

Abstract
For over a decade our group has been designing, preparing and evaluating bioactive, remineralizing composites based on amorphous calcium phosphate (ACP) fillers embedded in polymerized methacrylate resin matrices. In these studies a major focus has been on exploring structure-property relationships of the matrix phase of these composites on their anti-cariogenic potential. The main challenges were to gain a better understanding of polymer matrix/filler interfacial properties through controlling the surface properties of the fillers or through fine-tuning of the resin matrix. In this work, we describe the effect of chemical structure and composition of the resin matrices on some of the critical physicochemical properties of the copolymers and their ACP composites. Such structure-property studies are essential in formulating clinically effective products, and this knowledge base is likely to have strong impact on the future design of therapeutic materials, appropriate for mineral restoration in defective tooth structures.
Keywords: adhesion, amorphous calcium phosphate, degree of vinyl conversion, mechanical strength, methacrylate monomers, polymerization, structure-property relationship, water sorption
1. Introduction
Amorphous calcium phosphate (ACP) based dental materials have enhanced biocompatibility [1, 2] originating from the compositional similarity of ACP to tooth mineral, and potentially can repair damaged tooth structures due to the extended supply of calcium and phosphate ions they provide upon exposure to oral environments. These ions are essential building blocks needed for the mineral recovery of caries-damaged tooth hard tissues (enamel, dentin). When embedded in polymeric methacrylate matrices [3, 4], and exposed to an aqueous environment, these ACP composites release remineralizing ions in a sustained manner and at levels sufficient to promote formation of thermodynamically stable, apatitic tooth mineral [5, 6]. A mixture of crystalline calcium phosphates, i.e., tetracalcium phosphate and dicalcium phosphate anhydrous in a diluted phosphate solution, that hardens in situ and also forms apatite as the main product has been used as calcium phosphate cement (CPC) in restorative dentistry and orthopedics for hard tissue recovery since its introduction in 1986 [7]. Despite their multiple clinical applications, CPCs, even injectable types, still have drawbacks such as prolonged setting times, dispersion upon contact with blood or aqueous media and brittleness. One of the proposed ways for overcoming these drawbacks is by formulation of CPC/polymer composite cements [8, 9].
A critical problem with the acrylic polymeric dental composites of all types is the development of internal and interfacial stresses caused by the shrinkage of the matrix phase upon polymerization. In addition, remineralizing ACP composites have low fracture resistance under masticatory stresses due to their low strength and toughness. Major reasons for their inferior mechanical properties compared to the traditional glass-reinforced composites, are the lower elastic modulus and strength of ACP vs. glass or ceramic fillers, and the poor interfacial interaction between the bioactive ACP filler and the resin phase caused by the uncontrolled agglomeration of ACP particles and their uneven distribution throughout the composites [10]. To improve the ACP filler/polymer matrix interfacial properties (and, in turn, other composite properties), we focused our research on 1) controlling the agglomeration/distribution of ACP through filler surface-modification [11-13] and 2) fine-tuning the composition of the resin matrix [14-17]. The ultimate goal of these studies was to improve physicochemical properties of the material without compromising its remineralizing ability.
Typically dental resins contain a relatively viscous base monomer which minimizes polymerization shrinkage by virtue of its relatively large molecular volume and enhances the modulus of the cured polymer due to its relatively rigid structure, and a diluent co-monomer which reduces resin viscosity and improves handling properties and copolymer conversion due to its smaller molecular volume and greater flexibility [18]. The most commonly utilized copolymers are based on the base monomer 2,2-bis[p-(2‘-hydroxy-3‘-methacryloxypropoxy)phenyl]propane (Bis-GMA) and the diluent monomer triethylene glycol dimethacrylate (TEGDMA). The hydroxyl groups of Bis-GMA and the ethylene oxide segments of TEGDMA contribute to the relatively high water sorption (WS) of Bis-GMA/TEGDMA copolymers [19]. High concentrations of the more rigid structure of Bis-GMA typically result in monomer systems with relatively low degrees of cure or vinyl conversion (DVC) and low shrinkage. The relatively low cure efficiency at ambient temperatures and subsequent plasticization of Bis-GMA/TEGDMA copolymers by oral fluids (primarily water) affect the service life of these composites. Alternative base monomers and/or diluent monomers have been explored to overcome some of the known shortcomings of the Bis-GMA/TEGDMA copolymers. Dental polymers based on ethoxylated bisphenol A dimethacrylate (EBPADMA), a more hydrophobic analog of Bis- GMA with a higher molecular mass but with a more flexible structure and lower viscosity, reportedly show higher DVC and lower polymerization shrinkage than Bis-GMA/TEGDMA resins [20]. In photo-polymerization, urethane dimethacrylate (UDMA) monomer has been shown to be more reactive than Bis-GMA or EBPADMA [18].
In this study, a series of resins comprising Bis-GMA, EBPADMA or UDMA as base monomers were formulated with TEGDMA, a poly(ethylene glycol)-extended UDMA (PEG-U) and 2-hydroxyethyl methacrylate (HEMA) as diluent co-monomers. In addition, the surface active monomers zirconyl dimethacrylate (ZrDMA) or methacryloyloxyethyl phthalate (MEP) were used as monomeric additives in an attempt to promote adhesion to ACP. Introduction of ZrDMA and MEP into resin matrix was motivated by the ability of these monomers to improve coupling between the organic phase and the inorganic ACP filler (ZrDMA) and possibly also enhance adhesion of the composites to tooth structures (MEP) [5, 17, 21]. It is hypothesized that by appropriate resin modification a final consistency suitable for the incorporation of particulate ACP filler can be achieved and satisfactory levels of vinyl conversion (DVC) attained. It is also postulated that the experimental resins will exhibit sufficient water sorption (WS) and, consequently, generate sufficient release of mineral ions needed for tooth mineral recuperation and/or protection, without excessively compromising the mechanical stability of the composites, thereby making them inadequate for the intended dental applications. To test these hypotheses, extensive physicochemical evaluation of both the unfilled resins (copolymers) and their ACP composites was performed. Knowing the relationship(s) between the chemical structure and/or composition of the resin matrix and the critical properties of copolymers and their corresponding composites appears necessary when designing remineralizing ACP composites for different clinical applications.
2. Results and Discussion
2.1. Characteristics of ACP filler
Results of the physicochemical characterization of Zr-ACP filler used to formulate Bis-GMA/TEGDMA/HEMA/ZrDMA (BTHZ)/ACP, EBPADMA/TEGDMA/HEMA/MEP (ETHM)/ACP and UDMA/PEG-U/HEMA/MEP (UPHM)/ACP composites are summarized in Table 1.
Table 1.
Physicochemical properties of Zr-ACP filler used in the study.
| Property/method | Findings |
|---|---|
| Long-range structure (XRD) | Two diffuse broad bands in 2θ(°) = (4 to 60) region typical of ACP |
| Short-range structure (FTIR) | Two typical phosphate absorption bands at (1200 to 900) cm-1 and (630 to 500) cm-1 |
| Morphology, size (SEM) | Heterogeneous particle sizes, predominantly agglomerates ranging from approx. 5 μm to 20 μm; some larger than 100 μm |
| Particle size (PSD analysis) | Particle sizes ranging from submicron to > 100 μm in diameter; average calculated median diameter, dm = 7.4 μm |
| Water content (TGA) | Zr-ACP filler contained a mass fraction of (16.1 ± 2.0) % water |
As a result of being excessively agglomerated, Zr-ACP did not easily blend at 40 % mass fraction with any of the resins. Prolonged mixing (hand-spatulation) was required to obtain pastes with workable flowability/handling properties. This drawback can easily be circumvented by grinding or milling the ACP filler prior to their use in composite paste preparation. Ground or milled ACP blended easily at desired level with various resins yielding highly flowable pastes suitable for clinical uses [12, 17, 21]. Reducing the ACP’s particle size by grinding and/or milling inevitably affects properties that were assessed in this work, such as water sorption, ion release and mechanical strength of the composites. The effects of grinding and/or milling of the ACP on physicochemical and mechanical properties of the various methacrylate based ACP composites were extensively studied and the results of these investigations were reported previously [4, 5, 10-14, 21, 22].
2.2. Evaluation of Copolymers and Composites
As outlined in Table 2 (please, see the Experimental section), three different experimental resins were used as matrix phases in light-cure ACP-based pit and fissure sealant (BTHZ resin), orthodontic adhesive (ETHM resin) and endodontic sealer (UPHM resin). The differences in resin formulations were primarily guided by different clinical requirements regarding composite’s handling properties and the expected levels of DVC. The latter was shown to be dependent on both the type of the base monomer as well as the co-monomers used to form the resin matrix [5, 15, 22]. The results of the DVC screening of the unfilled resins (copolymers) and their Zr-ACP composites are compiled in Figure 1.
Table 2.
Composition (mass fraction %) of experimental resins evaluated in the study.
| Monomer/photoinitiator | BTHZ resin | ETHM resin | UPHM resin |
|---|---|---|---|
| Bis-GMA: 2,2-bis[p-(2’-hydroxy-3-methacryloxypropoxy)phenyl]propane | 35.5 | - | - |
| EBPADMA: ethoxylated bisphenol A dimethacrylate | - | 62.8 | - |
| UDMA: urethane dimethacrylate | - | - | 48.7 |
| TEGDMA: triethylene glycol dimethacrylate | 35.5 | 23.2 | - |
| HEMA: 2-hydroxyethyl methacrylate | 27.0 | 10.4 | 17.3 |
| PEG-U: poly(ethylene glycol)-extended UDMA | - | - | 30.0 |
| ZrDMA: zirconyl dimethacrylate | 1.0 | - | - |
| MEP: methacryloyloxyethyl phthalate | - | 2.6 | 3.0 |
| CQ: camphorquinone | 0.2 | 0.2 | - |
| 4EDMAB: ethyl-4-N,N-,dimethylaminobenzoate | 0.8 | 0.8 | - |
| IRGACURE 1850: bis(2,6-dimethoxybenzoyl)-2,4,4-triethylpentyl phosphine oxide & 1-hydroxycyclohexyl phenyl ketone | - | - | 1.0 |
Figure 1.
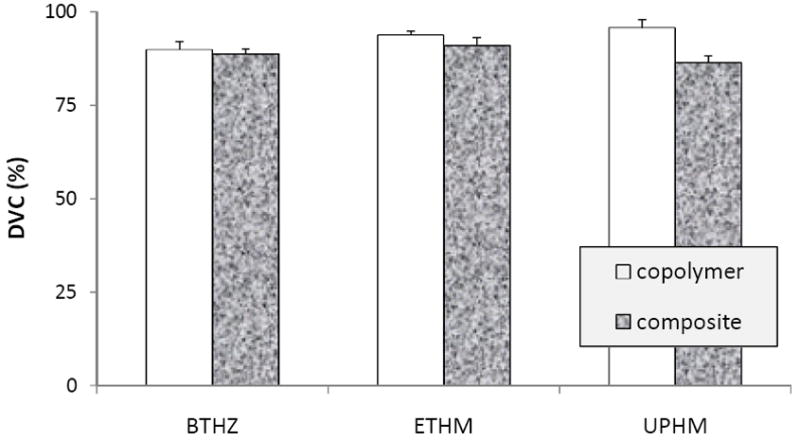
Degree of vinyl conversion [DVC; mean value + standard deviation (SD; indicated by bars)] of unfilled resins (copolymers) and their ACP composites. Number of specimens in each experimental group: 5 ≤ n ≤ 8.
All copolymers attained exceptionally high DVC values ranging from (89.9±2.2) % to (93.7±1.2) % at 24 h post-cure. DVC values of the corresponding composites were lower (ranging from (86.4±2.0) % to (91.0±2.2) %. While the differences in DVC values between the BTHZ and ETHM copolymers and their corresponding composites were slightly lower, they were not statistically significant. However, the difference between the UPHM copolymers and composites (reduction of 9.8 %) was significant. DVC attained in dental resins upon polymerization is generally strongly affected by the chemical structure of the monomers, composition, polymerization kinetics and their gel points. At ambient temperatures, the majority of dental resins based on viscous Bis-GMA, reaches the gel point on polymerization very quickly. As a result, the attained DVC values in such matrices are relatively low since the diffusion of monomer and other reactive species to the radical sites or pendant vinyl groups is hindered by the relatively immobilized polymer network. Bis-GMA/TEGDMA copolymers typically yield DVC values between 55 % and 75 % [23]. However, when Bis-GMA, EBPADMA or UDMA are blended with TEGDMA and HEMA, DVC values similar to the values reported in this study were obtained ((82.0 to 94.0) % for copolymers and (74 to 91) % for the corresponding ACP composites [16, 22]). The high DVC values attained in these polymeric matrices were attributed to the high diffusivity and monofunctionality of the HEMA monomer. Incorporating HEMA into BTHZ, ETHM and UPHM matrices is most likely the main reason for the high DVC values observed in this study. The effect of HEMA may have been enhanced further in UPHM resins by introducing a high molecular mass, oligomeric PEG-U monomer. Its favorable effect on DVC is attributed to the long and flexible structure between the terminal vinyl groups of this oligomer [17].
Reduction in DVC values in going from copolymer to composite has already been reported in similarly formulated resin composites [5, 14, 16]. It is attributed to the reduction in exotherm of resin polymerization by ACP filler. Other filler-related effects, such as the light scattering attenuation and air entrapment, both enhanced by the uneven distribution of large ACP agglomerates throughout the resin matrix, may also diminish DVC of composites.
While suggesting high biocompatibility of the experimental ACP materials [24], these exceptionally high DVC values attained in all types of composites, would unquestionably lead to high the volumetric contraction upon polymerization (usually called polymerization shrinkage). In UPHM composites this shrinkage can be as high as 7.1 % by volume [17] leading potentially to increased formation of strains and gaps in composite microstructure and at the composite/tooth interfaces, and ultimately resulting in microleakage. However, there is a reasonable possibility that this high polymerization shrinkage may be compensated by the hygroscopic expansion of the composites from water uptake as has already been documented for various types of resin composites [25-27].
Mass increases of copolymer and composite specimens following their immersion in saline are shown in Figure 2. Statistical analysis revealed that WSmax, i.e., the maximum, plateau WS values of copolymers decreased in the following order: UPHM > BTHZ > ETHM essentially following the order of decreasing affinity for water (relative hydrophilicity) of the base monomers UDMA ≥ Bis-GMA > EBPADMA. The WSmax of the composites showed, however, a different order: BTHZ/ACP > (UPHM/ACP, ETHM/ACP). The WS values of specimens reflect their net hydrophilicites, resulting from the water absorbed by the resin components (in the case of copolymers) or the sum of water absorbed by the resin phase and the ACP filler (in the case of ACP composites) [5, 21]. Within a given resin system, WS of the corresponding ACP composites containing the same ACP level decreases with decreasing sizes of the filler [5, 22]. Since ACP provides paths of diffusion and absorbs water itself, random and uncontrolled, i.e., heterogeneous dispersion of porous ACP agglomerates in the resins is responsible for the differences in the net water uptake of the composite specimens which deviate from the trend seen in copolymers.
Figure 2.
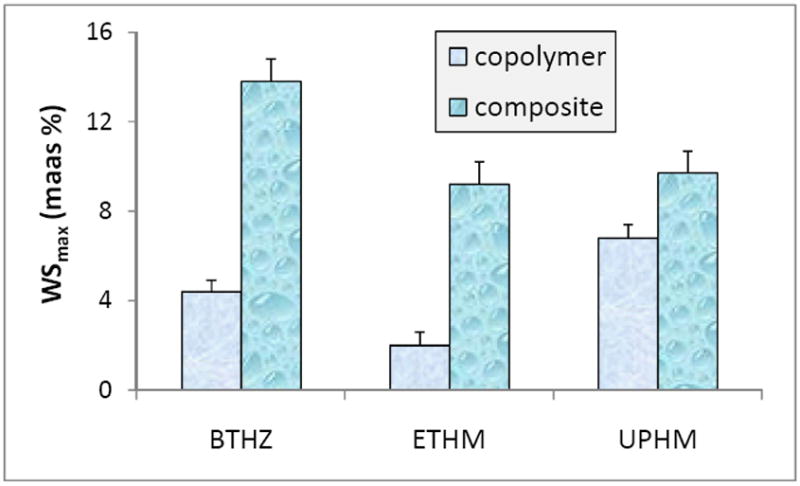
Maximum water sorption (WSmax; mean + SD) values in BTHZ, ETHM and UPHM copolymers and their ACP composites attained after 1 month immersion in buffered saline solutions at 23 °C. Number of specimens in each experimental group n = 5.
The overall WS of all three groups of composites ranging from 9.2 % to 13.8 % is equal to or exceeds the WS of UTHM specimens which were assessed for their volumetric expansion by calculating changes in specimens’s volume on the basis of the measured dimensional changes (cylindrical geometry [21]. The volume change or hygroscopic expansion of the UPHM/ACP specimens with WSmax of (9.7 ± 0.3) mass % was equal to (12.5 ± 1.5) vol %, a value that should be more than sufficient to compensate polymerization shrinkage (7.1 vol %) in those specimens. The same should apply to geometrically identical and equally immersed specimens of BTHZ/ACP and ETHM/ACP composites which exhibited WSmax equal to (ETHM/ACP) or higher (BTHZ/ACP) than their UPHM/ACP counterparts.
The total mass changes of the immersed composite specimens are defined by the nature of the polymer matrix network structure which controls the copolymer’s permeability to water and the rate of water sorption by the ACP filler. Catalyzed by WS and kinetically controlled by the internal pH, the resulting spontaneous, intra-composite ACP to apatite conversion leads to a substantial increase in solution concentration of calcium and phosphate ions with time of immersion and create solution conditions thermodynamically favorable for the re-precipitation of tooth mineral.
The solution concentration data recalculated in terms of supersaturation with respect to stoichiometric hydroxyapatite (Figure 3) clearly demonstrate that the attained supersaturations with all three types of composites were significantly above the minimum needed for remineralization to take place (ΔG° < 0), thus confirming their potential to regenerate tooth mineral when applied as remineralizing sealants and/or adhesives. Statistically, there was no difference between the calculated ΔG° values for BTHZ, ETHM and UPHM composites. It can, therefore, be concluded that the compositional changes in the resin phase did not compromise the remineralzing potential of the experimental ACP composites. Efficacy of ACP remineralizing composites has been verified under conditions replicating pH changes occurring in oral medium in two microradiographic studies of enamel lesions created in bovine [28] and human tooth specimens [6]. However, to overcome a significant gap that exists between the laboratory results and in vitro simulations on one side and the clinical applications on the other, testing of these experimental materials in clinical settings will be necessary.
Figure 3.
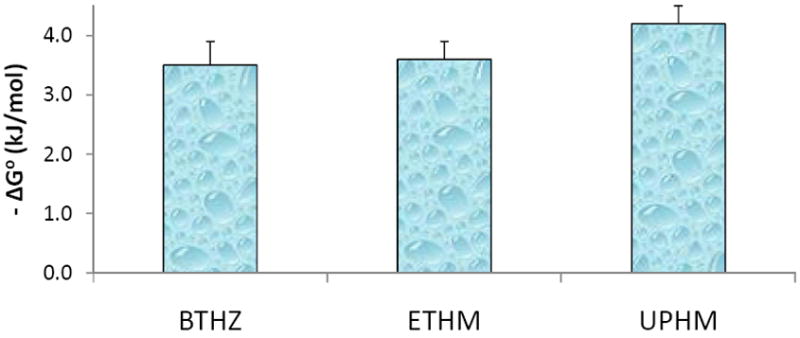
Thermodynamic stability expressed as Gibbs free energy (ΔG°) of the solutions containing maximum levels of calcium and phosphate ions released from BTHZ/ACP, ETHM/ACP and UPHM/ACP composites. Number of samples in each group n= 3. Negative ΔG° values indicate solutions supersaturated with respect to stoichiometric apatite.
The mechanical strength of dry and wet copolymer and composite specimens is compared in Figure 4. Although for some of the intended applications (specifically UPHM composite intended for use as endodontic sealer), a material’s mechanical strength may not be its most critical property and the specimen’s geometry does not effectively simulate the in vivo situations, the conventional strength testing was used in this study for comparison purposes. Regardless of the resin composition, unexpectedly large BFS data scattering was observed in the copolymer groups. This phenomenon is most likely due to a random introduction of defects (bubbles, voids) into copolymer disk specimens during their preparation and their exothermic photo-polymerization. Generally, copolymers exhibited significantly higher biaxial flexure strength (BFS) compared to their corresponding composites in both dry state (BFScopolymer = (1.8 to 3.3) × BFScomposite) and after immersion in saline solutions (BFScopolymer = (2.3 to 3.3) × BFScomposite). These lower BFS values regularly seen in composite specimens regardless of the resin matrix composition suggest that the level of ACP filler/resin matrix interlocking in the composites was inadequate to achieve better mechanical properties of these materials which, in turn, would mean that ACP-containing composites are too weak to be used as direct filling materials and their restorative potential should be restricted to dental applications where high mechanical strength is not the main requirement.
Figure 4.
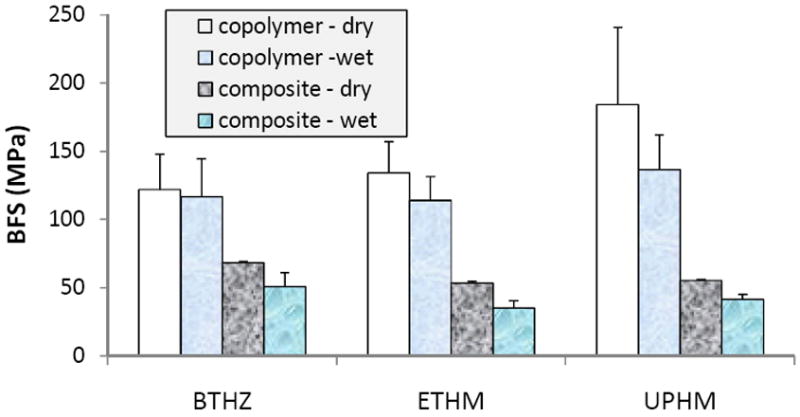
Biaxial flexure strength (BFS; mean value + SD) of dry (before immersion) and wet (after 1 mo immersion in buffered saline solution) copolymer and composite specimens. Number of specimens tested in each group 5 ≤ n ≤ 18.
The BFS values of BTHZ, ETHM and UPHM copolymers and their ACP composites could not be correlated with the relative hydrophilicity of base monomers or the net water affinity of the copolymers and/or composites. Reduction in BFS of copolymers in going from dry to wet state was minimal (4. 2%; BTHZ) or either apparent but statistically not significant (15.1 % (ETHM) and 25.9 % (UPHM)). In all there composite groups, a differential between the dry and wet values (between 25.0 % and 34.4 %) was statistically significant. It is possible that the elevated WS seen in all immersed composites may have caused additional weakening of the materials due to irreversible disruption of the polymer matrix via solvation, plasticization and/or rupture of weak inter-chain bonds [29-31], regardless of the resin matrix composition.
3. Experimental Section
3.1. Resin formulation, copolymer and composite specimen preparation
The experimental resins were formulated from commercial Bis-GMA, EBPADMA, UDMA, TEGDMA, PEG-U, HEMA, ZrDMA and MEP monomers (Esstech, Essington, PA, USA) which were used as received from the manufacturer, i.e., without additional purification. Resins were photo-activated with camphorquinone (CQ) and ethyl-4-N,N-,dimethylaminobenzoate (4EDMAB) (Sigma Aldrich, Milwaukee, WI, USA) or IRGACURE 1850 (Ciba Specialty Chemicals Corp., Tarrytown, NY, USA), a bis(acyl) phosphine oxide containing photo-initiator. Chemical structures of the monomers and photo-initiators are shown in Figure 5. After introducing the appropriate initiators to the monomer blends, the activated resin mixtures were magnetically stirred until fully homogenized. Resin compositions as well as the acronyms used throughout this manuscript are provided in Table 2. The composite pastes were prepared by hand spatulation by combining mass fractions of 40 % ACP filler and 60 % resin. The volume filler fraction of the experimental material was approximately 30 %. Its shade is B1 compared to the VITAPAN Lumin Vacuum shade guide (Vita Zahnfabrik, Bad Säckingen, Germany). The observed slightly higher translucency is attributed to the relatively low filler content by volume compared to commercial restorative composites. Copolymer and composite specimens alike were cured by irradiating disk-shaped specimens (cylindrical molds with an average (12.9 ± 0.1) mm diameter and (1.4 ± 0.1) mm thickness) for 60 s per side (Triad 2000; Dentsply International, York, PA, USA).
Figure 5.
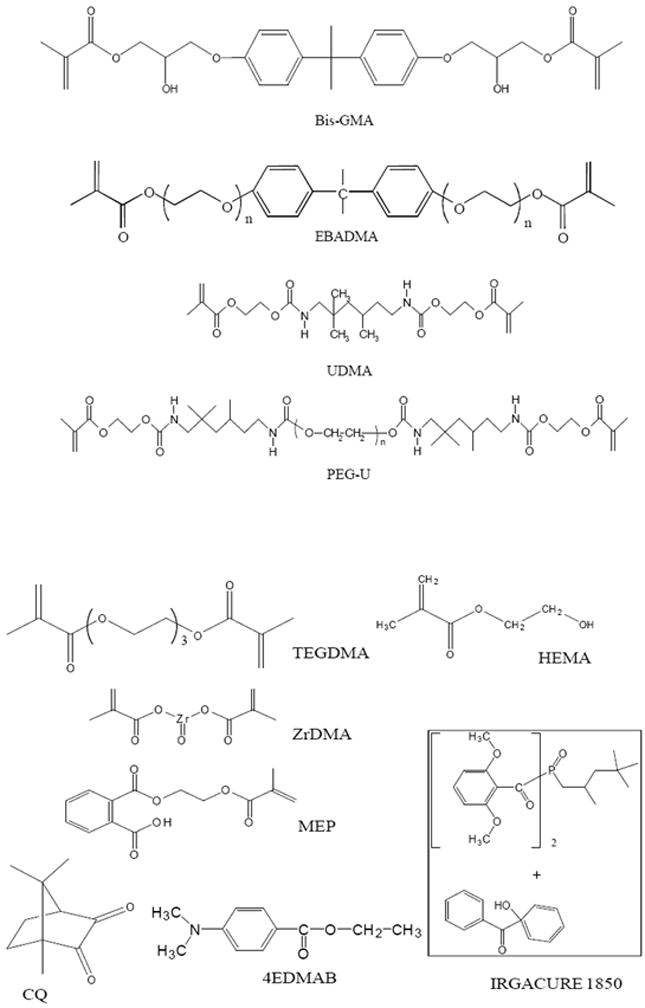
Chemical structure of monomers and photo-initiating systems used in the study. EBPADMA: n = 6-8.
3.2. ACP filler synthesis and characterization
Zirconia-hybridized ACP (Zr-ACP) was synthesized as originally reported in [3, 4]. The amorphous state of the filler ACP was verified by powder X-ray diffraction (XRD; Rigaku DMAX 2000 X-ray diffractometer; Rigaku/USA Inc., Danvers, MA, USA) and Fourier-transform spectroscopy (FTIR; Nicolet Magna-IR FTIR 550 spectrophotometer; Nicolet Instrumentations, Madison, WI, USA). XRD patterns were recorded from 4° to 60° 2θ with CuKα radiation (λ = 0.154 nm) at 40 kV and 40 mA. The samples were step-scanned in intervals of 0.010° 2θ at a scanning speed 1.000 deg/min. The FTIR spectra (4,000 cm-1 to 400 cm-1) were recorded using a KBr pellet technique (0.5 mg to 0.8 mg solid/400 mg KBr). Particle size distribution (PSD) of the filler dispersed in isopropanol and utrasonicated for 10 min at room temperature prior to the analysis was measured using particle size analyzer (CIS-100, Ankersmid Ltd., Yokneam, Israel). PSD analyses were performed in triplicate. Median particle size diameter (dm) determined from the volume size distribution histograms was taken as a primary indicator of the state of agglomeration of the ACP particulates (the higher the dm value, the more aggregated the ACP). Morphology of the filler was examined by scanning electron microscopy (SEM; JSM-5400 instrument, JEOL Inc. Peabody, MA, USA). The overall water content (mass fraction, %) of ACP fillers was determined by thermogravimetric analysis (Series 7 Thermal Analysis System, Perkin Elmer, Waltham, MA, USA).
3.3. Degree of vinyl conversion (DVC)
The DVC of copolymer and composite specimens was measured at 23 °C by near-infrared (NIR) spectroscopy [32]. NIR scans (Nicolet Magna 550, Nicolet Inc., Madison, WI, USA) were taken before photo cure and 24 h post-cure of composites with a thickness of approximately 3.0 mm and compared. Number of specimens varied between 5 and 8 per experimental group. DVC was defined as the % change in the integrated peak area of the 6165 cm-1 absorption band related to the first overtones of the = C-H stretching vibrations of the methacrylate vinyl group (= CH2) before and after photo-polymerization. It was calculated utilizing the following formula:
| (1) |
By measuring the thickness of monomer/polymer specimens, the need to use an invariant absorption band as an internal standard was circumvented.
3.4. Water Sorption (WS)
To determine WS profiles of copolymers and composites, their specimens were initially dried over anhydrous CaSO4 until a constant mass was achieved (± 0.1 mg). Specimens were then immersed in 25 mL of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered, pH=7.4, 0.13 mol/L sodium chloride (saline) solution at 23 °C and mass changes of dry-padded specimens were recorded at predetermined time intervals. The number of specimens tested in each experimental group n=5. The relative degree of WS of each individual specimen at any given time interval (t), expressed as a % mass fraction, was calculated using a simple equation:
| (2) |
where Wt represents a specimen mass at time t and Wo is the initial mass of dry specimen.
3.5. Release of Mineral Ions from Composites
Mineral ion release from ACP composite disk specimens was assessed at 23 °C, in magnetically stirred, HEPES-buffered (pH=7.4) 0.13 mol/L saline solutions (25 mL saline/specimen). Saline solution was replaced at predetermined time intervals (1 day, 2 days, 14 days, 1 month, 1.5 month, 2, month and 2.5 month of immersion). The kinetic changes in calcium and phosphate concentrations were determined by utilizing atomic emission spectroscopy (Prodigy High Dispersion ICP-OES, Teledyne Leeman Labs, Hudson, NH, USA). Number of specimens in each experimental group n = 3. The pooled data for all of the intervals were used to calculate the thermodynamic stability of the immersing solutions which were expressed as Gibbs free energy:
| (3) |
where IAP is the ion activity product for stoichiometric apatite, i.e., (Ca2+)10(PO4 3-)6(OH-)2, Ksp is the corresponding thermodynamic solubility product, R is the ideal gas constant, T is the absolute temperature and n is the number of ions in the IAP (n=18). Calculations were performed using the solution chemical equilibrium software program EQUIL (Micromath Scientific Software, Salt lake City, UT, USA).
3.6. Mechanical Testing of the Copolymers and Composites
To compare the mechanical strength of dry (kept in the air 24 h at 23 °C) and wet (after prolonged immersion in saline solution at 23 °C) copolymer and composite specimens, their biaxial flexure strength (BFS) was determined using a computer-controlled Universal Testing Machine (Instron 5500R, Instron Corp., Canton, MA, USA) operated by Instron Merlin Software series 9. BFS of disk specimens ((15.15±0.13) mm in diameter and (1.10±0.09) mm in thickness; number of specimen=5/group) was determined under a static load utilizing a piston-on-three-ball loading arrangement with a cross head speed of 0.5 mm/min. The failure stress was calculated according to the ASTM specification F394-78 [33].
3.7. Statistical Analysis
The results were analyzed by the analysis of variance (ANOVA; α = 0.05). Statistical significance of change values was obtained from two-tailed P values using the Holm-Sidak test for paired data. Statistical calculations were done by means of SigmaStat software (version 3.5; SPSS Inc., Chicago, IL, USA). One standard deviation (SD) is identified in this paper for comparative purposes as the estimated uncertainty of the measurements.
4. Conclusions
Copolymers and ACP composites fabricated from the experimental resins comprising Bis-GMA, EBPADMA or UDMA as base monomers, TEGDMA or PEG-U and HEMA as diluent monomers, and ZrDMA or MEP as surface active monomers attained high levels of vinyl conversion (DVC), relatively high water sorption (WS), desirable ion release profiles and moderate to low mechanical strength. High DVC values that suggest high biocompatibility of all experimental polymers and their composites were obtained by incorporating the highly diffusive and monofunctional HEMA monomer into BTHZ, ETHM and UPHM matrices or by introducing a high molecular mass, oligomeric PEG-U monomer into UPHM resins. Adequate release of mineral ions was attained in ACP composites formulated with all three types of resins to yield solution supersaturations required for the effective tooth remineralization. Relatively low mechanical strength of the composites is related their high WS and instability of the ACP/resin interfaces caused by the heterogeneous ACP distribution throughout the composites, and the lack of interfacial bonding of the filler phase and polymer matrix. While restricting the potential use of ACP composites as direct filling materials, their relatively low mechanical strength is not critical for the intended applications of ACP composite in low-stress bearing areas. This study reiterates the importance of understanding the structure-composition-property relationship(s) in remineralizing ACP composites when designing these complex materials for different clinical applications.
Acknowledgments
Reported work was supported by the National Institute of Dental and Craniofacial Research (NIDCR grant DE 13169), National Institute of Standards and Technology and American Dental Association Foundation. Generous contribution of the monomers utilized in this study from Esstech, Essington, PA, USA is gratefully acknowledged.
Abbreviations
- ACP
amorphous calcium phosphate
- ADAF
American Dental Association Foundation
- ANOVA
analysis of variance
- BFS
biaxial flexural strength
- Bis-GMA
2,2-bis[p-(2’-hydroxy-3’-methacryloxypropoxy)phenyl]propane
- BTHZ
Bis-GMA/TEGDMA/HEMA/ZrDMA resin
- CQ
camphorquinone
- dm
median diameter
- DVC
degree of vinyl conversion
- EBPADMA
ethoxylated bisphenol A dimethacrylate
- ETHM
EBPADMA/TEGDMA/HEMA/MEP resin
- 4EDMAB
ethyl-4-N,N-dimethylaminobenzoate
- FTIR
Fourier transform infrared spectroscopy
- ΔG°
Gibbs free energy
- HAP
hydroxyapatite
- HEMA
2-hydroxyethyl methacrylate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IAP
ion activity product
- IRGACURE 1850
bis(2,6-dimethoxybenzoyl)-2,4,4-triethylpentyl phosphine oxide & 1-hydroxycyclohexyl phenyl ketone
- MEP
methacryloyloxyethyl phthalate
- NIDCR
National Institute of Dental and Craniofacial Research
- NIR
near infrared
- NIST
National Institute of Standards and Technology
- PEG-U
poly(ethylene glycol) extended UDMA
- PRC
Paffenbarger Research Center
- PSD
particle size distribution
- SEM
scanning electron microscopy
- SD
standard deviation
- TEGDMA
triethylene glycol dimethacrylate
- TGA
thermogravimetric analysis
- UDMA
urethane dimethacrylate
- UPHM
UDMA/PEG-U/HEMA/MEP resin
- WS
water sorption
- XRD
X-ray diffraction
- ZrDMA
zirconyl dimethacrylate
Footnotes
Official contribution of NIST; not subject to copyrights in USA.
Disclaimer Certain commercial materials and equipment are identified in this article to specify the experimental procedure. In no instance does such identification imply recommendation or endorsement by the National Institute of Standards and Technology or American Dental Association Foundation, or that the material and equipment identified is necessarily the best available for the purpose.
References
- 1.Dorozhkin SV. Calcium Orthophosphates in Nature, Biology and Medicine. Materials. 2009;2:399–498. [Google Scholar]
- 2.Dorozhkin SV. Calcium Orthophosphate-based Biocomposites and Hybrid Materials. J Mater Sci. 2009;44:2343–2387. [Google Scholar]
- 3.Antonucci JM, Skrtic D, Hailer AW, Eanes ED. Bioactive Polymeric Composites Based on Hybrid Amorphous Calcium Phosphate. In: Ottenbrite RM, Kim SW, editors. Polymeric Drugs & Drug Delivery Systems. Technomics Publ. Co., Inc.; Lancaster PA, USA: 2000. pp. 301–310. [Google Scholar]
- 4.Skrtic D, Antonucci JM, Eanes ED, Eichmiller FC, Schumacher GE. Physicochemical Evaluation of Bioactive Polymeric Composites Based on Hybrid Amorphous Calcium Phosphates. J Biomed Mat Res (Appl Biomater) 2000;53:381–391. doi: 10.1002/1097-4636(2000)53:4<381::aid-jbm12>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 5.Skrtic D, Antonucci JM, Eanes ED. Amorphous Calcium Phosphate-based Bioactive Polymeric Composites for Mineralized Tissue Regeneration. J Res Natl Inst Stands Technol. 2003;108(3):167–182. doi: 10.6028/jres.108.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langhorst SE, O’Donnell JNR, Skrtic D. In vitro Remineralization Effectiveness of Polymeric ACP Composites: Quantitative Micro-radiographic Study. Dent Mater. 2009;25:884–891. doi: 10.1016/j.dental.2009.01.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown WE, Chow LC. Combination of Sparingly Soluble Calcium Phosphates in Slurries and Paste as Mineralizers and Cements. 4,612,053. US Patent. 1986
- 8.Chen WC, Ju PC, Wang JC, Hung CC, Lin JHC. Brittle and Ductile Adjustable Cement Derived from Calcium Phosphate Cement/Polyacrylic Acid Composites. Dent Mater. 2008;24:1616–1622. doi: 10.1016/j.dental.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 9.Wang JC, Ko CL, Hung CC, Tyan YC, Lai CH, Chen WC, Wang CK. Deriving Fast Setting Properties of Tetracalcium Phosphate/Dicalcium Phosphate Anhydrous Bone Cement with Nanocrystallites on the Reactant Surfaces. J Dent. 2010;38:158–165. doi: 10.1016/j.jdent.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Skrtic D, Antonucci JM, Eanes ED, Eidelman N. Dental Composites Based on Hybrid and Surface-modified Amorphous Calcium Phosphates. Biomaterials. 2004;25:1141–1150. doi: 10.1016/j.biomaterials.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 11.O’Donnell JNR, Antonucci JM, Skrtic D. Amorphous Calcium Phosphate Composites with Improved Mechanical Properties. J Bioact Compat Polym. 2006;21(3):169–184. doi: 10.1177/0883911506064476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SY, Regnault WF, Antonucci JM, Skrtic D. Effect of Particle Size of an Amorphous Calcium Phosphate Filler on the Mechanical Strength and Ion Release of Polymeric Composites. J Biomed Mater Res Part B: Appl Biomater. 2007;80B:11–17. doi: 10.1002/jbm.b.30561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antonucci JM, Liu DW, Skrtic D. Amorphous Calcium Phosphate Based Composites: Effect of Surfactants and Poly(ethylene oxide) on Filler and Composite Properties. J Disp Sci Technol. 2007;28(5):819–824. doi: 10.1080/01932690701346255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonucci JM, Skrtic D. Matrix Resin Effects on Selected Physicochemical Properties of Amorphous Calcium Phosphate Composites. J Bioact Compat Polym. 2005;20(1):29–49. [Google Scholar]
- 15.Skrtic D, Antonucci JM, Liu DW. Ethoxylated Bisphenol A Methacrylate-based Amorphous Calcium Phosphate Composites. Acta Biomaterialia. 2006;2(1):85–94. doi: 10.1016/j.actbio.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skrtic D, Antonucci JM. Effect of Composition of the Resin Phase on Vinyl Conversion of Amorphous Calcium Phosphate-based Composites. Polym Int. 2007;56:497–505. doi: 10.1002/pi.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Donnell JNR, Skrtic D. Degree of Vinyl Conversion, Polymerization Shrinkage and Stress Development in Experimental Endodontic Composites. J Biomim Biomater Tissue Eng. 2009;4:1–12. doi: 10.4028/www.scientific.net/JBBTE.4.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stansbury JW, Dickens SH. Network Formation and Compositional Drift During Photo-initiated Copolymerization of Dimethacrylate Monomers. Polymer. 2001;42:6363–6369. [Google Scholar]
- 19.Antonucci JM, Stansbury JW. Molecularly Designed Dental Polymers. In: Arshady R, editor. Desk Reference of Functional Polymers Syntheses and Applications. ACS; Washington DC, USA: 1997. pp. 719–738. [Google Scholar]
- 20.Antonucci JM, Stansbury JW, Venz S. Synthesis and Properties of Polyfluorinated Prepolymer Multifunctional Urethane Methacrylate. In: Gebelein CG, Dunn RL, editors. Progress in Biomedical Polymers. Plenum Press; New York, USA: 1990. pp. 121–131. [Google Scholar]
- 21.Johns JI, O’Donnell JNR, Skrtic D. Selected Physicochemical Properties of the Experimental Endodontic Sealer. J Mater Sci Mater Med. 2010;21:797–805. doi: 10.1007/s10856-009-3873-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skrtic D, Antonucci JM. Dental Composites Based on Amorphous Calcium Phosphate – Resin Composition/Physicochemical Properties Study. J Biomater Appl. 2007;21(4):375–393. doi: 10.1177/0885328206064823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowell LG, Stansbury JW, Syrpes DC, Bowman CN. Effects of Composition and Reactivity on the Reaction Kinetics of Dimethacrylate/Dimethacrylate Copolymerizations. Macromolecules. 1999;32(12):3913–3921. [Google Scholar]
- 24.Simon CG, Antonucci JM, Liu DW, Skrtic D. In vitro Cytotoxicity of Amorphous Calcium Phosphate Composites. J Bioact Compat Polym. 2005;20(3):279–295. [Google Scholar]
- 25.Momoi Y, McCabe JF. Hygroscopic Expansion of Resin Based Composites During 6 Months of Water Storage. Br Dent J. 1994;176:91–96. doi: 10.1038/sj.bdj.4808379. [DOI] [PubMed] [Google Scholar]
- 26.Huang C, Tay FR, Cheung GSP, Kei LH, Wei SHY, Pashley DH. Hygroscopic Expansion of a Compomer and a Composite on Artificial Gap Reduction. J Dent. 2002;30:359–362. doi: 10.1016/s0300-5712(01)00053-7. [DOI] [PubMed] [Google Scholar]
- 27.Anttila EJ, Krintilä OH, Laurila TK, Lassila LVJ, Vallitu PK, Hernberg RGR. Evaluation of Polymerization Shrinkage and Hygroscopic Expansion of Fiber-reinforced Biocomposites Using Optical Fiber Bragg Grating Sensors. Dent Mater. 2008;24:1720–1727. doi: 10.1016/j.dental.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Skrtic D, Hailer AW, Takagi S, Antonucci JM, Eanes ED. Quantitative Assessement of the Efficacy of Amorphous Calcium Phosphate/Methacrylate Composites in Remineralizing Caries-like Lesions Artificially Produced in Bovine Enamel. J Dent Res. 1996;75(9):1679–1686. doi: 10.1177/00220345960750091001. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Fiero JL, Aleman JV. Sorption of Water by Epoxide Prepolymers. Macromolecules. 1982;15:1145–1149. [Google Scholar]
- 30.Arima T, Hamada T, McCabe JF. The Effects of Cross-linking Agents on Some Properties of HEMA-based Resins. J Dent Res. 1995;74(9):1597–1601. doi: 10.1177/00220345950740091501. [DOI] [PubMed] [Google Scholar]
- 31.Skrtic D, Antonucci JM. Effect of Bifunctional Co-monomers on Mechanical Strength and Water Sorption of Amorphous Calcium Phosphate- and Silanized Glass-filled Bis-GMA-based Composites. Biomaterials. 2003;24(17):2881–2888. doi: 10.1016/s0142-9612(03)00119-4. [DOI] [PubMed] [Google Scholar]
- 32.Stansbury JW, Dickens SH. Determination of Double Bond Conversion in Dental Resins by Near Infrared Spectroscopy. Dent Mater. 2001;17:71–79. doi: 10.1016/s0109-5641(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 33.ASTM F394-78 (re-approved 1991) Standard Test Method for Biaxial Strength (Modulus of Rupture) of Ceramic Substrates [Google Scholar]