

Abstract
ATP-sensitive K+ (KATP) channels are the target of a number of pharmacological agents, blockers like hypoglycemic sulfonylureas and openers like the hypotensive cromakalim and diazoxide. These agents act on the channel regulatory subunit, the sulfonylurea receptor (SUR), which is an ABC protein with homologies to P-glycoprotein (P-gp). P-gp is a multidrug transporter expressed in tumor cells and in some healthy tissues. Because these two ABC proteins both exhibit multispecific recognition properties, we have tested whether SUR ligands could be substrates of P-gp. Interaction with P-gp was assayed by monitoring ATPase activity of P-gp-enriched vesicles. The blockers glibenclamide, tolbutamide, and meglitinide increased ATPase activity, with a rank order of potencies that correlated with their capacity to block KATP channels. P-gp ATPase activity was also increased by the openers SR47063 (a cromakalim analog), P1075 (a pinacidil analog), and diazoxide. Thus, these molecules bind to P-gp (although with lower affinities than for SUR) and are possibly transported by P-gp. Competition experiments among these molecules as well as with typical P-gp substrates revealed a structural similarity between drug binding domains in the two proteins. To rationalize the observed data, we addressed the molecular features of these proteins and compared structural models, computerized by homology from the recently solved structures of murine P-gp and bacterial ABC transporters MsbA and Sav1866. Considering the various residues experimentally assigned to be involved in drug binding, we uncovered several hot spots, which organized spatially in two main binding domains, selective for SR47063 and for glibenclamide, in matching regions of both P-gp and SUR.
Keywords: ABC Transporter, Drug Transport, Membrane Enzymes, Multidrug Transporters, Protein Structure, P-glycoprotein, Molecular Modeling, Sulfonylurea Receptor, KATP Channels, Potassium Channel Openers, Potassium Channel Blockers, MDR1/P-glycoprotein, Hot Spots
Introduction
ABC8 proteins form a large superfamily of mostly membrane proteins. They present as common characteristics a nucleotide binding domain able to hydrolyze ATP, which includes the sequence motifs Walker A and B and the signature C (1). As membrane active transporters, they are expressed in virtually all branches of the living reign, and they exhibit a very broad diversity of transported substrates, handled in either the direction of influx or efflux (2). In higher mammals, they are involved in various pathophysiological situations and genetic diseases (3).
The KATP channel results from the constitutive association of four pore-forming Kir6.x subunits and four regulatory SUR subunits (4, 5). In various combinations of the SUR isoforms, SUR1, SUR2A, and SUR2B, and the Kir6 isoforms, Kir6.1 and Kir6.2, these channels are present in most excitable cells, including neuronal, cardiac muscle, smooth muscle, and endocrine cells (6), where they serve to couple membrane electrical properties to intracellular metabolism.
SUR, a member of the ABCC/MRP (multidrug resistance-associated protein) subfamily of ABC proteins, is the site of action of numerous drugs that cause either closing (KATP channel blockers) or opening (KATP channel openers) of the Kir6.x potassium pore (7). Blockers include antidiabetic drugs, sulfonylureas like glibenclamide as well as non-sulfonylureas like meglitinide. Openers present interesting therapeutic opportunities, in particular for their antihypertensive and cardioprotective properties; a few, like diazoxide, pinacidil, nicorandil, and minoxidil, are in clinical use as treatment against hypertension, angina pectoris, and alopecia, respectively (8).
Among ABC proteins of higher organisms, SUR stands out because its only known function is that of a channel regulator, whereas most others are active (or passive in the case of CFTR) transporters. However, SUR possesses strong sequence homologies with other eukaryotic ABC proteins, like the multidrug resistance transporters MRP1 and P-glycoprotein (P-gp), suggesting that these proteins could share some structural and pharmacological properties (9). Indeed, SUR is the target of a panel of structurally unrelated ligands, like P-gp and other multidrug transporters. Multispecific drug recognition by P-gp is still a misunderstood molecular property despite a mass of published data, including extensive mutational analyses (10) as well as a pharmacophore model of some of its known ligands (11). Because binding of SUR ligands has also been linked to some particular residues (12, 13), it is of interest to compare the ligand recognition features of these two ABC proteins, with the aim of gaining new insights into both of them.
In addition, regulation of several ABC transporters by KATP channel modulators has been reported; CFTR is inhibited by glibenclamide and less potently by a few other KATP channel blockers and openers (14). Glibenclamide also inhibits MRP1 (15) and P-gp (16), and it appears to be a substrate of the latter. These observations are of interest because they can complete the pharmacological toolbox of researchers (glibenclamide is now the most widely used blocker of CFTR channels), and they can point to potential side effects and altered biodisposition. This is specially important for P-gp, which is an essential gatekeeper at the blood-brain barrier as well as in cancer cells (17, 18).
In this work, we have therefore focused on P-gp and examined whether representative compounds among KATP channel blockers and openers could interact with P-gp. As blockers, we used glibenclamide, as a reference, and its two moieties, tolbutamide and meglitinide; as openers, we used diazoxide, SR47063, a derivative of cromakalim, and P1075, a derivative of pinacidil. Our findings demonstrate that these molecules bind to and are possibly transported by P-gp. They imply a possible structural similarity between SUR and P-gp, which could extend to a mechanistic similarity, in line with our recent work (12, 19) suggesting that KATP channel openers and blockers interact with SUR much like transport substrates would, although intrinsic active transport by SUR has not yet been demonstrated.
In an effort to rationalize the observed data and in the absence of high resolution crystallographic data, we built new homology models of human P-gp and SUR, based on multiple alignments with the bacterial ABC transporters MsbA and Sav1866, whose structures have been solved by x-ray crystallography in different conformations (20, 21), and on mouse P-gp, whose structure has been recently solved in one conformation (22). Highlighting the various residues known to be implicated in drug binding specificity helped to delineate different hot spots for drug binding in the P-gp and SUR structures. Their three-dimensional arrangement revealed two main binding domains for the drugs verapamil/SR47063 and for vinblastine/glibenclamide in P-gp and their counterpart for binding of SR47063 and of glibenclamide in SUR.
In this work, we used structural models derived from comparative modeling because they offer the advantage, in contrast to crystallographic structures, of allowing interpretation of a huge amount of binding data and addressing the multispecific character of these ABC proteins. Homology modeling also allowed us to interpret the results in various protein conformations, in relation to the enzymatic turnover.
EXPERIMENTAL PROCEDURES
P-glycoprotein-containing Membrane Vesicle Preparation
The MDR cell line used, DC-3F/ADX, was selected from spontaneously transformed Chinese hamster lung fibroblast DC-3F on the basis of resistance to actinomycin D. This resistance is due to overexpression of the pgp1 gene. The DC-3F/ADX cells and DC-3F cells, their drug-sensitive parental counterparts, were cultured, harvested by scraping, and washed in PBS in the presence of antiproteases. The cells in suspension were then disrupted by sonication. The resulting centrifuged supernatant was layered on a sucrose cushion to isolate the total membrane vesicle fraction. Membrane protein concentrations were determined by the Bradford method. In vesicles prepared from DC-3F/ADX, P-glycoprotein accounts for about 12–15% of total membrane proteins, whereas P-gp cannot be detected in the control vesicles prepared from the DC-3F cells (23). Experiments were performed using DC-3F cells as a control and DC-3F/ADX cells as a P-gp test. Because SUR displays ATPase activity modulated by its ligands (19), our results could be tainted by the unlikely presence of different amounts of SUR in DC-3F and DC-3F/ADX cells. The effect would be negligible, however, because the ATPase activity of SUR is >100-fold less than that of P-gp (24–26).
Measurement of ATPase Activity
The experiments are based on the enzymatic determination of the ATPase activity of native membrane vesicles containing high amounts of P-glycoprotein, measured at 37 °C by using a standard coupled enzyme assay, comprising an ATP-regenerating system and continuous spectrophotometric detection of NADH absorbance at 340 nm, which allows the monitoring of NADH consumption in the reaction medium, which is stoichiometric to ADP production.
The reaction medium contained 30 mm Tris-HCl (pH 7.5), 100 mm NaCl, 10 mm KCl, 2 mm MgCl2, 1 mm dithiothreitol, 1 mm MgATP, supplemented with 0.1 mg/ml pyruvate kinase, 1 mm phosphoenol pyruvate, 0.1 mg/ml lactate dehydrogenase, and 0.5 mm NADH. ATPase activity was determined with about 20 μg/ml membrane proteins. The assay medium was supplemented with 10 mm sodium azide, 0.5 mm ouabain, and 1 mm EGTA, to inhibit the H+-ATPases, Na+/K+-ATPase, and Ca2+-ATPases, respectively. The residual ATPase activity measured in the presence of the specific inhibitors of these enzymes and in the absence of any added drug was essentially due to the basal activity of P-glycoprotein. MgATP concentration was kept throughout the experiments at the subsaturating level of 1 mm because we have previously reported that the Km value of P-gp for MgATP remains essentially unchanged and independent of transported substrates (23, 27, 28). Shown error bars represent S.E. values, computed from measurements performed in 5–14 experiments using up to four distinct vesicle preparations. Model fitting to the ATPase activity values normalized to the basal values was done with Origin 5.0 software using a standard Hill equation,
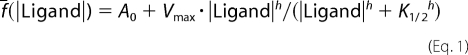 |
where A0 is the basal activity in the absence of ligand, K½ is the concentration for half-maximal activation (or inhibition), and h (or −h) is the Hill coefficient.
Although activity versus concentration curves often show an inhibition at the highest concentrations, we fitted these curves using only one high affinity specific binding site because (i) this inhibition may be due to a nonspecific membrane perturbation by amphiphilic drugs, rather than to a low affinity specific binding site, and (ii) curve fitting using a two-site binding model would require twice as many parameters as a single-site model, producing more uncertainty and no more qualitative information about the mutual relationships exhibited by the compounds tested. (See, for example, the parameters determined in Fig. 4 compared with those in supplemental Fig. S1, A and B, the two last fits depending on the level of the maximal inhibition expected (experimentally unattainable in the usable drug concentration range).) Overall, the single-site binding model yielded estimates of the apparent affinities of various P-gp substrates consistent with both the K½ values characterizing basal ATPase activity stimulation and the Ki values characterizing the inhibition of ATPase activity stimulated by reference P-gp substrates (11, 27–29).
FIGURE 4.

Modulation of the progesterone-induced ATPase activity by KATP channel openers and blockers. A, effects of progesterone in the absence (squares) or presence of 10 (diamonds), 100 μm (inverted triangles), or 300 μm (triangles) glibenclamide. Fits shown as dashed lines yielded K½, h, and Vmax of 17 μm, 2.1, and 66% in control; 12 μm, 3, and 32% with 10 μm glibenclamide; 1.8 μm, 1, and 32% with 100 μm glibenclamide; and 1 μm, 1.5, and 11% with 300 μm glibenclamide. B, effects of progesterone in the absence (squares) or presence of 1 mm diazoxide (circles), 1 mm P1075 (triangles), or 40 μm SR47063 (diamonds). Fits shown as dashed lines yielded K½, h, and Vmax of 17 μm, 2.1, and 66% in control; 12 μm, 2.5, and 76% with diazoxide; 7 μm, 1.5, and 40% with P1075; and 15 μm, 2, and 39% with SR47063.
Chemicals
To test a range of concentrations for a given compound, aliquots were sequentially added to the same optical cuvette under continuous stirring. Dilution effects were taken into account for the final ATPase activity calculations. Chemicals (from Sigma except where stated) and stock solution concentrations were as follows: diazoxide, 100 mm; glibenclamide, 100 mm; meglitinide (4-[2-(5-chloro-2-methoxybenzamido)ethyl]benzoic acid; Aventis (Frankfurt, Germany)), 100 mm; P1075 (N-cyano-N′-(1,1-dimethylpropyl)-N′-3-pyridylguanidine; Leo Pharmaceutical Products (Copenhagen, Denmark)), 150 mm; progesterone, 30 mm; SR47063 (4-(2-cyanimino-1,2-dihydro-1-pyridyl)-2,2-dimethyl-6-nitrochromene; Sanofi Recherche (Montpellier, France)), 150 mm; tolbutamide, 100 mm; verapamil, 150 mm; vinblastine, 100 mm. All stocks were prepared in DMSO except for progesterone, which was dissolved in ethanol. Compounds were added to the reaction medium directly from stock solutions or from dilutions either in DMSO or ethanol or in distilled water. The maximal amount of DMSO added was ∼2% for the last point of the dose-response measurements of the lowest affinity compounds. The effect of such concentrations of vehicle was assessed by testing increasing amounts of DMSO on control vesicles (with and without the non-P-gp ATPase inhibitors mentioned above) and on P-gp-containing/enriched vesicles (with and without the P-gp substrate verapamil) (data not shown). In all cases, DMSO, at the concentrations reached in our experiments, did not modify ATPase activity in any significant manner. At higher concentrations, the effects remained moderate because 6% DMSO augmented P-gp ATPase activity by 6% (n = 4).
Absorbance spectra for all tested compounds (data not shown) revealed that only SR47063, which has an absorbance peak at 328 nm, absorbed significantly at our assay wavelength of 340 nm. The resulting interference decreased the signal/noise ratio at the highest SR47063 concentrations used but would not alter the slope of the signal, which is the parameter used to measure the ATPase rate.
Phylogenetic Analyses
Amino acid sequences of 42 human and 2 bacterial, Sav1866 (accession number Q99T13) and MsbA (accession number A1A9J1), transmembrane ABC proteins were used for a phylogenetic reconstruction. The Protein Knowledgebase (UniProtKB; available on the World Wide Web) (30)) and the TCDB (Transport Classification Database) (available on the World Wide Web) (31) were used to retrieve fasta files, Swiss-Prot accession codes, synonyms, and TCDB classification numbers (see supplemental Table S1).
Because some ABC proteins are monomeric (full-size transporters) whereas others are dimeric (half-size transporters), and because some of them exhibit an additional N-terminal TMD0 domain, we used the “Conserved Domain Database” of NCBI (available on the World Wide Web) (32) to detect the respective consensus regions in the proteins. This roughly corresponds to one monomer of the dimeric proteins (plus or minus a dozen residues), and amino acid sequences with a size of about 600–650 residues were selected for the phylogenetic study, thereby excluding the TMD0 sequences (supplemental Table S1). Multiple-amino acid sequence alignment was performed with DIALIGN (33).
A maximum likelihood tree was constructed using PhyML (34), and we made 100 bootstrap replicates (35, 36) to assess confidence in the results. Indeed, this statistic technique is used to improve the robustness of a constructed tree and to inform about its reliability. As a general rule, if the bootstrap value for a given branch is found to be higher than 90%, then the topology of that branch is considered as “correct.” The phylogenetic tree was edited using TreeViewX (available on the World Wide Web) (37); in this representation, each branch length reflects a statistical distance between the considered sequence and its “common ancestor.”
Homology Modeling
The alignment of SUR, P-gp, Sav1866, and MsbA, shown in supplemental Fig. S2, was edited using Jalview Software (38). The secondary structures, derived from the three-dimensional model of P-gp, were added manually above the alignment.
Template Selection
PSI-BLAST (39) against the Protein Data Bank was used to select the templates for the homology modeling of P-gp and SUR1. This analysis confirmed that MsbA and Sav1866 have the closest sequences to these two mammalian ABC proteins. MsbA and Sav1866 share 28–31% identity with P-gp and 20–21% with SUR1 (see the legend to supplemental Fig. S2). After the withdrawal of the first crystallographic MsbA structures, the new structures deposited were proposed to represent three distinct putative functional conformations of this representative ABC transporter (20), the open inward-facing, the closed inward-facing, and the outward-facing conformations. They were thus used as templates to rebuild three structural models for the human P-gp (see Table 1); the outward-facing conformation also used Sav1866 as a template, allowing a two-template homology modeling. The recent crystallographic structure of mouse P-gp allowed us to rebuild another model for the human P-gp in the open inward-facing conformation based on a single pairwise alignment (87% sequence identity between human and mouse P-gp). For SUR1, only the outward-facing conformation was built using a two-template reconstruction from both St-MsbA and Sav1866 (Table 1), hence giving a higher confidence than a monotemplate homology modeling.
TABLE 1.
Reconstructed P-gp models and their corresponding templates
Vc, Vibrio cholera; Ec, Escherichia coli; St, Salmonella typhimirium; Sa, Staphylococcus aureus.
| Templates (PDB code) | Resolution | Models |
|---|---|---|
| Å | ||
| Ec-MsbA (3B5W) | 5.3 | Pgp (open inward-facing conformation) |
| Vc-MsbA (3B5X) | 5.5 | Pgp (closed inward-facing conformation) |
| Sav1866 (2HYD) | 3.0 | Pgp (outward-facing conformation) |
| St-MsbA (3B60) | 3.7 | Pgp (outward-facing conformation) |
| Mouse Pgp (3G5U) | 3.8 | Pgp (open inward-facing conformation) |
| Sav1866 (2HYD) | 3.0 | SUR1 (outward-facing conformation) |
| St-MsbA (3B60) | 3.7 | SUR1 (outward-facing conformation) |
Model Generation
For each modeling procedure, 10 models were generated with the Modeler 9v6 software (40), with refinement settings “slow” and the “automodel” procedure. The templates, which were either the N- and C-terminal halves of the mouse P-gp structure or half-size bacterial transporters, do not contain the domain TMD0, the first intracellular loop and the linker region. Hence, these regions were excluded from modeling.
Model Selection
A scoring program was applied to each generated model, and the best scored model among the 10 was selected. We have used a combination of the scoring programs available under Modeler, GA341 score and DOPE (Discrete Optimized Protein Energy) score.
Structural Model Assessment
The stereochemical qualities of the models were checked using programs for analyzing and validating protein structures: Procheck (41), Whatcheck (42), ProSA web (43), Molprobity (44), and QMEAN (45).
Rebuilding Structural Models in a Lipid Environment
The three modeled P-gp conformations were placed in a “native-like” environment in order to analyze the interactions of the protein with model phospholipids arranged in a double layer membrane. We chose 1-palmitoyl-2-oleoyl-phosphatidylcholine because it is the most prominent lipid in mammalian cell membranes. The procedure included a first step of solvation, realized by the Solvate program (available on the World Wide Web), and then successive steps allowed by the VMD (Visual Molecular Dynamics) software (46), using the “solvation box” and the “membrane builder” procedures.
Ligand Molecular Modeling and Alignment
The structures of glibenclamide and SR47063 were minimized and aligned under SYBYL (Tripos, St. Louis, MO) with the structures of vinblastine and verapamil, respectively, using procedures and data from Garrigues et al. (11). Hydrophobicity and hydrogen bond potential areas were calculated using the MOLCAD routine of SYBYL. The compounds were superimposed manually, and consensus groups were selected. The compounds were then aligned using the multifit command of SYBYL.
RESULTS
Experimental Assay Validation
Throughout this work, the ATPase activity of P-gp-enriched vesicles was used as a reporter of the function of native P-gp. This assay has been validated thoroughly in earlier works (11, 28). To further ascertain its suitability in our experimental settings, tests were performed using the P-gp substrates verapamil, progesterone, and vinblastine (Fig. 1A and Table 2). Our results are fully consistent, qualitatively and quantitatively, with the expected effects of these substrates on P-gp ATPase activity (28). Verapamil and progesterone increased ATPase activity, with a K½ of 1.3 and 17 μm, respectively, with no obvious cooperativity (Hill coefficients nearing 1). This activation declined at higher concentrations, a phenomenon that could be consistent with the presence of both high and low affinity sites as well as with a nonspecific perturbation due to the insertion of an amphiphilic compound into the membrane bilayer. Vinblastine inhibited the basal ATPase activity of P-gp-enriched vesicles down to a level close to that of the control vesicles. Because control vesicles were unaffected by these three compounds, it appears that the ∼70% additional ATPase activity measured in P-gp-enriched vesicles arose mainly from the presence of P-gp proteins.
FIGURE 1.

Drug concentration dependence of stimulated P-gp ATPase activity. A, effect of standard P-gp ligands on ATPase activity using P-gp-containing vesicles. The ATPase activity of P-gp-devoid (control, open symbols) and P-gp-enriched (filled symbols) native membrane vesicles was measured over a range of concentrations for three well characterized P-gp substrates: verapamil (squares), progesterone (triangles), and vinblastine (circles). Dashed lines represent fits of the Hill equation to the initial ligand-induced changes in ATPase activity of P-gp-enriched vesicles. Concentrations for half-maximal effect (K½), Hill coefficient (h), and Vmax were 1.3 μm, 0.94, and 73% for verapamil; 31 μm, 1.35, and 53% for vinblastine; and 17 μm, 2.1, and 66% for progesterone. Basal activity of P-gp-containing vesicles (i.e. 100% after normalization) was 120–300 nmol/min/mg of membrane protein, whereas it was ∼30 nmol/min/mg in control vesicles. When not visible, error bars were smaller than the symbols. B, effect of SUR ligands that block KATP channels on ATPase activity using P-gp-containing vesicles, showing that SUR blockers are P-gp substrates with the same order of potency. ATPase activity of control (open symbols) and P-gp-enriched (filled symbols) vesicles was measured over a range of concentrations for the KATP channel blockers, glibenclamide (squares), tolbutamide (triangles), and meglitinide (circles). The chemical structures of these compounds are illustrated on the right. Dashed lines represent fits of the Hill equation to the initial ligand-induced increases in ATPase activity of P-gp-enriched vesicles. K½, h, and Vmax were 11 μm, 1.2, and 73% for glibenclamide; 276 μm, 0.75, and 70% for meglitinide; and 3 mm, 0.68, and 70% for tolbutamide. Basal activities of P-gp-enriched vesicles used for the glibenclamide, tolbutamide, and meglitinide assays were on average 126, 96, and 114 nmol/min/mg of membrane protein, respectively. C, effect of SUR ligands that open KATP channels on ATPase activity using P-gp-containing vesicles, showing that SUR openers are low affinity P-gp substrates. ATPase activity of control (open symbols) and P-gp-enriched (filled symbols) vesicles was measured over a range of concentrations for the KATP channel openers, SR47063 (squares), P1075 (triangles), and diazoxide (circles). The chemical structures of these compounds are illustrated on the right. Dashed lines represent fits of the Hill equation to the ligand-induced increases in ATPase activity of P-gp-enriched vesicles. K½, h, and Vmax were 36 μm, 1.3, and 55% for SR47063; 610 μm, 1, and 63% for diazoxide; and 1.5 mm, 1.2, and 101% for P1075. Basal activities of P-gp-enriched vesicles used for the SR47063, P1075, and diazoxide assays were on average 150, 138, and 114 nmol/min/mg of membrane protein, respectively.
TABLE 2.
Enzymological parameters from P-gp ATPase activity measurements shown in Figs. 1–5
K½, the half-activating concentration of the tested compound on P-gp basal ATPase activity; the minus sign in parentheses indicates that it is the half-inhibiting concentration because there is no apparent activation. Ki, the half-inhibiting concentration of the tested compound on P-gp-stimulated ATPase activity, as determined from the concentration dependence curves of the substrate indicated in brackets. ND, not determined. ME, mutually exclusive (i.e. the mutual relationship between two drug substrates revealed by a competitive inhibiting effect on P-gp ATPase activity). NME, non-mutually exclusive (i.e. the mutual relationship between two drug substrates revealed by either a non-competitive inhibiting effect or an additive effect or a positive allosteric effect on P-gp ATPase activity). VRP, verapamil; PRG, progesterone; VBL, vinblastine; GLB, glibenclamide; MGL, meglitinide; TLB, tolbutamide; DZX, diazoxide.
| Drug studied at increasing concentrations | K½ | Ki | Mutual relationships (ME versus NME) |
|---|---|---|---|
| μm | μm | ||
| Reference Pgp substrates | |||
| VRP | 1.3 | ∼1 (from GLB) | NME with GLB |
| PRG | 17 | ND | ND |
| VBL | 31 (−) | ∼1 (from GLB) | ME with GLB |
| SUR blockers | |||
| GLB | 11 | ∼10 (from VRP), 310 (from PRG) | NME with VRP, PRG |
| MGL | 280 | ND | NME with GLB |
| TLB | 3000 | ND | NME with GLB |
| SUR openers | |||
| SR47063 | 36 | 350 (from VRP), >40 (from PRG) | ME with VRP |
| NME with PRG | |||
| P1075 | 1500 | ND | NME with GLB, VRP, PRG |
| DZX | 610 | ND | NME with GLB, VRP, PRG |
KATP Channel Blockers Stimulate P-gp ATPase
Inhibitors of KATP channels include sulfonylureas as well as non-sulfonylurea compounds, all known to act through direct binding to SUR (47). Evidence has been published that the prototypical sulfonylurea glibenclamide is a P-gp substrate able to block, at 100 μm, transport of colchicine by P-gp (16). The data from Fig. 1B are in agreement with this finding in that they show that glibenclamide augments the ATPase activity with a maximum attained near 100 μm. The dose-effect curve resembles that obtained in Fig. 1A with verapamil.
Two other blockers were examined: meglitinide, which represents the non-sulfonylurea half of glibenclamide, and tolbutamide, the sulfonylurea half of glibenclamide. Both increased ATPase activity although with lower affinities (Fig. 1B). Although the maximum activation could not be reached, because this would have required excessive concentrations and excessive levels of the vehicle DMSO, Hill equation fits to the data gave maximum activation levels of ∼70% for the two compounds, a value similar to that achieved with the other ATPase activators, glibenclamide, verapamil, and progesterone.
KATP Channel Openers Stimulate P-gp ATPase
KATP channel openers form an extended family of seemingly unrelated molecules that bind to SUR at a site distinct from the sulfonylurea binding site and allosterically cause channel opening (7). We tested three representative openers: diazoxide; SR47063, a derivative of cromakalim; and P1075, a derivative of pinacidil. Diazoxide targets both SUR1 and SUR2 isoforms (48), whereas SR47063 and P1075 target only SUR2 isoforms through a common site separate from that of diazoxide.
All three compounds caused an increase in ATPase activity of P-gp-containing vesicles but not control vesicles (Fig. 1C). P1075 caused the largest increase (Vmax = 101%) but had the lowest affinity (K½ = 1.5 mm). Diazoxide also had a low affinity of 0.6 mm. The affinity of SR47063 was highest at 36 μm, but its effect was more modest (Vmax = 55%). These latter values probably underestimate the actual effects of SR47063 because the higher concentrations of SR47063 had an inhibitory effect on the contaminant sources of ATPase activity that appeared when the compound was tested in the control vesicles devoid of P-gp (Fig. 1C). It should be noted that the effect of SR47063 was modest in the experiments using the full concentration range but could be more sizeable in separate single-concentration experiments as in the experiment of Figs. 3 and 4.
FIGURE 3.

Interactions between verapamil and KATP channel modulators. A, verapamil-induced ATPase activity measured in the absence (squares) or presence of 2 (triangles) or 10 μm (diamonds) glibenclamide. Dashed lines represent fits of the Hill equation to the data points. K½, h, and Vmax were 1.3 μm, 0.94, and 76% in control; 1.4 μm, 0.9, and 49% with 2 μm glibenclamide; and 1.6 μm, 1.7, and 27% with 10 μm glibenclamide added. B, glibenclamide-induced ATPase activity measured in the absence (squares) and presence of 0.1 (circles), 1 (diamonds), or 10 μm (triangles) verapamil. Dashed lines represent fits of the Hill equation to the experimental data. K½, h, and Vmax were 11 μm, 1.2, and 73% in control; 10.7 μm, 1.3, and 62% with 0.1 μm verapamil; and 6.4 μm, 1.8, and 28% with 1 μm verapamil. C, verapamil-induced ATPase activity measured in the absence (squares) or presence of the KATP channel openers SR47063 (50 μm) (circles), P1075 (1 mm) (triangles), or diazoxide (1 mm) (diamonds). Fits shown as dashed lines yielded K½, h, and Vmax of 1.3 μm, 0.94, and 76% in control; 1.9 μm, 1.4, and 41% with SR47063; 1.2 μm, 0.9, and 72% with P1075; and 0.95 μm, 1.2, and 77% with diazoxide.
Cumulative Effects of Glibenclamide and Other KATP Channel Modulators
Binding assays on SUR using radiolabeled glibenclamide have shown that most KATP channel modulators displace glibenclamide binding (9). We therefore performed experiments designed to reveal interaction between glibenclamide and our other test compounds. Full concentration-effect curves for glibenclamide were repeated in the presence of millimolar concentrations of the KATP channel blockers tolbutamide and meglitinide and the openers diazoxide and P1075. Fig. 2 shows that the glibenclamide curve was merely shifted up by these compounds without an increase in apparent affinity (i.e. K½ value). Thus, effects on P-gp were cumulative without hints of the kind of interactions observed with SUR (49, 50). Surprisingly, neither meglitinide nor tolbutamide competed with glibenclamide despite their very similar chemical structures.
FIGURE 2.

Effect of various SUR ligands on the glibenclamide-induced stimulated P-gp ATPase activity, showing a lack of “cross-talk” between glibenclamide and other KATP blockers and openers. The effects of glibenclamide on P-gp ATPase activity were measured in the absence (squares) and presence of other KATP channel modifiers, meglitinide (1 mm) (inverted triangles), tolbutamide (3 mm) (diamonds), P1075 (1 mm) (triangles), or diazoxide (1 mm) (circles). Dashed lines represent fits of the Hill equation to the glibenclamide-induced increases in ATPase activity. K½, h, and Vmax were 11 μm, 1.2, and 73% in control; 11 μm, 0.9, and 78% with meglitinide added; 6.7 μm, 1.1, and 94% with tolbutamide; 6.5 μm, 1.1, and 81% with P1075; and 9.8 μm, 1.3, and 69% with diazoxide.
Interactions between Glibenclamide and Established P-gp Substrates
The compounds verapamil, progesterone, and vinblastine are typical P-gp substrates (Fig. 1A) presumed to bind at different positions within the P-gp binding pocket (28, 51). As such, they constitute useful test drugs to characterize the binding of novel P-gp ligands, such as those reported above. Each drug was therefore assayed in combination with selected KATP channel modulators.
Experiments combining glibenclamide and verapamil, shown in Fig. 3, A and B, demonstrate that glibenclamide inhibited the verapamil-induced increase in ATPase activity, and vice versa. This inhibition was not accompanied by a significant increase in the K½ value of the control substance, implying a non-competitive interaction. Indeed, a “pure” competitive inhibition by 10 μm glibenclamide or by 1 μm verapamil, concentrations corresponding to their respective affinities, should have induced a 2-fold increase of K½ value for verapamil and glibenclamide, respectively. The dose dependence of the inhibition correlated well with the respective K½ values of glibenclamide and verapamil observed for basal ATPase stimulation (Fig. 1, A and B). Indeed, the K½ value for verapamil activation of basal ATPase was close to 1 μm, and the same concentration was sufficient to reduce by half the glibenclamide-induced increase in ATPase activity (Fig. 3B). Conversely, the K½ value for glibenclamide activation of basal ATPase was near 10 μm, and Fig. 3A shows that, in the presence of that concentration of glibenclamide, the verapamil-induced increase in ATPase activity was roughly half what it was in control. This correlation suggests that a single glibenclamide binding site mediates both activation of basal ATPase and inhibition of verapamil-induced ATPase.
A similar observation was made when glibenclamide was tested against progesterone, as shown in Fig. 4A; glibenclamide inhibited the effects of progesterone with a half-maximal concentration close to 10 μm. Curve fitting of the ATPase activity versus progesterone concentration data yielded K½ values decreasing from 17 μm in the absence of glibenclamide to a non-significantly different 12 μm in 10 μm glibenclamide. Lower K½ values of 1.8 and 1 μm were obtained in 100 and 300 μm glibenclamide, respectively, but the shallow nature of the curves makes these values too uncertain to consider this decrease as significant and infer cooperativity rather than non-competitive inhibition.
The effects of vinblastine on the ATPase versus glibenclamide concentration curve (Fig. 5A) were also inhibitory, with a dose-dependent increase in K½ (from 11 μm in control to 19 μm at 1 μm vinblastine), suggestive of a weak competitive interaction. Vinblastine inhibited the ATPase stimulation elicited by glibenclamide with full inhibition achieved at ∼10 μm vinblastine, a concentration that, by itself, did not affect basal activity of P-gp (see Fig. 1A). Half-maximal effect was observed at ∼1 μm, which matches values previously determined from the inhibition by vinblastine of other drug-stimulated ATPase activities: verapamil (28), nicardipine (29), and indinavir (52).
FIGURE 5.

Inhibition by vinblastine of ATPase stimulation produced by glibenclamide and SR47063. A, effects of glibenclamide on P-gp ATPase activity measured in the absence (squares) or presence of 0.1 μm (triangles), 1 μm (diamonds), or 10 μm (circles) vinblastine. Dashed lines represent fits of the Hill equation to the glibenclamide-induced increases in ATPase activity. K½, h, and Vmax were 11 μm, 1.2, and 73% in control; 15 μm, 0.9, and 70% with 0.1 μm vinblastine; and 19 μm, 1.2, and 39% with 1 μm vinblastine. A fit was not possible for 10 μm vinblastine, but Vmax was estimated as ∼5%. B, effects of SR47063 on P-gp ATPase activity measured in the absence (squares) and presence of 10 μm (circles) vinblastine.
Interactions between KATP Channel Openers and Established P-gp Substrates
The KATP channel openers diazoxide and P1075, unlike glibenclamide, shifted upward the ATPase versus verapamil concentration curve without modifying significantly the K½ of verapamil (Fig. 3C). We therefore conclude that the ATPase stimulation by these openers and verapamil were mostly additive, suggesting separate binding sites on P-gp. SR47063 increased the K½ of verapamil from 1.1 to 1.9 μm, a slight change, considering the scattering of data points and it caused a decrease in Vmax from 76% to 41%. Although this Vmax decrease could reflect saturation of the verapamil effect due to the large stimulation already elicited by SR47063 in those particular experiments, these results suggest a weak competitive inhibition between verapamil and SR47063. Experiments with progesterone (Fig. 4B) revealed that stimulation by diazoxide and progesterone were additive, whereas P1075 and SR47063 produced a decrease in the Vmax of progesterone from 66% to 40% with a reduction in K½ with P1075 that appeared less significant because of experimental data uncertainties. This indicates a difference in the mechanism of action of diazoxide and P1075 or SR47063.
Finally, as with glibenclamide, 10 μm vinblastine fully inhibited the ATPase stimulation produced by SR47063 (Fig. 5B), although the data with lower concentrations of vinblastine (not shown) did not permit quantification of changes in K½ indicative of competitive or non-competitive interactions.
Analysis of Molecular Recognition Elements in Glibenclamide and SR47063 Structures
Using SYBYL software, we identified hydrophilic (i.e. electron donor and acceptor groups) and hydrophobic (i.e. alkyl and aromatic groups) elements contained in the molecular structures of the two P-gp ligands, glibenclamide and SR47063. SR47063, which exhibits a competitive interaction with verapamil, has in common with verapamil an electron donor group (CN) and an aromatic group (Fig. 6A). Glibenclamide, which exhibits a competitive interaction with vinblastine, presents five parts in common with vinblastine when considering their three-dimensional alignment: two aromatic cycles, two hydrogen donor groups (NH), and one electron donor group (CO and SO2) (Fig. 6B). We have previously shown that multispecific ligand interaction with P-gp relies on the recognition of a three-dimensional pattern of consensus hydrophilic and hydrophobic elements presented by these ligands, independently of their precise chemical structures (11). Furthermore, multidrug recognition capacity is amplified by the existence of different three-dimensional patterns, each defining a pharmacophore on P-gp. Indeed, according to the multipharmacophoric model of the multidrug recognition domain of P-gp established on the basis of mutual interactions between several P-gp ligands, verapamil and vinblastine were identified as representatives of two distinct pharmacophores, pharmacophores I and II, respectively (11). The combined enzymological and structural evidence presented here suggests that SR47063 and glibenclamide belong to the pharmacophores I and II, respectively (Fig. 6C). Indeed, the aromatic and the electron donor groups of SR47063 and of glibenclamide correspond to the consensus elements defining these two pharmacophores. These data about pharmacophoric-type recognition processes by P-gp have prompted us to investigate the structural features of the P-gp protein involved in the selective molecular recognition of its various ligands.
FIGURE 6.

Pharmacophoric model of glibenclamide and SR47063. A, three-dimensional alignment of SR47063 (blue sticks) and verapamil (red sticks) with surfaces highlighting the aligned aromatic groups (green) and the electron donor CN group (blue) on both molecules. B, two-dimensional structures of verapamil and SR47063 showing the common aromatic groups in green and electron donor groups in blue. C, three-dimensional alignment of glibenclamide (yellow sticks) and vinblastine (cyan sticks) with surfaces showing the aligned aromatic groups (green), the hydrogen donor NH/OH groups (red), and the electron donor CO/CO2 groups (blue) on both molecules. D, two-dimensional structures of glibenclamide and vinblastine showing the common aromatic groups in green, hydrogen donor groups in red, and electron donor groups in blue. E, schematic representation of the functional interactions between the binding sites on P-gp for KATP channel blockers (glibenclamide (GLB), meglitinide (MGL), and tolbutamide (TLB) in dark gray), openers (SR47063 (SR), diazoxide (DZX), and P1075 in light gray), and known P-gp substrates (verapamil (VRP), vinblastine (VBL), and progesterone (PRG) in white). Solid arrow lines represent mutual exclusiveness indicated by the competitive inhibition observed between glibenclamide and vinblastine and between SR47063 and verapamil. Connecting lines represent binding to separate sites: either mutual destabilization reflected by observed non-competitive inhibition, mutual stabilization reflected by observed positive allosteric effect, or a lack of recorded interactions reflected by additive effects. The interaction between SR47063 and vinblastine is shown exclusively by a simple proximity because our data are insufficient to assess the competitive or non-competitive nature of the observed inhibition. The dotted boxes represent the two pharmacophores characterized previously (11).
Phylogenetic Study of Membrane ABC Proteins
The phylogenetic tree of a set of membrane ABC proteins composed of the membrane human ABC transporters and two bacterial ABC transporters of known structure is shown in Fig. 7. Its branches split along the five human subfamilies, A, B, C, D, and G, previously defined on the basis of sequence similarities signing divergent evolution (3). Using either the N- or C-terminal sequence of the monomeric full-size ABC transporters in the alignment did not change the topology of the tree (not shown). Based on the high bootstrap values, the phylogenetic tree could be interpreted with good confidence. Two distinct “clusters” are found. They represent ABCA-G and ABCB-C-D. It appears that P-gp (ABCB subfamily) and SUR1/2 (ABCC subfamily) belong to the same cluster. Moreover, inside this cluster, the ABCC branch is closer to the ABCB than the other branches in terms of sequence evolution, as indicated by the length of the (horizontal) line defining these branches.
FIGURE 7.

A phylogenetic tree depicting the relationships between the human membrane ABC transporters and two bacterial ABC transporters. This maximum likelihood tree is based on 44 amino acid sequences (42 human and two bacterial). Five principal branches are distinguishable: ABCB branch (red hook), ABCA branch (blue hook), ABCC branch (brown hook), ABCD branch (green hook), and ABCG branch (orange hook). P-gp (ABCB1), MsbA, and Sav1866 are highlighted in a red box. SUR1 (ABCC8) is shown in a brown box. Only the bootstrap values higher or equal to 90% are shown.
Interestingly, the two multispecific bacterial transporters MsbA and Sav1866 belong to the ABCB branch, in close proximity to P-gp (ABCB1) as well as ABCB4 (MDR3) and ABCB11 (BSEP). This confirms that MsbA and Sav1866 can be considered as suitable templates for modeling P-gp structure by homology.
Molecular Modeling of P-gp
Selection and Assessment of the Final Models
All of the modeled structures generated, for P-gp as well as for SUR, were validated by GA341 scores of >0.9. The selection of the “best” models (one for each conformation) was performed using the DOPE score, although we are aware that it was initially designed for globular soluble proteins and is applied to membrane proteins for lack of more suitable tools. The quantitative assessment of the selected models is presented in supplemental Table S2. Even for the two inward-facing conformations, constructed from the Ec- and Vc-MsbA low resolution Cα positions-only structures, all models show very satisfying quality, as evidenced by Ramachandran plots that show in all cases that >90% of the residues are in favorable or allowed regions.
Structural Model of P-gp
From the three conformations of the bacterial ABC transporters (two nucleotide-free “apo conformations” (“open inward-facing” and “closed inward-facing”) and one nucleotide-bound (“outward-facing”)), we generated three different P-gp structural models that could reflect different enzymological states of the active transporter during its catalytic turnover (see coordinate files in Protein Data Bank format in the supplemental material).
The open inward-facing conformation model (supplemental Fig. S3), derived from the template Ec-MsbA (20), displays the following features: (i) the two NBDs are far apart (distance between P-loop 1 (Ser429) and P-loop 2 (Ser1072) ∼48.4 Å, mean distance between P-loop and LSGGQ segment (Leu531/Leu1176) ∼65.4 Å); (ii) the transmembrane helices TM1-2 and TM4-5 are not exactly symmetrical compared with their C-terminal counterparts, helices TM7-8 and TM10-11, respectively; (iii) TM4-5/10-11 are intertwined between the two halves of the protein; (iv) all of the helices of the transmembrane domain are in continuity with their corresponding intracellular helices; (v) these helices, oblique with respect to the membrane plane, form an internal cavity (“chamber”) lined by TM4, -6, -7, -10, and -12 at the level of the cytosolic leaflet of the membrane.
During the course of this work, the publication of an x-ray structure for murine P-gp (87% identity with human P-gp) in the open inward-facing conformation (22), with a better resolution than that of Ec-MsbA and that of the previously published structures obtained by electron microscopy (53, 54), although solved “by halves” and phased on MsbA, allowed us to generate another model of this conformation with a better reliability (Fig. 8A). For this reason, we will only consider this model in the following. Compared with P-gp modeled from Ec-MsbA, the main difference is that the two NBDs are closer (distance between P-loop 1 and P-loop 2 ∼35.0 Å, mean distance between P-loop and LSGGQ segment ∼23.7 Å).
FIGURE 8.

Side views of structural models of P-gp in a 1-palmitoyl-2-oleoyl-phosphatidylcholine membrane. A, open inward-facing conformation structural model, rebuilt from murine P-gp. B, closed inward-facing conformation structural model, rebuilt from Vibrio cholera MsbA. C, outward-facing conformation structural model, rebuilt from Salmonella typhimirium MsbA and Staphylococcus aureus Sav1866. D, same as C after 90° rotation around the symmetrical axis of the protein.
The closed inward-facing conformation model (Fig. 8B), derived from the template Vc-MsbA (20), differs from the open inward-facing models by the following features: (i) the two NBDs are in close proximity (distance between P-loop 1 and P-loop 2 ∼8.0 Å but mean distance between P-loop and LSGGQ segment ∼36.5 Å); (ii) the transmembrane helices are perpendicular to the membrane plane and no longer form a “chamber.”
The outward-facing conformation models (Fig. 8, C and D), built from templates St-MsbA (20) and Sav1866 (21), differ from the inward-facing models: (i) the two NBDs are associated (distance between P-loop 1 and P-loop 2 ∼23.5 Å; mean distance between P-loop and LSGGQ segment ∼11.6 Å, indicating a rotation of the NBDs from their position in the closed inward-facing conformation); (ii) the transmembrane helices are oblique with respect to the membrane plane, with an orientation opposite to that in the open inward-facing conformation, and form an extracellularly accessible pocket in the phospholipid membrane that is best seen upon rotation of the model (Fig. 8D). At variance with our model, the model of the same conformation elaborated by O'Mara and Tieleman (55) presents a region, TM1-EC1-TM2, that extends in the extracellular medium with a marked asymmetry between the two halves of the protein. The model of Ravna et al. (56) also predicts a slight asymmetry between the two halves of the protein, which is not apparent in our model. For an unobstructed view of the protein structure, the models are presented devoid of their membrane embedding in supplemental Fig. S4.
Three-dimensional Localization of P-gp Residues Involved in Substrate Recognition
The P-gp models permit spatial localization of 52 residues (Table 3) reported to be involved in substrate recognition, as previously collected from point mutagenesis and chemical labeling data both for human and for mouse P-gp (10, 57) as well as from crystallographic data (22). In Fig. 9A, displaying a P-gp model in the open inward-facing conformation, the top view representation reveals that the 52 residues are not distributed uniformly around the internal chamber, suggesting that the different helices do not participate equally in drug recognition. It is noteworthy that all important residues (i) are placed at the level of the phospholipid membrane, which is determined with a good confidence because it results from a simulation of the molecular interactions between the lipids and the protein (and not from the prediction from a simple hydropathy plot), and (ii) are oriented toward the interior of the protein cavity, the so-called internal chamber. In addition, in a given TM helix (except TM11), important residues are not uniformly distributed but appear clustered at the level of either the cytosolic or the exoplasmic leaflet. This non-uniform distribution reinforces the notion of “hot spots for drug binding” previously developed by Shilling et al. (10) (see below). Finally, the open inward-facing conformation of P-gp being the drug binding-competent form of the protein, the drug recognition specificity of all of the experimentally documented residues allows the identification of binding hot spots for verapamil (TM4E, TM5E, TM6E, TM7E, TM10C, TM11C, and TM12E) and for vinblastine (TM1C, TM6E, TM11C, and TM11E), where the E and C suffixes designate the exoplasmic and cytosolic halves of the transmembrane helices, respectively.
TABLE 3.
Transmembrane localization of P-gp residues involved in substrate recognition
P-gp residues identified from point mutagenesis, chemical labeling (10, 57), or crystallographic data (22). The equivalent residues belonging to MsbA, Sav1866, and SUR1 are identified thanks to the primary sequences alignment represented in supplemental Fig. S2.
| P-gp | MsbA | SUR1 | Sav1866 | References | |
|---|---|---|---|---|---|
| N-terminal | |||||
| TM1 | His61 | Ile28 | Gly16 | Phe17 | 10 |
| Gly64 | Gly31 | Cys19 | Ile20 | 10 | |
| Leu65 | Ile32 | Ile20 | Ile21 | 10 | |
| Met69 | Leu36 | Val24 | Ile25 | 22 | |
| TM4 | Ser222 | Ala175 | Ile467 | Phe171 | 22 |
| TM5 | Leu304 | Leu257 | Ala549 | Thr253 | 22 |
| Ile306 | Ala259 | Pro551 | Thr255 | 10 | |
| Tyr307 | Ser260 | Ile552 | Asp256 | 22 | |
| TM6 | Phe335 | Phe288 | Leu580 | Val284 | 10 |
| Phe336 | Ser289 | Ser581 | Gly285 | 22 | |
| Val338 | Met291 | Phe583 | Leu287 | 10 | |
| Leu339 | Ile292 | His584 | Glu288 | 22 | |
| Ile340 | Ala293 | Ile585 | Leu289 | 22 | |
| Gly341 | Leu294 | Leu586 | Leu290 | 10 | |
| Ala342 | Met295 | Val587 | Phe291 | 22 | |
| Phe343 | Arg296 | Thr588 | Gly292 | 22 | |
| Ala354 | Gln307 | Ser599 | Thr303 | 10 | |
| C-terminal | |||||
| TM7 | Gln725 | Asn37 | Leu1028 | Pro34 | 57 |
| Pro726 | Ala38 | Val1027 | Leu35 | 57 | |
| Phe728 | Ser40 | Ile1031 | Ile37 | 57 | |
| Ala729 | Asp41 | Asp1030 | Lys38 | 57 | |
| Phe732 | Met44 | Leu1035 | Ile41 | 22 | |
| TM8 | Leu762 | Leu54 | Leu1070 | Phe70 | 22 |
| TM9 | Thr837 | Val147 | Ser1143 | Trp141 | 22 |
| Ala841 | Ala151 | Leu1147 | Ile145 | 10 | |
| Leu843 | Ile153 | Cys1149 | Ile147 | 10 | |
| TM10 | Pro866 | Pro176 | Ile1172 | Pro172 | 10 |
| Ile868 | Val178 | Cys1174 | Tyr174 | 22 | |
| Ala869 | Ser179 | Tyr1175 | Ile175 | 10 | |
| Gly872 | Ile182 | Gln1178 | Val178 | 22 | |
| TM11 | Ile937 | Ala247 | Ala1243 | Trp243 | 10 |
| Phe938 | Ser248 | Asn1244 | Asn244 | 10 | |
| Phe942 | Asp252 | Glu1248 | Phe248 | 10 | |
| Ser943 | Pro253 | Val1249 | Ala249 | 10 | |
| Thr945 | Ile255 | Met1251 | Phe251 | 10 | |
| Gln946 | Gln256 | Glu1252 | Asn252 | 10 | |
| Tyr950 | Ser260 | Ala1256 | Asp256 | 10 | |
| Ser952 | Ala262 | Val1258 | Gly258 | 10 | |
| Tyr953 | Leu263 | Val1259 | Pro259 | 10 | |
| Phe957 | Leu267 | Ala1263 | Ile263 | 10 | |
| TM12 | Leu975 | Thr285 | Gly1281 | Ala281 | 10 |
| Phe978 | Phe288 | Leu1284 | Val284 | 10 | |
| Ser979 | Ser289 | Thr1285 | Gly285 | 22 | |
| Val981 | Met291 | Ala1287 | Leu287 | 10 | |
| Val982 | Ile292 | Leu1288 | Glu288 | 10 | |
| Phe983 | Ala293 | Met1289 | Leu289 | 10 | |
| Gly984 | Leu294 | Val1290 | Leu290 | 22 | |
| Ala985 | Met295 | Ser1291 | Phe291 | 22 | |
| Met986 | Arg296 | Asn1292 | Gly292 | 22 | |
| Gly989 | Lys299 | Asn1295 | Arg295 | 22 | |
| Gln990 | Ser300 | Trp1296 | Arg296 | 22 | |
| Ser993 | Asn303 | Arg1299 | Ala299 | 22 | |
FIGURE 9.

Magnification and top view of the membrane region of P-gp conformations. Black, hot spots of the transmembrane helix 1C; orange, hot spots of the transmembrane helix 11C; brown, hot spots of the transmembrane helix 10C; red, hot spots of the transmembrane helix 12E; marine, hot spots of the transmembrane helix 11E; magenta, hot spots of the transmembrane helix 7E; green, hot spots of the transmembrane helix 6E; blue, hot spots of the transmembrane helix 5E; purple blue, hot spots of the transmembrane helix 4E. A, open inward-facing conformation. B, closed inward-facing conformation. C, outward-facing conformation.
In Fig. 9B, the same residues are highlighted in the model of the closed inward-facing conformation. As a consequence of helix motion, they now even more clearly segregate between “lower” and “upper” hot spots, reminiscent of the “hot spot model” proposed by Shilling et al. (10), especially when considering the top view of the structure.
In the outward-facing conformation of Fig. 9C, the important residues are also distributed between “lower” and “upper” hot spots. We can thus divide these hot spots into four classes: (i) “cytosolic N-terminal”, located in TM1; (ii) “cytosolic C-terminal” located in TM10-11; (iii) “exoplasmic N-terminal,” located in TM4-5-6; and (iv) “exoplasmic C-terminal,” located in TM7-11-12. These four hot spot classes include 49 of the 52 selected residues. The three outsider residues (354, 937, and 938) are rather isolated and located at the membrane interface. From the inward-facing conformations to the outward-facing one, there is a significant reorientation of the residues of helices TM1-6-7-12 due to a rotation of these helices consistent with recent interpretation of crystallographic data (58).
Molecular Modeling of SUR
SUR1 in the outward-facing conformation was modeled with the St-MsbA and Sav1866 templates (see coordinate files in the Protein Data Bank format in the supplemental material). This model (Fig. 10A) based on ∼20% sequence identity was of good quality in terms of ProSA and QMEAN scores and Ramachandran analysis (supplemental Table S2). It reveals the position in the transmembrane domains of the residues previously shown to be involved in the binding of KATP channel openers (12), namely Thr1285 and Met1289, and blockers (13), namely Ser1237 (Fig. 10B). Their orientation toward the interior of the protein appears consistent with their presumed role. As already apparent in the alignment of supplemental Fig. S2, these residues, located in the upper (TM12E) and the lower part of the transmembrane domain (Fig. 10C), correspond to P-gp residues of the above characterized hot spots.
FIGURE 10.

SUR1 structural model and three-dimensional alignment of outward-facing models of P-gp and SUR1. A, outward-facing conformation model of SUR1. B, top view of three-dimensional transmembrane location of residues involved in the binding of KATP channel openers (Thr1285 and Met1289) and blockers (Ser1237). C, structural alignment of TM11 (P-gp)/TM16 (SUR1) and TM12 (P-gp)/TM17 (SUR1). SUR1 residues involved in opener and blocker binding are represented and labeled in green, P-gp TM12E hot spots are in red, and P-gp TM11 residue is in blue.
DISCUSSION
Evidence has been reported that the sulfonylurea glibenclamide is a substrate of P-gp (16). We show here, using an ATPase measurement assay, that other ligands of the ABC protein SUR that act as KATP channel blockers and openers are also able to interact with the ABC protein P-gp. P-gp is a multidrug transporter responsible for the MDR phenotype of some tumor cells, and as such it is able to recognize and transport a broad panel of structurally unrelated molecules. The multispecific recognition of various substrates by P-gp constitutes the first step in the enzymatic cycle leading to the ATP hydrolysis-coupled expelling of the drugs out of the cell. However, multidrug recognition by P-gp, as well as by SUR, remains mysterious at the molecular level even after the publication of several structures of multidrug transporters (20–22). Here we show that useful insight can be gained by comparing the binding properties and structures of these two rather homologous ABC proteins. Our strategy was to gather new information on shared ligands, develop models based on the latest high resolution ABC protein structures, and analyze the large body of existing functional, biochemical, and mutagenetic data in order to identify the structural determinants of drug recognition by both P-gp and SUR. This permitted the discovery and characterization of hot spots for drug binding consistent with a multipharmacophoric model with a similar architecture for P-gp and SUR.
KATP Channel Blockers and P-glycoprotein
As representative KATP channel blockers, we tested glibenclamide, tolbutamide, and meglitinide. The sulfonylurea glibenclamide was the most potent (K½ ∼11 μm) at stimulating ATPase activity of P-gp. Meglitinide, which represents the non-sulfonylurea half of glibenclamide, also had a clear effect (K½ ∼300 μm). Tolbutamide, the sulfonylurea half of glibenclamide, was the weakest agonist (K½ ∼3 mm). The Hill coefficients for all three molecules were close to 1, suggesting a single binding site linked to ATPase activation.
Although concentrations for half-maximal stimulation of P-gp ATPase were lower by 2–3 orders of magnitude than those necessary to bind to SUR and block KATP channels, the rank order of potency of these compounds was the same for P-gp and SUR1 (47, 49). There appeared to be no obvious interactions between these three compounds in modulating P-gp ATPase because when tolbutamide or meglitinide, at high concentrations, was combined with glibenclamide, the resulting increase in ATPase activity was merely the sum of the increases elicited by each drug with no hint of competition. This finding could be considered as surprising because it rules out any overlap between the binding sites of these three molecules despite the large degree of resemblance of their chemical structures because the benzoic acid derivative meglitinide and the sulfonylurea tolbutamide roughly represent the two halves of the glibenclamide molecule. This observation is reminiscent of what has been reported previously on P-gp when studying steroids (27) and dihydropyridines (29). It reveals the versatility of the possible interactions of the P-gp drug binding domain with the various hydrophilic/hydrophobic recognition elements of its ligands. To explain the much higher channel blocking affinity of glibenclamide on SUR1 and its very slow dissociation rate, it has been postulated that SUR1 possesses a benzoic acid site and a sulfonylurea site, both sites being separate but close enough to allow the binding of a single molecule of glibenclamide (13, 49). Such a scheme cannot apply to P-gp because it predicts a competitive behavior between glibenclamide and tolbutamide or meglitinide that we have not observed. Instead, evidence points to separate non-interacting sites on P-gp for each of the three molecules. This conclusion implies that one should not discard the hypothesis that SUR could also harbor distinct although mutually exclusive sites for these molecules (see below).
To examine the relations between the site of action of glibenclamide on P-gp and the sites of established ligands of P-gp, we performed experiments combining both glibenclamide and either verapamil, progesterone, or vinblastine. Results are recapitulated in Fig. 6C. Verapamil reduced the glibenclamide-induced increase in ATPase activity in a dose-dependent manner without significantly shifting the glibenclamide K½. Antagonism of activation by glibenclamide and increase in basal ATPase activity were both half-maximal at about 1 μm verapamil, indicating that the same verapamil binding site mediates the two phenomena. The opposite experiment, effect of glibenclamide on verapamil-induced activity, similarly supports the existence of a single binding site for glibenclamide on P-gp. These mutually non-competitive inhibitions show that glibenclamide and verapamil bind to different, although allosterically connected, sites. The same could be concluded from the observed non-competitive antagonism of the progesterone-induced ATPase activity by glibenclamide.
Vinblastine also antagonized the glibenclamide-stimulated ATPase activity much like it did for the verapamil-stimulated ATPase (28, 51). Full inhibition was obtained at a vinblastine concentration of 10 μm that had by itself little impact on P-gp basal ATPase. Half-inhibition of the glibenclamide-stimulated ATPase activity was reached at ∼1 μm vinblastine, whereas half-inhibition of the basal activity was at 30 μm. There was also a moderate (about 2-fold) increase by 1 μm vinblastine of the K½ of glibenclamide, indicative of a competition between vinblastine and glibenclamide. Assuming a 1:1 stoichiometry for this mutual exclusion leads to a Ki value for vinblastine antagonism of glibenclamide stimulation of about 1 μm, which reinforces the existence of a high affinity vinblastine binding site on P-gp, consistent with previous observations on the inhibition by vinblastine of P-gp ATPase stimulated by various P-gp substrates (28, 29, 52). The observed inhibition of the basal ATPase activity by higher vinblastine concentrations is thus probably of a similar nature as the inhibition observed for verapamil above 50 μm or progesterone above 100 μm. The competition between glibenclamide and vinblastine could arise from a partial site overlap, leading to mutually exclusive steric constraints because they possess the common hydrophilic/hydrophobic recognition elements of pharmacophore II (11) and probably bind to the same structural determinants of P-gp (see below). The absence of competition between glibenclamide and verapamil, whereas verapamil binds close to vinblastine and displays with it mutual exclusion by steric constraints without a common recognition element (11), can be explained by the fact that glibenclamide molecule is smaller than vinblastine (Fig. 6C).
KATP Channel Openers and P-glycoprotein
Three prototypical KATP channel openers were tested: SR47063, an analog of cromakalim; P1075, an analog of pinacidil; and diazoxide. The former two are known ligands of the SUR2 isoform, probably sharing overlapping binding sites (12), whereas the latter targets another site on both SUR1 and SUR2 isoforms (48). SR47063, P1075, and diazoxide augmented P-gp ATPase activity with K½ values of 36 μm, 1.5 mm, and 0.6 mm, respectively, and Hill coefficients near 1, indicative of a single binding site on P-gp for each of them. In comparison, radioligand binding assays on the SUR2B isoform (26) have yielded dissociation constants of ∼300 nm for levcromakalim, a molecule close in structure and activity to SR47063; ∼10 nm for P1075; and ∼15 μm for diazoxide. Values for the SUR2A isoforms were about 5-fold greater, whereas for the SUR1 isoform, a dissociation constant of ∼150 μm was registered for diazoxide and in the 1 mm range for the other two openers. Whatever the SUR isoform considered, there appears to be therefore little correlation between SUR and P-gp in terms of affinities for openers and rank order of potencies.
Binding experiments on various SUR isoforms have shown an apparent competition between KATP channel openers and blockers (for a review, see Ref. 7). Such competition was not seen with P-gp between the two openers tested, P1075 and diazoxide, and glibenclamide because the effects of these openers on the glibenclamide-stimulated ATPase activity were strictly cumulative. We infer that sites for openers and blockers are located in different regions of P-gp, as they probably are for SUR (50).
Combined experiments with verapamil indicate no evidence of competition between verapamil and P1075 or diazoxide. This suggests no overlap between the sites for P1075 and diazoxide and verapamil. Further tests showed a non-competitive inhibition of progesterone-stimulated activity by P1075 or SR47063 but no interaction between progesterone and diazoxide. Therefore, diazoxide and P1075/SR47063 interact differently with P-gp than they do with SUR. The experiment concerning SR47063 points to a moderate competitive inhibition between verapamil and SR47063 and to a clear inhibition by vinblastine of the SR47063-stimulated activity, although the type of inhibition between these two drugs could not be determined. The existence of common recognition elements in the molecular structures of SR47063 and verapamil indicates that SR47063 may belong to the pharmacophore I, according to Garrigues et al. (11). This is reinforced by the close structural resemblance of SR47063 with rhodamine 123, a compound for which recognition by P-gp has been determined to be competitive with verapamil9 and to correspond to pharmacophore I.10 Due to the incomplete overlap with verapamil, SR47063 could sterically interfere with vinblastine, but this is difficult to ascertain experimentally. Fig. 6C summarizes these observations in relation to the two pharmacophores described previously on P-gp. The data suggest a striking functional homology between SUR and P-gp for recognition of the tested ligands, especially glibenclamide and SR47063.
Implications for the Molecular Mechanisms of Multispecific Ligand Recognition
Our results demonstrate a correlation between SUR and P-gp in terms of shared ligands. Does this correlation arise from the promiscuous nature of these ligands, which have a tendency to lack specificity (glibenclamide, for instance, has been postulated to be a universal ABC protein ligand (16)), or from the promiscuous nature of the target sites or from a true structural homology between the target proteins, SUR and P-gp? Because the structural data on eukaryotic ABC proteins are limited to the recently published structure of mouse P-gp in one conformation (22), we performed an in silico structural modeling of P-gp and SUR, based on the various crystallographic structures of the bacterial ABC transporters MsbA and Sav1866. To validate this choice of template homologues, we performed a phylogenetic analysis of a large set of membrane ABC proteins using a global analysis method based on a maximum likelihood algorithm that is less dependent on varying sequence lengths and that provides a quantitative measure of protein homology (59). This analysis positioned MsbA and Sav1866 close to P-gp in the ABCB branch of the phylogenetic tree. In addition, the SUR-containing ABCC branch was the closest to the ABCB branch. This supports the choice of MsbA and Sav1866 as structural homologues. Furthermore, the various satisfactory quantitative assessments of the models obtained for P-gp as well as for SUR confirmed the pertinence of this choice. Indeed, the models correctly predict (i) the intramembrane position of the various residues known to be involved in substrate recognition and (ii) the orientation of their side chains toward the interior of the protein, where they delimit a variably sized “chamber,” depending on the conformation of the protein.
In the case of P-gp, we modeled three different conformations, open inward-facing, closed inward-facing, and outward-facing, which are supposed to represent successive states of the enzyme during its transport cycle (20). The comparison between the open conformation and the two others revealed that residues involved in substrate recognition are not spatially distributed evenly but segregate between several hot spots mainly located either in the “lower”/“cytosolic” or “upper”/“exoplasmic” region of the phospholipid membrane. Analysis of the abundant mutagenetic and chemical labeling data for verapamil and vinblastine binding shows that the set of hot spots 4E/5E/6E/7E/10C/11C/12E could constitute anchoring points for verapamil and the ligands belonging to its competition class; similarly, the set of hot spots 1C/6E/11C/11E could constitute anchoring points for vinblastine and its competition class. Thus, these two sets of hot spots correspond to pharmacophores I and II, respectively (11), which provides a valuable link between the enzymological analysis and the structural analysis that reinforces the multisite model for the molecular mechanisms involved in multispecific drug recognition. This rough description of the drug recognition by P-gp obviously cannot replace a deep and careful analysis of ligand binding using in silico docking techniques, which would require a massive effort beyond the scope of this report. However, this allows us to propose the following interpretations. (i) The two hot spots, 6E and 11C, shared by verapamil and vinblastine, could represent the common point between the two pharmacophores that we have previously reported (11). (ii) The higher number of hot spots forming the verapamil recognition pattern could indicate that P-gp is able to bind simultaneously two verapamil molecules, of limited size, as observed for QZ-59SSS (22), but only one vinblastine molecule, which is much larger. (iii) The residues that interact with the two peptides QZ-59RRR and QZ-59SSS in the crystallographic structure of P-gp are mostly located outside the hot spots, which is not surprising because these peculiar ligands do not share the hydrophilic/hydrophobic recognition elements of either verapamil or vinblastine (not shown). (iv) By comparing the three P-gp conformations, it appears that some of the hot spots do not move within the membrane plane (TM6, -7, -8, and -12), whereas others do (TM1, -4, -5, -10, and -11); this is true for both sets of hot spots linked to verapamil and vinblastine and is consistent with the required affinity alteration for the transported substrate during the enzymatic turnover of a typical active transporter. (v) The location of the hot spots within the membrane bilayer, found both at the level of the cytosolic leaflet and the exoplasmic leaflet, is consistent with the two dominant functional models proposed in the literature for P-gp (i.e. a membrane floppase and a “vacuum-cleaner” expelling the transported substrate directly out of the membrane).
In the case of SUR1, we built a structural model for the outward-facing conformation, which is supposed to be the physiologically relevant enzymatic state because it is the nucleotide-bound form. SR47063 binding to SUR1 involves a protein region that corresponds to the “upper sites” of P-gp, namely residues Thr1285 and Met1289 (9, 12), that are equivalent to P-gp residues Ser979 and Phe983. Glibenclamide binding to SUR1 involves Ser1237 (13), equivalent to P-gp Ser931 located at the membrane interface just beyond the “lower sites,” which are compatible with the size of the average conformation of free glibenclamide (18 Å). Thus, the two ligands present binding sites located in corresponding regions of SUR and P-gp when aligned. As a whole, these two multispecific ABC membrane proteins appear to harbor a common architecture of binding sites for a pattern of structurally diverse ligands.
Because the openers can augment the ATPase activity of SUR2 (19), we have hypothesized that these ligands could act as “pseudotransport” substrates of SUR2 (12). The finding that the openers SR47063 and P1075 stimulate the ATPase activity of P-gp suggests not only structural but also functional homology between P-gp and SUR.
Beyond these similarities, SUR and P-gp display distinct characteristics for drug recognition and binding: (i) the variety of ligands of P-gp exceeds that of SUR (60–62); (ii) P-gp binds larger ligands (up to 1250 Da (60)) than SUR (up to 500 Da for glibenclamide (62)); (iii) the smallest P-gp ligands (in the size range of SUR ligands) have poor affinities (beyond the micromolar range (11)), whereas the smallest SUR ligands can have high affinity; (iv) P-gp ligands often display non-competitive interactions except if they have sufficient size (>∼700 Da) (11), whereas SUR ligands are generally always competitive one with each other, even when they bind distinct sites (47, 63). All of these features can be reconciled in a model in which the volume of the “cavity” forming the P-gp multidrug binding domain is much larger than that presented by SUR. Schematically, in P-gp, the multidrug binding domain forms a continuous multispecific pocket, whereas in SUR, it appears rather restrained to (at least) two discrete multispecific sites, although the two multidrug binding domains appear to be built around common hot spots.
The phylogenetic analysis of the membrane ABC proteins, supported by the high values of bootstrap at several branches, shows the relative proximity of the various subfamilies. This analysis supports a close structural and functional relationship between P-gp, representative of the ABCB branch, and SUR, representative of the ABCC branch. In contrast, this cannot be extrapolated readily to the D branch, and a fortiori to the more remote A and G branches.
Therapeutical Implications
Several KATP channel blockers and openers are in clinical use for a variety of conditions, including diabetes and hypertension (8, 64). Because P-gp is present and active at the intestinal and blood-brain barriers as well as in biliary and urinary elimination systems (17, 18), knowing if and how a molecule is transported by P-gp is useful in anticipating pharmacokinetics and tissue distribution and potential side effects due to drug-drug interactions. In this work, we have measured the effects of blockers and openers on P-gp ATPase activity. The correlation between ATPase activity and transport is complex and not yet fully resolved (65, 66). Molecules that augment P-gp ATPase activity are likely to be transported, although ATPase activation does not necessarily imply a net transport (67). Of the molecules tested here, direct evidence of transport has only been supplied for glibenclamide (16). Such evidence would be difficult to obtain for the other molecules, which are either not available in labeled form or have weaker affinities. If transport indeed occurs, one can infer that the biodisponibility of these KATP channel modulators could be altered and that interferences with P-gp transport of co-administered drugs could lead to drug-drug interaction and eventual side effects. This is expected to be a significant factor for compounds that have comparable affinities for P-gp and their intended target, SUR. This is the case for SR47063, which activates muscle KATP channels at micromolar concentrations (68) and interacts with P-gp at the same concentrations. In contrast, the other compounds tested have much greater affinities for KATP channels than for P-gp, as exemplified by glibenclamide, which blocks pancreatic KATP channels at nanomolar concentrations and activates P-gp ATPase activity at micromolar concentrations. We may anticipate that the capacity of these compounds to act across barriers should be high.
Supplementary Material
Acknowledgments
We are grateful to Dr. P. Gautier (Sanofi Recherche, Montpellier, France) for the generous gift of SR 47063, Dr. L. Billerup (Leo Pharmaceutical Products, Copenhagen, Denmark) for P1075, and Dr. J. Pünter (Aventis Pharma, Frankfurt, Germany) for meglitinide.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and S2 and Figs. S1–S4.
S. Martin and S. Orlowski, unpublished results.
N. Loiseau and S. Orlowski, unpublished results.
- ABC
- ATP-binding cassette
- KATP channel
- ATP-sensitive K+
- SUR
- sulfonylurea receptor
- TM(D)
- transmembrane (domain)
- NBD
- nucleotide binding domain
- P-gp
- P-glycoprotein.
REFERENCES
- 1. Higgins C. F. (1992) Annu. Rev. Cell Biol. 8, 67–113 [DOI] [PubMed] [Google Scholar]
- 2. Holland I. B., Blight M. A. (1999) J. Mol. Biol. 293, 381–399 [DOI] [PubMed] [Google Scholar]
- 3. Vasiliou V., Vasiliou K., Nebert D. W. (2009) Hum. Genomics 3, 281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Inagaki N., Gonoi T., Clement J. P., 4th, Namba N., Inazawa J., Gonzalez G., Aguilar-Bryan L., Seino S., Bryan J. (1995) Science 270, 1166–1170 [DOI] [PubMed] [Google Scholar]
- 5. Antcliff J. F., Haider S., Proks P., Sansom M. S., Ashcroft F. M. (2005) EMBO J. 24, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi N. Q., Ye B., Makielski J. C. (2005) J Mol. Cell Cardiol. 39, 51–60 [DOI] [PubMed] [Google Scholar]
- 7. Moreau C., Prost A. L., Dérand R., Vivaudou M. (2005) J Mol. Cell Cardiol. 38, 951–963 [DOI] [PubMed] [Google Scholar]
- 8. Jahangir A., Terzic A. (2005) J. Mol. Cell Cardiol. 39, 99–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moreau C., Gally F., Jacquet-Bouix H., Vivaudou M. (2005) Mol. Pharmacol. 67, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 10. Shilling R. A., Venter H., Velamakanni S., Bapna A., Woebking B., Shahi S., van Veen H. W. (2006) Trends Pharmacol. Sci. 27, 195–203 [DOI] [PubMed] [Google Scholar]
- 11. Garrigues A., Loiseau N., Delaforge M., Ferté J., Garrigos M., André F., Orlowski S. (2002) Mol. Pharmacol. 62, 1288–1298 [DOI] [PubMed] [Google Scholar]
- 12. Moreau C., Jacquet H., Prost A. L., D'hahan N., Vivaudou M. (2000) EMBO J. 19, 6644–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ashfield R., Gribble F. M., Ashcroft S. J., Ashcroft F. M. (1999) Diabetes 48, 1341–1347 [DOI] [PubMed] [Google Scholar]
- 14. Sheppard D. N., Welsh M. J. (1992) J Gen. Physiol. 100, 573–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Payen L., Delugin L., Courtois A., Trinquart Y., Guillouzo A., Fardel O. (2001) Br. J. Pharmacol. 132, 778–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Golstein P. E., Boom A., van Geffel J., Jacobs P., Masereel B., Beauwens R. (1999) Pflugers. Arch. 437, 652–660 [DOI] [PubMed] [Google Scholar]
- 17. Fromm M. F. (2004) Trends Pharmacol. Sci. 25, 423–429 [DOI] [PubMed] [Google Scholar]
- 18. Ambudkar S. V., Dey S., Hrycyna C. A., Ramachandra M., Pastan I., Gottesman M. M. (1999) Annu. Rev. Pharmacol. Toxicol. 39, 361–398 [DOI] [PubMed] [Google Scholar]
- 19. Bienengraeber M., Alekseev A. E., Abraham M. R., Carrasco A. J., Moreau C., Vivaudou M., Dzeja P. P., Terzic A. (2000) FASEB J. 14, 1943–1952 [DOI] [PubMed] [Google Scholar]
- 20. Ward A., Reyes C. L., Yu J., Roth C. B., Chang G. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dawson R. J., Locher K. P. (2006) Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 22. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garrigos M., Belehradek J., Jr., Mir L. M., Orlowski S. (1993) Biochem. Biophys. Res. Commun. 196, 1034–1041 [DOI] [PubMed] [Google Scholar]
- 24. de Wet H., Fotinou C., Amad N., Dreger M., Ashcroft F. M. (2010) FEBS J. 277, 2654–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Wet H., Mikhailov M. V., Fotinou C., Dreger M., Craig T. J., Vénien-Bryan C., Ashcroft F. M. (2007) FEBS J. 274, 3532–3544 [DOI] [PubMed] [Google Scholar]
- 26. Schwanstecher M., Sieverding C., Dörschner H., Gross I., Aguilar-Bryan L., Schwanstecher C., Bryan J. (1998) EMBO J. 17, 5529–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orlowski S., Mir L. M., Belehradek J., Jr., Garrigos M. (1996) Biochem. J. 317, 515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garrigos M., Mir L. M., Orlowski S. (1997) Eur. J. Biochem. 244, 664–673 [DOI] [PubMed] [Google Scholar]
- 29. Pascaud C., Garrigos M., Orlowski S. (1998) Biochem. J. 333, 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bairoch A., Apweiler R., Wu C. H., Barker W. C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Martin M. J., Natale D. A., O'Donovan C., Redaschi N., Yeh L. S. (2005) Nucleic Acids Res. 33, D154–D159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Le T., Tseng T. T., Saier M. H., Jr. (1999) Mol. Membr. Biol. 16, 173–179 [DOI] [PubMed] [Google Scholar]
- 32. Marchler-Bauer A., Anderson J. B., DeWeese-Scott C., Fedorova N. D., Geer L. Y., He S., Hurwitz D. I., Jackson J. D., Jacobs A. R., Lanczycki C. J., Liebert C. A., Liu C., Madej T., Marchler G. H., Mazumder R., Nikolskaya A. N., Panchenko A. R., Rao B. S., Shoemaker B. A., Simonyan V., Song J. S., Thiessen P. A., Vasudevan S., Wang Y., Yamashita R. A., Yin J. J., Bryant S. H. (2003) Nucleic Acids Res. 31, 383–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morgenstern B., Dress A., Werner T. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 12098–12103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guindon S., Gascuel O. (2003) Syst. Biol. 52, 696–704 [DOI] [PubMed] [Google Scholar]
- 35. Felsenstein J. (1985) Evolution 39, 783–791 [DOI] [PubMed] [Google Scholar]
- 36. Efron B. (1982) The Jackknife, the Bootstrap and Other Resampling Plans, Society for Industrial and Applied Mathematics, Philadelphia, PA [Google Scholar]
- 37. Page R. D. (1996) Comput. Appl. Biosci. 12, 357–358 [DOI] [PubMed] [Google Scholar]
- 38. Clamp M., Cuff J., Searle S. M., Barton G. J. (2004) Bioinformatics 20, 426–427 [DOI] [PubMed] [Google Scholar]
- 39. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sali A., Potterton L., Yuan F., van Vlijmen H., Karplus M. (1995) Proteins 23, 318–326 [DOI] [PubMed] [Google Scholar]
- 41. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 42. Hooft R. W., Vriend G., Sander C., Abola E. E. (1996) Nature 381, 272. [DOI] [PubMed] [Google Scholar]
- 43. Sippl M. J. (1993) Proteins 17, 355–362 [DOI] [PubMed] [Google Scholar]
- 44. Davis I. W., Murray L. W., Richardson J. S., Richardson D. C. (2004) Nucleic Acids Res. 32, W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Benkert P., Tosatto S. C., Schomburg D. (2008) Proteins 71, 261–277 [DOI] [PubMed] [Google Scholar]
- 46. Humphrey W., Dalke A., Schulten K. (1996) J Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 47. Dörschner H., Brekardin E., Uhde I., Schwanstecher C., Schwanstecher M. (1999) Mol. Pharmacol. 55, 1060–1066 [DOI] [PubMed] [Google Scholar]
- 48. D'hahan N., Moreau C., Prost A. L., Jacquet H., Alekseev A. E., Terzic A., Vivaudou M. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 12162–12167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gribble F. M., Tucker S. J., Seino S., Ashcroft F. M. (1998) Diabetes 47, 1412–1418 [DOI] [PubMed] [Google Scholar]
- 50. Gribble F. M., Reimann F. (2002) Biochem. Soc. Trans. 30, 333–339 [DOI] [PubMed] [Google Scholar]
- 51. Litman T., Zeuthen T., Skovsgaard T., Stein W. D. (1997) Biochim. Biophys. Acta. 1361, 169–176 [DOI] [PubMed] [Google Scholar]
- 52. Megard I., Garrigues A., Orlowski S., Jorajuria S., Clayette P., Ezan E., Mabondzo A. (2002) Brain Res. 927, 153–167 [DOI] [PubMed] [Google Scholar]
- 53. Rosenberg M. F., Kamis A. B., Callaghan R., Higgins C. F., Ford R. C. (2003) J. Biol. Chem. 278, 8294–8299 [DOI] [PubMed] [Google Scholar]
- 54. Lee J. Y., Urbatsch I. L., Senior A. E., Wilkens S. (2008) J. Biol. Chem. 283, 5769–5779 [DOI] [PubMed] [Google Scholar]
- 55. O'Mara M. L., Tieleman D. P. (2007) FEBS Lett. 581, 4217–4222 [DOI] [PubMed] [Google Scholar]
- 56. Ravna A. W., Sylte I., Sager G. (2007) Theor. Biol. Med. Model. 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Loo T. W., Bartlett M. C., Clarke D. M. (2006) Biochem. J. 399, 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gutmann D. A., Ward A., Urbatsch I. L., Chang G., van Veen H. W. (2010) Trends Biochem. Sci. 35, 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Felsenstein J. (1981) J. Mol. Evol. 17, 368–376 [DOI] [PubMed] [Google Scholar]
- 60. Avendaño C., Menéndez J. C. (2002) Curr. Med. Chem. 9, 159–193 [DOI] [PubMed] [Google Scholar]
- 61. Bryan J., Crane A., Vila-Carriles W. H., Babenko A. P., Aguilar-Bryan L. (2005) Curr. Pharm. Des. 11, 2699–2716 [DOI] [PubMed] [Google Scholar]
- 62. Coghlan M. J., Carroll W. A., Gopalakrishnan M. (2001) J Med. Chem. 44, 1627–1653 [DOI] [PubMed] [Google Scholar]
- 63. Löffler-Walz C., Quast U. (1998) Br. J. Pharmacol. 123, 1395–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nagashima K., Takahashi A., Ikeda H., Hamasaki A., Kuwamura N., Yamada Y., Seino Y. (2004) Diabetes Res. Clin. Pract. 66, Suppl. 1, S75–S78 [DOI] [PubMed] [Google Scholar]
- 65. Sharom F. J. (1997) J. Membr. Biol. 160, 161–175 [DOI] [PubMed] [Google Scholar]
- 66. Al-Shawi M. K., Polar M. K., Omote H., Figler R. A. (2003) J. Biol. Chem. 278, 52629–52640 [DOI] [PubMed] [Google Scholar]
- 67. Polli J. W., Wring S. A., Humphreys J. E., Huang L., Morgan J. B., Webster L. O., Serabjit-Singh C. S. (2001) J. Pharmacol. Exp. Ther. 299, 620–628 [PubMed] [Google Scholar]
- 68. Forestier C., Pierrard J., Vivaudou M. (1996) J Gen. Physiol. 107, 489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.