

Abstract
The de novo synthesis of compatible solutes is an essential part of the cellular osmotic stress response. Upon an osmotic challenge, it is regulated by the immediate biochemical activation of preformed enzymes and by activation of gene expression. Whereas the transcriptional response has been investigated intensively, the mechanisms by which enzymes are activated in osmotic stress situations are still elusive. Here, we address this topic for the moderately halotolerant cyanobacterium Synechocystis sp. PCC 6803, which synthesizes glucosylglycerol as a compatible solute. The key enzyme of the glucosylglycerol pathway (GgpS) is inhibited by nucleic acids in a sequence- and length-independent manner. The protein binds DNA, RNA, and heparin via a salt-dependent electrostatic interaction with the negatively charged backbone of the polyanions. Mechanistically, DNA binding to the enzyme causes noncompetitive inhibition of GgpS activity. The interaction of the enzyme and nucleic acids under in vivo conditions is indicated by the co-purification of both after cross-linking in Synechocystis cells. We propose a novel mechanism of activity regulation by the nonspecific salt-dependent binding of an enzyme to nucleic acids.
Keywords: DNA-binding Protein, Enzyme Inhibitors, Heparin-binding Protein, Metabolic Regulation, Protein-Nucleic Acid Interaction, Synechocystis, Compatible Solutes, Glucosylglycerol, Osmoregulation, Osmosensing
Introduction
All living bacterial cells maintain a proper turgor pressure as the driving force for cell growth (1). Turgor is caused by water influx into the cell, which is driven by the different water potentials of the cytoplasm and the cell's surrounding. The osmotic homeostasis is challenged by a change in water availability, which is caused mainly by drought and/or increasing external salt concentrations. Cells exposed to increased salt concentrations have to cope with two major problems: the maintenance of a low internal ion concentration despite high concentration gradients across the cytoplasmic membrane and the increase in the internal osmotic potential by the efflux of water (2). Under these conditions, the accumulation of compatible solutes was found to be essential in most bacteria (3, 4). Compatible solutes are low molecular weight compounds like sugars or amino acids and their derivatives, which are accumulated in high concentrations to establish the required osmotic potential without perturbation of the cellular metabolism (5). Additionally, they are able to protect cellular macromolecules directly by stabilizing their hydration shell (6). The accumulation of compatible solutes is achieved by either de novo synthesis or uptake. Both processes are part of the immediate response to osmotic stress and comprise the direct activation of preformed enzymes. Synthesis of trehalose in Escherichia coli is regulated by modulation of enzyme activities, as is proline synthesis in Bacillus subtilis and synthesis of glucosylglycerol in the cyanobacterial model strain Synechocystis sp. PCC 6803 (7–9). Following the rapid activation of enzymes, their activity must be tuned during the subsequent acclimatization to adjust the osmotic potential of the cytoplasm in relation to the external stress condition. However, mechanisms that cause activation and regulation of enzymes involved in the synthesis of compatible solutes are unknown (3).
Here, we focus on the synthesis of glucosylglycerol (GG) in Synechocystis sp. PCC 6803 (hereafter Synechocystis; Reaction 1).
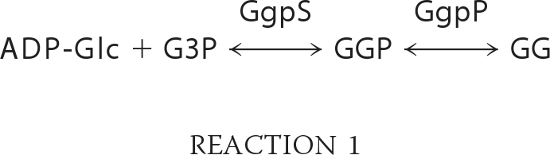 |
The key enzyme is the glucosylglycerol-phosphate synthase (GgpS), which uses ADP-glucose (ADP-Glc) and glycerol 3-phosphate (G3P) as precursors for the formation of glucosylglycerol phosphate (GGP), a reaction of second order. The intermediate is dephosphorylated by the second enzyme of the pathway, the glucosylglycerol-phosphate phosphatase (GgpP) (9). Cells unable to accumulate glucosylglycerol are not able to survive at higher salt concentrations (9). When de novo protein synthesis has been inhibited, glucosylglycerol synthesis is activated immediately following an increase in salt concentration, which demonstrates that the enzymes of the glucosylglycerol pathway are preformed but inactive in Synechocystis cells under low salt conditions (9). We report the identification of nucleic acids as inhibitory factors and address the biochemical mechanism of inhibition. We describe the nonspecific salt-dependent binding of the GgpS protein to nucleic acids and the concomitant noncompetitive inhibition of enzyme activity and propose a novel model for GgpS regulation under in vivo conditions.
EXPERIMENTAL PROCEDURES
Strains and Culture Conditions
Synechocystis sp. strain PCC 6803 was cultivated photoautotrophically in BG11 medium buffered with 20 mm TES4 at pH 8.0 and aerated by CO2-enriched air (5%) and continuous light (150 microeinsteins m−2 s−1) at 30 °C. The construction and cultivation of the Synechocystis ggpS+ mutant have been described previously (10). Analogous to the ggpS+ strain, a ggpS_strep+ strain was constructed to produce a GgpS protein harboring a C-terminal Strep-tag. The Strep-tag-encoding sequence was introduced at the C terminus using appropriate 3′-primers for amplification of the ggpS gene. E. coli cells were grown in LB medium at 37 °C in shaker flasks. The strain DH5αmcr (11) was used for cloning, and the strain BL21(DE3) (12) was used for expression.
Expression, Purification, and Activity Measurement of the GgpS Enzyme
For heterologous expression, the ggpS gene was cloned into the vector pASK-IBA3 (IBA GmbH, Göttingen, Germany) according to the supplier's manual. Expression of the ggpS-strep gene was induced by the addition of anhydrotetracycline (2 μg/ml) at an A600 of 1, and cells were harvested 6 h later by centrifugation at 5000 × g for 5 min. The cells were resuspended in wash buffer (100 mm Tris-HCl, pH 8.0, and 150 mm NaCl) and disrupted by passing three times through a French pressure cell press (SLM-AMINCO) at 18,000 p.s.i. After centrifugation at 15,000 × g for 20 min, the supernatant was subjected to Strep-tag purification (IBA GmbH) according to the manufacturer's protocol. The purified GgpS enzyme was frozen in liquid nitrogen and stored at −80 °C until further use. The activity of the GgpS enzyme and the glucosylglycerol content was determined as described previously (13). For analyzing the impact of nucleic acids on GgpS stability, the enzyme was incubated with or without DNA (1 μm) at 30 and 4 °C, and the activity was determined upon the addition of 200 mm NaCl. Glycerol 3-phosphate and ADP-glucose for enzyme assays were purchased from Sigma. Salmon sperm DNA was obtained from Invitrogen, and heparin was purchased from Sigma. All other chemicals not provided by the aforementioned kit suppliers were of analytical grade and purchased from Carl-Roth GmbH Co. KG (Karlsruhe, Germany).
Preparation and Treatment of Synechocystis Cell Extracts
Cells were collected by centrifugation at 20,000 × g for 5 min, and the pellet was resuspended in buffer A (20 mm Tris maleate, pH 7.5, and 10 mm MgCl2) and disrupted using FastPrep® treatment (Thermo Savant) using glass beads after the addition of 0.1 mg/ml DNase I (Roche Diagnostics). After centrifugation at 20,000 × g for 30 min, the supernatant was applied to a PD10 desalting column (GE Healthcare) to remove low molecular weight compounds. The protein content was estimated using Bradford reagents and adjusted to 10 μg/μl. Detection of the GgpS protein was performed as described previously (10). The nucleic acid content of samples was determined photometrically. To remove proteins, proteinase K (400 μg/ml; New England Biolabs) was applied (24 h, 40 °C) and inactivated (20 min, 95 °C), and the result was checked by Coomassie Blue staining after separation of proteins by SDS-PAGE and determination of the protein concentration according Bradford (14).
Limited Proteolysis
Purified GgpS protein or BSA as a control (each at 200 μg/μl) was incubated with salmon sperm DNA (2.5 μg/μl) at 37 °C and trypsin (20 ng/μl). At different time points, aliquots (20 μl) were taken, sample buffer (2% SDS and 1% β-mercaptoethanol) was added, and samples were incubated at 98 °C for 10 min. For SDS-PAGE analysis, 1.5 μg of proteins/lane were applied.
Strep Protein Interaction Experiment
The assay was basically performed as described previously (15). Briefly, the Synechocystis ggpS_strep+ strain expressing the ggpS-strep gene under the control of a constitutively active psbA2 promoter (10) was grown in the presence of 18 or 554 mm NaCl. At an A750 of 1, 1% formaldehyde was added, and after 20 min, the cells were harvested by centrifugation at 2000 × g for 10 min. The cell pellet was resuspended, washed, and disrupted using a French® press (see above). After centrifugation at 15,000 × g for 20 min, GgpS-Strep purification was performed using a Strep-Tactin-Sepharose column (see above). Samples were subjected to SDS-PAGE analysis, and the nucleic acid content was determined photometrically. For detection of the GgpS protein, an anti-GgpS antibody or an anti-Strep-tag antibody was applied.
EMSA
A DNA fragment (233 bp) was amplified by PCR using primers M13uni (GTAAAACGACGGCCAGT) and M13rev (CAGGAAACAGCTATGAC) and the circular plasmid pDRIVE (Qiagen, Hilden, Germany) as a template. An RNA fragment (250 nucleotides) was obtained by in vitro transcription of a Synechocystis rnpB gene fragment as described previously (10). For the assays, nucleic acids (35 nm DNA and 70 nm RNA) were incubated with GgpS protein (4.4 μm) in the absence or presence of substrates and/or NaCl (200 mm) at room temperature for 5 min, and after the addition of glycerol (10% final concentration), samples were subjected to agarose gel electrophoresis (2%, 80 V). In cases in which NaCl was applied, the agarose gel and the running buffer contained NaCl (200 mm). Alternatively, 35 nm DNA was incubated with increasing protein concentrations (0–850 nm). Visualization of nucleic acids was done by ethidium bromide staining, and band intensities were determined densitometrically (PC-BAS software, Raytest).
Heparin and DNA Binding Assay
GgpS binding to heparin was analyzed by incubation of 210 μg of purified GgpS protein in buffer A with 300 μl of HiTrapTM heparin-Sepharose (GE Healthcare). The matrix was thoroughly washed with 20 ml of buffer A, and the protein was eluted with buffer A as well as increasing salt concentrations from 0 to 2 m NaCl. The elution fractions were separated by SDS-PAGE, and the protein was visualized by Coomassie Blue staining. All experiments were performed at least in triplicate, and means ± S.D. of a typical example are shown.
RESULTS
GgpS Activity Modulation in Vitro and Identification of the Inhibitory Factor
To address the regulation of GgpS, the protein was heterologously expressed as a Strep-tagged variant in E. coli and purified to apparent homogeneity (data not shown). At first, independence of the activity of the purified GgpS protein on the salt concentration was confirmed by performing an enzyme assay in the presence of low (∼10 mm) and high (up to 200 mm) NaCl concentrations (Fig. 1). Furthermore, an inhibition of GgpS activity by Synechocystis cell extract of ∼90% was observed (Fig. 1). The subsequent addition of 200 mm NaCl resulted in almost complete reactivation of the GgpS enzyme (Fig. 1). Several strategies were applied to identify the putative interaction partner of GgpS in Synechocystis cell extracts. To re-evaluate the hypothesis of a protein-protein interaction being responsible for GgpS inactivation (16), proteins were removed from the cell extract by boiling and subsequent proteinase K treatment. In an enzyme assay, however, the same degree of inhibition of GgpS after the addition of protein-free cell extract was observed (Fig. 1). We concluded that the inhibition of GgpS under in vitro conditions is mediated in a protein-independent manner.
FIGURE 1.

Impact of salt and cell extract on the activity of the GgpS protein. The activity of the purified GgpS protein was determined in the absence or presence of salt (200 mm NaCl), and inhibition of GgpS activity by Synechocystis cell extract was tested before (Syn) and after boiling for 30 min at 100 °C (Syn boiled) or after proteinase K treatment (Syn ProtK). Inhibition of GgpS by nucleic acids was tested by the addition of salmon sperm DNA (2.5 μg/μl).
Assuming that a low molecular weight compound could act as an inhibitory factor, preparative HPLC separation of Synechocystis cell extract was conducted, but no particular fraction containing the inhibitor was obtained (data not shown). Furthermore, interaction of the GgpS protein with remaining lipids and/or membranes in the cell extract was investigated; however, the GgpS activity was not affected by the addition of polar lipid extracts of E. coli (data not shown). Although the cell extracts were treated with DNase I, small DNA fragments were still present (∼1.5 μg μl−1) and may affect GgpS activity. To test the hypothesis that these remaining DNA fragments may have been affecting GgpS activity, protein-free salmon sperm DNA (∼900-bp fragments) was added to the enzyme assay. Interestingly, in the presence of these DNA fragments at 2.5 μg/μl, the GgpS activity was found to be reduced to ∼1% (Fig. 1). Upon the addition of 200 mm NaCl, this inhibition was abolished. Thus, the same pattern of salt-dependent reversible inhibition of the GgpS activity by Synechocystis cell extract was demonstrated with DNA fragments, suggesting that DNA acts as an inhibitory factor for the GgpS enzyme.
Biochemical Characterization of GgpS Inhibition by DNA
The dose dependence of enzyme inhibition by DNA was analyzed by applying artificial DNA fragments of ∼0.9 kb (Fig. 2). A strong inhibition was observed with a K50 value of 67 ± 6.8 nm (R2 = 0.996). In the presence of high DNA concentrations, no complete inhibition was observed with a remaining activity of 11 ± 1.4% (Fig. 2). In the presence of 200 mm NaCl, no inhibitory effect of the nucleic acids was observed, as already described. For investigation of the length and sequence specificity of the inhibitory nucleic acids, single-stranded DNA probes consisting of six adenosine, thymidine, or cytidine residues were applied in a standard enzyme assay. The hexanucleotides were added at a concentration of 4 mm and caused inhibition of GgpS activity (Fig. 2). In the presence of 200 mm NaCl, inhibition was abolished, indicating that even small single-stranded DNA fragments can inhibit GgpS activity in a length- and sequence-independent but salt-dependent manner. Subsequently, the kinetic parameters of the GgpS enzyme activity were determined in the absence or presence of inhibitory DNA concentrations. Km and Vmax values were derived from the Michaelis-Menten plots by nonlinear regression (Fig. 3 and Table 1). For the substrate ADP-glucose, a Km of ∼0.5 mm under all conditions was found, whereas the Vmax value dropped from 354 to 217 nmol/s/mg of protein at increasing DNA concentrations. For the substrate glycerol 3-phosphate, again a constant Km value of ∼0.7 mm was found, whereas the Vmax value shifted from 411 to 275 nmol/s/mg of protein upon the addition of increasing DNA concentrations. The different Vmax and comparable Km values indicate a noncompetitive inhibition of GgpS by nucleic acids for both substrates (Table 1). The Ki value was estimated to be ∼2 μm in the case of both substrates.
FIGURE 2.

Impact of the DNA concentration, length, and sequence on GgpS activity. GgpS activity was assayed in the presence of artificial double-stranded DNA fragments or single-stranded DNA probes (inset). The double-stranded DNA probes were ∼0.9 kb, and 1 μm equals 0.625 μg/μl DNA. As single-stranded DNA probes, hexanucleotides consisting of six adenosine (A), thymidine (T), or cytosine (C) nucleotides were applied (4 mm) in the absence (black bars) or presence (gray bars) of 200 mm NaCl. Co, control (no DNA addition).
FIGURE 3.

Impact of nucleic acids on kinetic parameters of the GgpS enzyme. The GgpS activity was determined in the absence (▵) and presence of 0.02 μg (○) or 0.05 μg (♢) of double-stranded DNA and increasing substrate concentrations (ADP-glucose (ADP-Gluc; A) or glycerol 3-phosphate (G3P; B)). The results are presented in a Michaelis-Menten plot.
TABLE 1.
Kinetic parameters (Km and Vmax) of the GgpS enzyme in the absence or presence of inhibitory DNA concentrations
For details, see the legend of Fig. 3.
| Km | Vmax | R2 | |
|---|---|---|---|
| mm | nanokatals/mg of protein | ||
| ADP-glucose | |||
| 0 nm DNA | 0.48 ± 0.07 | 354 ± 18 | 0.99 |
| 0.4 nm DNA | 0.51 ± 0.06 | 319 ± 14 | 0.99 |
| 1 nm DNA | 0.41 ± 0.04 | 217 ± 7 | 0.99 |
| Glycerol 3-phosphate | |||
| 0 nm DNA | 0.71 ± 0.07 | 411 ± 16 | 0.99 |
| 0.4 nm DNA | 0.72 ± 0.13 | 357 ± 27 | 0.99 |
| 1 nm DNA | 0.75 ± 0.09 | 275 ± 14 | 0.99 |
Furthermore, the GgpS activity was determined in the presence of increasing DNA and salt concentrations (Fig. 4). Again, GgpS activity was not affected by salt concentrations up to 200 mm NaCl, but at low salt concentrations, a significantly decreased enzyme activity was found. Without the addition of NaCl, GgpS activity was decreased to 15% in the presence of 0.5–4.3 μm (0.3–2.5 μg/μl) DNA. At increasing salt concentrations, GgpS activity was regained, with lower amounts of NaCl required for the reactivation of GgpS at low DNA concentrations (Fig. 4). At 200 mm NaCl, GgpS inhibition was abolished even in the presence of 4.3 μm DNA. The results indicate that GgpS activity can be modulated by the DNA and salt concentrations in an antagonistic manner. K50 values of inhibition were found to be 51 mm NaCl at 0.5 μm DNA, 57 mm NaCl at 1 μm DNA, 66 mm NaCl at 2.1 μm DNA, and 84 mm NaCl at 4.3 μm DNA, demonstrating a linear relationship between the salt concentration and GgpS inactivation by nucleic acid (Fig. 4).
FIGURE 4.

Salt-dependent inhibition of GgpS activity by nucleic acids. The GgpS activity was tested at indicated salt concentrations in the absence (○) or presence of 0.5 μm (▵), 1 μm (□), 2.1 μm (♢), or 4.3 μm (▿) double-stranded DNA. The data were fitted according to sigmoid functions, and K50 values for the release of inhibition by NaCl at the different DNA concentrations are shown in the inset.
Binding of Nucleic Acids and Heparin to the GgpS Enzyme
To investigate whether inhibition of GgpS activity by DNA depends on the binding of nucleic acids to the protein, EMSAs were performed. According to the observation that the GgpS protein activity was inhibited by random DNA fragments in a sequence- and length-independent manner, artificial 233-bp DNA fragments obtained by PCR as described under “Experimental Procedures” were applied at 35 nm (Fig. 5). In the presence of 4.4 μm GgpS, a defined DNA band was found to be retarded during electrophoresis, confirming binding of GgpS molecules to the DNA fragments. In the presence of 5 μg of BSA, no shift in the DNA fragment was observed (data not shown). In addition, the same experiment was performed in the presence of 200 mm NaCl, and no DNA shift was detected (Fig. 5). At lower protein concentrations, less DNA was shifted, indicating the binding of a multitude but different number of enzyme molecules per DNA fragment, causing the observation of a DNA smear. However, binding occurred in a protein concentration-dependent manner (Fig. 5). At a GgpS/DNA molar ratio of 8, 50% of the DNA fragment was bound; at a GgpS/DNA ratio of 18, no free DNA was found (Fig. 5). In an additional EMSA, it was demonstrated that the GgpS enzyme also bound RNA (Fig. 5). Again, in the presence of high salt concentrations, binding of RNA to GgpS was abolished, pointing to an electrostatic interaction. Furthermore, the independence of length or sequence implies a common mechanism of binding, namely the interaction with the negatively charged backbone of nucleic acids. To test this hypothesis, the binding of GgpS to heparin, which resembles the backbone structure of nucleic acids, was investigated. Purified GgpS protein was bound to heparin-Sepharose beads, and the release of the protein was dependent on the salt concentration. At 0–500 mm NaCl, only traces of the loaded GgpS protein was eluted, but at 2 m NaCl, the majority of the loaded GgpS protein was eluted (Fig. 6A). Furthermore, the determination of GgpS activity in the presence of heparin revealed a strong inhibition at 10 μg of heparin (Fig. 6B). As with our observations with nucleic acids, this inhibition was abolished in the presence of NaCl (Fig. 6B). In conclusion, salt-dependent DNA/RNA and heparin binding, as well as inhibition of GgpS activity, indicates that the protein interacts with the nucleic acid backbone via an electrostatic interaction.
FIGURE 5.

Binding of GgpS to nucleic acids. EMSAs were performed using purified GgpS protein, a 233-bp DNA fragment obtained by PCR, or a 250-nucleotide RNA fragment obtained by in vitro transcription. Incubation and agarose gel electrophoresis were performed at low or high salt concentrations of 0 and 200 mm NaCl, respectively. GgpS (4.4 μm) and DNA (35 nm) or RNA (70 nm) were applied (A), or DNA (35 nm) was incubated with increasing GgpS concentrations (0–850 nm; B). The amount of free DNA at all of GgpS/DNA molar ratios was quantified by densitometry (C). The positions of shifted and nonshifted nucleic acids are indicated by arrows. kbp, kilobase pairs; knt, kilonucleotides.
FIGURE 6.

Heparin binding to GgpS and impact on enzyme activity. A, the binding of the GgpS protein to heparin was proven by affinity purification using 210 μg of recombinant purified GgpS protein and the indicated NaCl concentrations for elution. M, marker; In, input. B, the impact of heparin on GgpS enzyme activity was analyzed in the absence (○) and presence (▵) of 200 mm NaCl.
Subsequently, the question of whether the conformation of the GgpS enzyme was affected by DNA binding was addressed. GgpS protein was subjected to degradation by trypsin in the absence or presence of DNA concentrations that are sufficient for complete inhibition of the GgpS enzyme activity, and the occurrence of degradation products was followed (supplemental Fig. S1). As a control, degradation of BSA in the absence and presence of DNA was analyzed, but no difference in the degradation pattern was observed under these conditions (data not shown). For the GgpS enzyme, degradation was slower in the presence of DNA, indicating an altered accessibility for the protease in the presence of DNA (supplemental Fig. S1). This implies that GgpS could be protected by DNA interaction. However, the thermostability of GgpS was not improved in the presence of DNA (data not shown). Finally, the impact of glycerol 3-phosphate and ADP-glucose binding to GgpS on protein-DNA interaction was studied. The addition of glycerol 3-phosphate did not alter the electromobility of the protein-DNA complex, whereas the addition of ADP-glucose caused a reduced electromobility pointing to the binding of more GgpS molecules to the DNA (supplemental Fig. S2). The addition of ADP had no comparable impact, excluding that the presence of a charged molecule affected GgpS conformation and/or GgpS-DNA interaction.
GgpS-Nucleic Acid Interaction under in Vivo Conditions
To address GgpS-nucleic acid interaction in vivo, the Strep protein interaction experiment method was applied by expression of the ggpS-strep gene in Synechocystis and performance of cross-linking by formaldehyde in the absence or presence of NaCl. After purification of the GgpS-Strep protein, the concentration of co-purified nucleic acids was determined. In samples not treated with formaldehyde, no DNA was detected. After formaldehyde treatment of cells, however, 0.077 μg/μl protein and 0.303 μg/μl nucleic acids were co-purified in the absence of salt. In the presence of salt, 0.090 μg/μl protein and 0.135 μg/μl nucleic acids were co-purified. The results corroborate the findings obtained in the in vitro studies and indicate an interaction of GgpS with nucleic acids under in vivo conditions.
DISCUSSION
The regulation of GgpS enzyme activity by an unknown factor present in Synechocystis cell extracts was described >20 years ago, and many attempts have since been undertaken to identify this factor (9, 17, 18). We can now prove that it is not a protein but rather the binding of nucleic acids in the cell extract that is responsible for GgpS inactivation. Our findings concur with all previous observations made by ourselves and others. (i) The regulatory potential of the cell extract could not be removed by boiling or by precipitation with acetone. (ii) Separation of the cell extract by HPLC caused dilution of nucleic acids by differential retention of heterogeneous DNA fragments. (iii) Purification of native GgpS enzyme by ion exchange chromatography caused removal of nucleic acids and resulted in maximal salt-independent activity (18). (iv) A comparable extent of inhibition of GgpS activity by adding cell extract (1.5 μg/μl nucleic acids) or the corresponding amount of DNA was observed. (v) The degradation pattern during proteolysis was identical after adding cell extract or DNA (supplemental Fig. S1 and data not shown). In conclusion, our results demonstrate that the nucleic acids, still present in the cell extract upon DNase I treatment, indeed cause the inhibition of GgpS activity and represent the thus far unidentified inhibitor.
GgpS Protein Belongs to the Large Family of Polyanion-binding Proteins
Based on its ability to bind to nucleic acids and heparin, GgpS belongs to the large family of polyanion-binding proteins also known as heparin-binding proteins (19, 20). More than 100 such proteins have been identified. They interact with cellular polyanions like DNA, RNA, actin, tubulin, and polyphosphates (19). The interaction of heparin-binding proteins and polyanions is nonspecific and has been demonstrated on the structural level for FGFs (19, 21). The binding to polyanions depends on the electrostatic interaction of positively charged protein surface areas and negatively charged molecules. The addition of salt interferes with this interaction and releases the polyanion from the protein surface. GgpS binds salt-dependently to heparin, RNA, and well as DNA probes of different length and sequence, indicating that the binding is nonspecific and does occur via electrostatic interactions with the backbone of the nucleic acids, as observed for other polyanion-binding proteins (19). Additionally, the ion dependence of GgpS activation in the presence of cell extract was found to follow the Hofmeister series, indicating a positive correlation between activation and the chaotropicity of the respective ion (16). Because of the nonspecific binding of GgpS to polyanions, polyphosphate could, in principle, be an alternative binding partner. However, under our experimental conditions, Synechocystis cells do not accumulate polyphosphate (22). Therefore, we assume that nucleic acids represent the only relevant binding partners for GgpS under in vivo conditions. In comparison, the DNA concentration applied under in vitro conditions (up to 2.5 μg/μl) is smaller than the concentration of nucleic acids found in Synechocystis cells (11–15 μg/μl), indicating the relevance of the inhibitory effect under in vivo conditions (23).
Binding of Nucleic Acids to GgpS Causes Noncompetitive Inhibition
GgpS enzyme activity measurements in the presence of inhibitory concentrations of DNA revealed a noncompetitive inhibition for the substrates glycerol 3-phosphate and ADP-glucose. In conclusion, substrate binding at the active site of the GgpS enzyme was not impaired. This noncompetitive inhibition is in agreement with binding of inhibitor to the substrate-free GgpS protein, proven by the EMSAs. Binding of nucleic acids to GgpS seems not to induce a strong conformational change. In total, 55 recognition sites for trypsin are present in the GgpS sequence, and it seems likely that, upon a strong conformational change, a different fragmentation pattern could be observed because alternative sites are accessible to the trypsin treatment. In conclusion, binding of DNA seems to protect the GgpS enzyme from proteolysis by shielding. The assumption that no conformational change occurs upon DNA binding is in agreement with the lack of protection of GgpS from heat inactivation by DNA interaction.
Interestingly, binding of ADP-glucose to GgpS induces a conformational change because binding of DNA to GgpS was stronger in the presence of this substrate. This implies that, upon ADP-glucose binding to GgpS, the affinity or the number of DNA interaction sites was altered. In conclusion, GgpS-DNA interaction could be of even more relevance under in vivo conditions.
The most likely putative binding sites at the GgpS protein surface are the exposed basic amino acids. A sequence comparison with the trehalose-phosphate synthase protein of E. coli, for which the three-dimensional structure was solved (24), and the trehalose-phosphate synthase enzymes of Mycobacterium tuberculosis and Propionibacterium freudenreichii, both of which have been previously identified as polyanion-binding proteins or as affected by heparin (25, 26), revealed a high degree of similarity between the GgpS and trehalose-phosphate synthase sequences, particularly with respect to conservation of residues in the catalytic center (24). Interestingly, basic amino acids (Lys, Arg, and His) are mostly accessible in trehalose-phosphate synthase, or they are predicted to be accessible in GgpS and are therefore potentially able to contribute to nucleic acid or heparin binding (supplemental Fig. S3). In addition, salt stress-dependent regulation of trehalose-phosphate synthase enzyme activity could be achieved by a comparable mechanism.
Internal Salt Concentration Can Serve as a Trigger for GgpS Regulation
We propose that the inactivation of GgpS occurs through its binding to nucleic acids at low salt concentrations also in Synechocystis cells. Upon a sudden increase in the external salt concentration, the influx of ions causes liberation of GgpS and thereby its activation. Accumulation of glucosylglycerol facilitates salt stress acclimatization, including the decrease in the internal ion concentration, which results in rebinding of GgpS to nucleic acids and thereby its inactivation. Accordingly, the GgpS protein has to be present already under low salt conditions, which is the case (23), and the internal salt concentration serves as a trigger for activation and regulation of glucosylglycerol synthesis. Correspondingly, an influx of sodium and chloride ions was observed in the cyanobacterial strains Synechococcus 6311 and Synechocystis PCC 6714 upon an increase in the external salt concentration (27, 28). During acclimatization, the internal ion concentration remains at higher levels than in cells grown under low salt conditions (23, 27, 28). These observations are consistent with our model of activation of Synechocystis GgpS enzyme by the salt-dependent liberation from nucleic acids. Application of non-ionic osmotic stress conditions to Synechocystis cells, e.g. by the addition of sorbitol, did not cause activation of GgpS, and consequently, no glucosylglycerol accumulated because it is not accompanied by the influx of high amounts of ions (13). In summary, our novel model of stress-dependent enzyme activity regulation by the electrostatic interaction of a protein with nucleic acids may represent a common mechanism used by bacteria in the immediate osmotic stress response.
Supplementary Material
Acknowledgments
We thank Anja Wittmann for excellent technical assistance and Reinhard Krämer and Martin Hagemann for support, comments, and discussion.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- TES
- N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid.
REFERENCES
- 1. Empadinhas N., da Costa M. S. (2008) Int. Microbiol. 11, 151–161 [PubMed] [Google Scholar]
- 2. Bremer E., Krämer R. (2000) in Bacterial Stress Responses (Storz G., Hengge-Aronis R, eds) pp. 79–97, ASM Press, Washington, D. C. [Google Scholar]
- 3. Wood J. M. (2007) Methods Enzymol. 428, 77–107 [DOI] [PubMed] [Google Scholar]
- 4. da Costa M. S., Santos H., Galinski E. A. (1998) Adv. Biochem. Eng. Biotechnol. 61, 117–153 [DOI] [PubMed] [Google Scholar]
- 5. Borowitzka L. J., Demmerle S., Mackay M. A., Norton R. S. (1980) Science 210, 650–651 [DOI] [PubMed] [Google Scholar]
- 6. Hincha D. K., Hagemann M. (2004) Biochem. J. 383, 277–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dinnbier U., Limpinsel E., Schmid R., Bakker E. P. (1988) Arch. Microbiol. 150, 348–357 [DOI] [PubMed] [Google Scholar]
- 8. Whatmore A. M., Chudek J. A., Reed R. H. (1990) J Gen. Microbiol. 136, 2527–2535 [DOI] [PubMed] [Google Scholar]
- 9. Hagemann M., Erdmann N. (1994) Microbiology 140, 1427–1431 [Google Scholar]
- 10. Stirnberg M., Fulda S., Huckauf J., Hagemann M., Krämer R., Marin K. (2007) Mol. Microbiol. 63, 86–102 [DOI] [PubMed] [Google Scholar]
- 11. Grant S. G., Jessee J., Bloom F. R., Hanahan D. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 4645–4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Studier F. W., Moffatt B. A. (1986) J. Mol. Biol. 189, 113–130 [DOI] [PubMed] [Google Scholar]
- 13. Marin K., Stirnberg M., Eisenhut M., Krämer R., Hagemann M. (2006) Microbiology 152, 2023–2030 [DOI] [PubMed] [Google Scholar]
- 14. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 15. Herzberg C., Weidinger L. A., Dörrbecker B., Hübner S., Stülke J., Commichau F. M. (2007) Proteomics 7, 4032–4035 [DOI] [PubMed] [Google Scholar]
- 16. Schoor A., Hagemann M., Erdmann N. (1999) Arch. Microbiol. 171, 101–106 [DOI] [PubMed] [Google Scholar]
- 17. Hagemann M., Schoor A., Erdmann N. (1996) J. Plant Physiol. 149, 746–752 [Google Scholar]
- 18. Hagemann M., Effmert U., Kerstan T., Schoor A., Erdmann N. (2001) Curr. Microbiol. 43, 278–283 [DOI] [PubMed] [Google Scholar]
- 19. Jones L. S., Yazzie B., Middaugh C. R. (2004) Mol. Cell. Proteomics 3, 746–769 [DOI] [PubMed] [Google Scholar]
- 20. Conrad H. E. (1998) Heparin-binding Proteins, Academic Press, San Diego, CA [Google Scholar]
- 21. Yeh B. K., Eliseenkova A. V., Plotnikov A. N., Green D., Pinnell J., Polat T., Gritli-Linde A., Linhardt R. J., Mohammadi M. (2002) Mol. Cell. Biol. 22, 7184–7192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morohoshi T., Maruo T., Shirai Y., Kato J., Ikeda T., Takiguchi N., Ohtake H., Kuroda A. (2002) Appl. Environ. Microbiol. 68, 4107–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hagemann M., Fulda S., Schubert H. (1994) Curr. Microbiol. 28, 201–207 [Google Scholar]
- 24. Gibson R. P., Turkenburg J. P., Charnock S. J., Lloyd R., Davies G. J. (2002) Chem. Biol. 9, 1337–1346 [DOI] [PubMed] [Google Scholar]
- 25. Pan Y. T., Carroll J. D., Elbein A. D. (2002) Eur. J. Biochem. 269, 6091–6100 [DOI] [PubMed] [Google Scholar]
- 26. Cardoso F. S., Castro R. F., Borges N., Santos H. (2007) Microbiology 153, 270–280 [DOI] [PubMed] [Google Scholar]
- 27. Reed R. H., Warr S. R. C., Richardson D. L., Moore D. J., Stewart W. D. (1985) FEMS Microbiol. Lett. 28, 225–229 [Google Scholar]
- 28. Blumwald E., Mehlhorn R. J., Packer L. (1983) Plant Physiol. 73, 377–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.