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed J.L., Vo T.D., Palsson B.O. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biol. 2003;4:R54. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segrè D., Vitkup D., Church G.M. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. USA. 2002;99:15112–15117. doi: 10.1073/pnas.232349399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shlomi T., Berkman O., Ruppin E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc. Natl. Acad. Sci. USA. 2005;102:7695–7700. doi: 10.1073/pnas.0406346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edwards J.S., Palsson B.O. The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. USA. 2000;97:5528–5533. doi: 10.1073/pnas.97.10.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Covert M.W., Schilling C.H., Palsson B. Regulation of gene expression in flux balance models of metabolism. J. Theor. Biol. 2001;213:73–88. doi: 10.1006/jtbi.2001.2405. [DOI] [PubMed] [Google Scholar]
- 47.Burgard A.P., Nikolaev E.V., Maranas C.D. Flux coupling analysis of genome-scale metabolic network reconstructions. Genome Res. 2004;14:301–312. doi: 10.1101/gr.1926504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smallbone K., Simeonidis E. Flux balance analysis: a geometric perspective. J. Theor. Biol. 2009;258:311–315. doi: 10.1016/j.jtbi.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 49.Orth J.D., Fleming R.M.T., Palsson B.O. Reconstruction and use of microbial metabolic networks: the core Escherichia coli metabolic model as an educational guide. In: Böck A., Curtiss III R., …, D. Ussery, editors. EcoSal—Escherichia coli and Salmonella: Cellular and Molecular Biology. ASM Press; Washington, DC: 2009. [DOI] [PubMed] [Google Scholar]
- 50.Thiele I., Vo T.D., Palsson B.Ø. Expanded metabolic reconstruction of Helicobacter pylori (iIT341 GSM/GPR): an in silico genome-scale characterization of single- and double-deletion mutants. J. Bacteriol. 2005;187:5818–5830. doi: 10.1128/JB.187.16.5818-5830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becker S.A., Palsson B.O. Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: an initial draft to the two-dimensional annotation. BMC Microbiol. 2005;5:8. doi: 10.1186/1471-2180-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feist A.M., Henry C.S., Palsson B.Ø. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duarte N.C., Becker S.A., Palsson B.Ø. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA. 2007;104:1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henry C.S., DeJongh M., Stevens R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 2010;28:977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 55.Chang R.L., Xie L., Palsson B.Ø. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLOS Comput. Biol. 2010;6:e1000938. doi: 10.1371/journal.pcbi.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lewis N.E., Schramm G., Palsson B.Ø. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010;28:1279–1285. doi: 10.1038/nbt.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.