

Abstract
The E6 protein of human papillomavirus (HPV) exhibits complex interaction patterns with several host proteins, and their roles in HPV-mediated oncogenesis have proved challenging to study. Here we use several biophysical techniques to explore the binding of E6 to the three PDZ domains of the tumor suppressor protein synapse-associated protein 97 (SAP97). All of the potential binding sites in SAP97 bind E6 with micromolar affinity. The dissociation rate constants govern the different affinities of HPV16 and HPV18 E6 for SAP97. Unexpectedly, binding is not mutually exclusive, and all three PDZ domains can simultaneously bind E6. Intriguingly, this quaternary complex has the same apparent hydrodynamic volume as the unliganded PDZ region, suggesting that a conformational change occurs in the PDZ region upon binding, a conclusion supported by kinetic experiments. Using NMR, we discovered a new mode of interaction between E6 and PDZ: a subset of residues distal to the canonical binding pocket in the PDZ2 domain exhibited noncanonical interactions with the E6 protein. This is consistent with a larger proportion of the protein surface defining binding specificity, as compared with that reported previously.
Keywords: Protein Domains, Protein Structure, Protein-Protein Interactions, Tumor Suppressor, Viral Protein, E6 Protein, PDZ Domain
Introduction
The E6 protein of certain human papillomaviruses (HPVs)4 (1–3) plays an active role in the development and pathogenesis of several types of cancers, with cervical cancer being the most prevalent type (4, 5). HPV uses its E6 and E7 proteins, which interact with and inhibit several key proteins, to hijack the erstwhile highly controlled cellular environment (4, 6–8). HPVs are broadly divided into two main groups: “high risk” and “low risk,” based on their occurrence in cervical cancer (9), and HPV16 and 18 are the two most common high risk types of HPV. One of the hallmarks of HPV pathogenesis is the E6-mediated inactivation of the p53 tumor suppressor protein. However, a number of studies have shown the existence of a p53-independent mechanism that leads to uncontrolled cell growth (10–12). For example, the E6 protein of the high risk (but not the low risk type) HPV interacts with the PDZ (PSD-95/Discs large/ZO-1) domains of several proteins such as SAP97 (synapse-associated protein 97), hScrib, MAGI, and MUPP1, through a conserved C-terminal motif (13–17). These PDZ-containing proteins participate in the maintenance of cell-cell contacts and cell polarity and are often found at tight and adherent junctions (18). The tumor suppressor protein SAP97 contains a series of three consecutive PDZ domains (PDZ1, PDZ2, and PDZ3), one Src homology 3 domain, and one guanylate kinase-like domain. The mechanism by which the E6 protein interacts with these three PDZ domains is not well understood. It has been shown through cell pulldown assays that only the PDZ2 domain of SAP97 interacts with the E6 protein of HPV16, whereas the E6 protein of HPV18 interacts with all three PDZ domains (13, 15). Biochemical characterization of the PDZ-E6 interaction have shown that discrimination between the E6 proteins of HPV16 and HPV18 is due to the C-terminal amino acid, which is Leu-151 in HPV16 and Val-158 in HPV18 (19). Because PDZ domains are often organized in arrays, as in SAP97 (20), this poses the question: how do such repeats affect the binding to the HPV E6 protein, if at all? For example, steric occlusion, allostery, or cooperativity in binding could regulate SAP97-E6 interactions and further contribute to differential specificity toward HPV E6 variants.
To dissect these issues, we analyzed the interaction between HPV E6 and six SAP97 PDZ domain constructs: PDZ1; PDZ2; PDZ3; the tandem constructs PDZ12 and PDZ23; and PDZ123, which contains all three concatenated PDZ domains. We used the complete C-terminal domain of E6 and the entire PDZ region of SAP97 and combined both equilibrium and kinetic experiments in solution (21, 22).
MATERIALS AND METHODS
cDNA Constructs
PDZ domain constructs used in this study contained either 12 extra residues (MHHHHHLVPRGS) (PDZ1, PDZ2, and PDZ3) or 10 extra residues (MHHHHHPRGS) (PDZ12, PDZ23, and PDZ123) at the N terminus. The E6WT (C-terminal domain of HPV16 E6) and mutant (E6L151V) contained four Cys to Ser mutations as described previously (23). The cDNA coding for E6WT (residues 80–151) was amplified by PCR and subcloned as a fusion with an Escherichia coli lipoyl domain (with a thrombin digestion site) in a modified pRSET vector (Invitrogen). The point mutation L151V (referred to hereafter as E6L151V) was introduced in the cDNA of E6 using inverse PCR. The cDNA coding for PDZ1 (residues 220–311), PDZ2 (residues 311–407), PDZ3 (residues 461–553), PDZ12 (residues 220–407) (PDZ1 and PDZ2 in tandem), PDZ23 (residues 311–553) (PDZ2 and PDZ3 in tandem), and PDZ123 (residues 220–553) (a concatenation of PDZ1, PDZ2, and PDZ3) of SAP-97 were amplified by PCR and subcloned as a His-tagged fusion in a modified pRSET vector (Invitrogen). To avoid cysteine disulfide bridge formation, Cys-275 and Cys-378 in the PDZ1 and the PDZ2 domains, respectively, were mutated to alanine.
Expression and Purification
Expression of PDZ domains was as described previously (24, 25). For NMR experiments, cells containing the plasmid of PDZ2 were grown in M9 minimal medium with 15NH4Cl and/or [13C]d-glucose as the sole source for nitrogen and carbon, respectively. The E6WT and E6L151V proteins were expressed as lipoyl domain fusion protein (see “cDNA Constructs”). Expression was done overnight at 25 °C. The cells were harvested by spinning at 7,000 × g for 10 min and resuspended in purification buffer (50 mm Tris/HCl, 400 mm NaCl for PDZ domains and 50 mm Tris/HCl, 400 mm NaCl, 0.2% 2-mercaptoethanol for E6 proteins). The cells were lysed by sonication and thereafter centrifuged at 35,000 × g for 1 h. The supernatant was filtered and loaded onto nickel (II)-charged chelating Sepharose FF column (Amersham Biosciences), equilibrated with purification buffer as above, and washed with 400 ml of the same buffer. The bound protein was eluted with 250 mm imidazole, pH 7.9, in 0.2% 2-mercaptoethanol in aliquots of 8 ml. Fractions containing proteins were pooled and further purified as follows: PDZ proteins were concentrated and purified on G-50 Sephadex (GE Healthcare) (PDZ1, PDZ2, and PDZ3) or G-200 Superdex (GE Healthcare) (PDZ12, PSZ23, and PDZ123) gel filtration chromatography column equilibrated with 100 mm potassium phosphate, pH 7.0. E6 proteins were first purified on an anion exchange column equilibrated with 50 mm Tris/HCl, pH 8.5, in 0.2% 2-mercaptoenthanol. Pure lipo-E6 was eluted at a gradient of 0–500 mm NaCl, 50 mm Tris/HCl, pH 8.5, in 0.2% 2-mercaptoethanol. The lipoyl domain from the fractions containing purified lipo-E6 was then digested out with thrombin for 3 h at 37 °C, filtered, and loaded on a cation exchange column equilibrated with 50 mm Tris/HCl, pH 7.5, in 0.2% 2-mercaptoethanol. Pure E6 was eluted by a gradient of 0–500 mm NaCl in 50 mm Tris/HCl, pH 8.5, 0.2% 2-mercaptoethanol. The purity of the PDZ and E6 proteins were checked on SDS-PAGE stained with Coomassie Brilliant Blue, and their identity was confirmed by MALDI-TOF mass spectrometry. The amount of zinc bound to E6 was determined by a commercially available inactively coupled plasma atomic emission spectroscopy platform (ALS Scandinavia AB). Zinc was present in approximately a 1:1 molar ratio with the E6 protein, as reported previously (26). The protein concentrations were determined by amino acid analysis. The stability of and secondary structure of the expressed proteins were assessed using far UV CD on a Jasco J-810 Spectropolarimeter. CD spectra were recoded between 200 and 260 nm at 25 °C with 20–40 μm protein in 50 mm potassium phosphate, pH 7.5.
Isothermal Titration Calorimetry (ITC), Fluorometric, and Stopped Flow Experiments
Calorimetric and fluorometric experiments were performed at 10 °C in 50 mm potassium phosphate buffer, pH 7.5. E6 protein (twenty 2-μl injections at 180-s intervals; stirring speed of 1000 rpm) was titrated into the PDZ solution using a microcalorimeter (ITC200; Microcal). The experiments were designed so that the c values were within 1–1000 (c value = Ka × [Protein] × N, where Ka is the equilibrium association constant, [Protein] is the protein concentration, and N is the stoichiometry of the binding event). Heats of dilution were initially determined by titrating the E6 into buffer and buffer into PDZ protein, respectively. ORIGIN 7.0 (Microcal) was used to determine the thermodynamic properties of ligand binding using nonlinear least squares fitting assuming a single-site model. All of the values were the averages of two to five individual experiments. Equilibrium fluorometric measurements were performed by measuring the increase in tryptophan fluorescence upon binding in an SLM 4800 spectrofluorimeter (SLM Instruments). Excitation was at 290 nm, and emission was at 320–360 nm. To determine the equilibrium constants for the E6-PDZ interaction, PDZ concentration was varied while keeping the concentration of E6 protein constant at 3 μm. The data were then fitted to the standard equation for equilibrium binding to obtain the Kd. All of the stopped flow binding experiments were done on an SX-20MV stopped flow spectrometer (Applied Photophysics, Leatherhead, UK). Fluorescence was monitored using the increase in tryptophan emission (excitation, 295 nm; emission, 330 ± 30 nm). To determine the rate constants for the E6-PDZ interaction, the PDZ concentration was varied at a constant concentration of E6 (3 μm). When estimating the amount of binding sites in concatenated PDZ domains, the E6 concentration was varied at a constant concentration of PDZ (3 μm). Kinetic traces from time-resolved E6-PDZ binding experiments were fitted to single and double exponential functions,
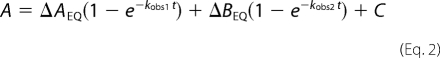 |
where A is the signal recorded with time t, ΔAEQ and ΔBEQ, are the amplitudes of the respective phase, and kobs is the observed rate constants. The kobs values were plotted versus PDZ or E6 concentration and fitted to the general equation for reversible association of two molecules (27, 28).
kon is the association or on rate constant, koff is the dissociation or off rate constant, and [A]0 and n are the initial concentrations of the varied and constants species, respectively.
Size Exclusion Chromatography-Multi-angle Laser Light Scattering (SEC-MALS)
MALS measurements were performed using 50 mm sodium phosphate buffer, pH 7.5 (ionic strength corrected to 150 mm using NaCl). SEC-MALS measurements were executed using a Shimadzu HPLC (to facilitate analytical SEC) daisy-chained with a Wyatt Technologies Dawn HeleosII multi-angle light scattering detector and Optilab dRX refractometer (Wyatt Tech). A column oven combined with the use of Peltier-controlled autosampler, light scattering detector and refractometer effected temperature thermostatting to 20 °C (± 0.1 °C) throughout the set-up. Test injections of 1 mg/ml BSA solutions were used to determine the delay volumes between instruments and the effects of band-broadening therein, using the Astra software as per the manufacturer's recommendations (Wyatt Tech).
Two different analytical size exclusion chromatography columns were used in these studies: (i) a 8 × 300-mm silica-based KW802.5 size exclusion chromatography column (Shodex) for studies of PDZ12, PDZ23, PDZ123, and complexes with E6L151V and (ii) a polymer-based 10 × 300-mm Superdex 75 column (GE Healthcare) to study the apo form of E6. SEC-MALS measurements typically involved a 25–50-μl injection of a given protein at a flow rate of 0.5 ml/min. Ligand-free forms of PDZ12, PDZ23, and PDZ123 were loaded onto SEC columns at a final protein concentration of 233 μm, whereas free E6L151V was studied over a wide concentration range of 500–1340 μm. PDZ123-E6L151V complexes were formed by incubating ∼120 μm PDZ123 with either a 2- or 3-fold molar excess of apo-E6L151V, for at least 2 h at 20 °C prior to SEC-MALS measurements.
Molar masses were determined by measuring the intensity of scattered light at 18 different scattering angles, as a function of protein concentration and elution volume (from the SEC column). Thereafter, the intrinsic instrumental base line for each data channel was subtracted, and the molar mass across a given two-dimensional slice of the elution profile was determined using the ASTRA software (Wyatt Tech). To ensure robust data fitting, for each elution peak the apparent molar mass was determined as a function of the width of the fitted window, and also for the front end, the center and trailing edge of the peaks were compared. In general, the studied materials were highly homogeneous and mono-disperse, with the measured molar masses being highly reproducible and independent of the data window used for curve fitting. The exception to this was the unbound form of E6L151V, which tended to form heterogeneous, higher order oligomers, and aggregates, as reported previously (29, 30).
NMR Titration
15N-1H titration experiments were acquired on Varian INOVA 600 and 800 MHz spectrometers equipped with cryogenically cooled and room temperature probes, respectively, at 310 K in 50 mm potassium phosphate pH 6.9. Protein samples were dissolved in 10% D2O, and two-dimensional 15N HSQC spectra for PDZ2 (300 μm) were recorded with increasing concentrations of E6L151V or E6WT (0–290 μm). For assignment purposes 15N NOESY-HSQC, 15N TOCSY-HSQC, HNCACB, CBCA(CO)NH, HN(CA)CO, and HNCO experiments were recorded for the unbound PDZ2 using pulse field gradient enhanced NMR spectroscopy. The data processing and analysis were done with NMRpipe (31) and Sparky NMR assignment and integration software, respectively. Backbone assignments for the free state were essentially complete. Residues Val-313, Leu-329, and Asn-376 could, however, not be assigned, presumably because of severe line broadening. 15N and 1HN assignments for the bound states were obtained by following the peaks in the titration experiments. Analysis and calculation of chemical shift changes were performed using Equation 4 and as described (33).
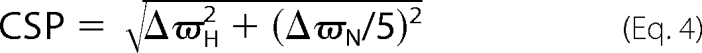 |
CSP is the combined chemical shift change, ΔϖH is the change for 1H, and ΔϖN is the change for 15N in units of ppm. NMR assignment data has been deposited in the BioMagResBank (accession number 17373).
RESULTS
E6 Binds to All Three PDZ Domains of SAP97
First we investigated whether each PDZ domain, PDZ1, PDZ2, and PDZ3, individually binds to the C-terminal domain of HPV16 E6. Standard equilibrium binding and time-resolved stopped flow experiments were used to determine affinity constants for these interactions. The E6 protein contains a tryptophan at position 132, which serves as a convenient fluorescent reporter group for binding studies. An increase in tryptophan fluorescence was recorded when either E6WT or E6L151V was mixed with PDZ1, PDZ2, and PDZ3, respectively. Experiments with increasing concentrations of PDZ resulted in saturation of the fluorescence signal at high concentration of PDZ, showing that each of PDZ1, PDZ2, and PDZ3 interacts with E6WT in vitro (not shown). The E6L151V mutant has a higher affinity toward the PDZ domains as compared with the E6WT and has similar immortalization properties as the HPV18 derived E6 protein (19). Thus, we decided to use this mutant as a mimic for HPV18 E6 for subsequent experiments. We performed ITC binding experiments to independently characterize E6L151V-PDZ interactions and to probe the thermodynamics of binding (Table 1 and Fig. 1). As shown in Table 1, the binding was enthalpy-driven for PDZ2 and PDZ3, whereas for PDZ1, a favorable entropy was observed. Among the three PDZ domains of SAP97, PDZ3 had the highest affinity for the E6L151V. Importantly, when performing ITC experiments on the tandem constructs PDZ12, PDZ23, and PDZ123, the binding stoichiometries agreed well with a model where each PDZ domain can bind one E6L151V molecule (Table 1).
TABLE 1.
Isothermal titration calorimetric data for the interaction between E6L151V and the respective PDZ domains
Stoichiometry from stopped flow is also included (see “Results”).
| Protein | ΔH | −TΔS | Kda | ΔG | Stoichiometry | Stoichiometry, stopped flow |
|---|---|---|---|---|---|---|
| kcal mol−1 | kcal mol−1 | μm | kcal mol−1 | |||
| PDZ1 | −5.6 ± 0.4 | −1.8 ± 0.5 | 1.7 ± 0.4 | −7.5 ± 0.1 | 1.0 ± 0.1 | 1.5 |
| PDZ2 | −9.3 ± 0.3 | 0.9 ± 0.3 | 0.34 ± 0.02 | −8.3 ± 0.1 | 1.3 ± 0.1 | 0.7 |
| PDZ3 | −9.9 ± 0.2 | 0.7 ± 0.3 | 0.08 ± 0.01 | −9.2 ± 0.3 | 0.8 ± 0.1 | 0.8 |
| PDZ12 | −9.4 ± 0.5 | 1.4 ± 0.5 | 0.7 ± 0.01a | −8.0 ± 0.1 | 1.7 ± 0.1 | 2.4 |
| PDZ23 | −9.7 ± 0.1 | 0.7 ± 0.1 | 0.1 ± 0.01a | −9.0 ± 0.1 | 2.4 ± 0.1 | 1.7 |
| PDZ123 | −9.5 ± 0.6 | 1.4 ± 0.6 | 0.5 ± 0.01a | −8.2 ± 0.1 | 3.6 ± 0.2 | 2.8 |
a Apparent average Kd over all binding sites.
FIGURE 1.

Equilibrium binding experiments. a–c, ITC data for PDZ3 (a), PDZ12 (b), and PDZ123 (c) with E6L151V at 10 °C. Top panels, raw data. Bottom panels, integrated titration curves. See Table 1 for fitted parameters. d and e, SEC-MALS analysis of PDZ123 in the presence and absence of a 3-fold molar excess of HPV E6. d, the elution volumes from an analytical Superdex 75 10/30 column were very similar for the apo- and saturated PZD123-E6 complex (red and black traces, respectively). For clarity, the UV signals of PDZ123 peaks are normalized to have similar intensities. The additional peak at ∼19.4 ml corresponds to unbound HPV E6 (and reductants in the E6 samples used to prevent thiol-mediated oligomerization). e, SEC-MALS was used to determine the molar mass across the peaks for the apo- and saturated PZD123-E6 complex (trace colors are as in d). PDZ123 exists as a discrete monomer, even at high protein concentrations (>8 mg/ml). Despite having a similar elution volume to apo-PDZ123, the saturated PDZ123-E6 complex has a significantly increased molar mass. This mass difference was consistent with each PDZ123 binding three E6 molecules, which agrees with the number of binding sites available and independent ITC and kinetic measurements (Table 1).
Compaction of SAP97 PDZ123 Occurs upon Complex Formation with E6
To investigate whether any conformational changes were reflected in changes in hydrodynamic volume on E6L151V-PDZ complex formation and to directly measure the stoichiometry of binding, we combined size exclusion chromatography with SEC-MALS. SEC-MALS combines the resolving power of SEC, with the ability of MALS to provide accurate, absolute measurements of molecular mass as different biomolecules elute from the SEC column. Purified PDZ12, PDZ23, and PDZ123 preparations each eluted as single peaks from analytical SEC columns (see “Materials and Methods” and Fig. 1d). In each case, the measured molar mass using SEC-MALS was very close to the theoretical mass for a monomer, and there was no evidence for mass heterogeneity across the eluted peak (Table 2). For these concatenated PDZ domains, the relationship between elution volume and molar mass was that typically expected for globular proteins, with the larger proteins eluting before smaller ones: PDZ123 (measured mass, ∼36.8 kDa) eluted first followed by PDZ23 (measured mass, ∼27.0 kDa) then PDZ12 (measured mass, ∼21.6 kDa). Thus, each PDZ concatenamer behaves as a homogeneous, stable monomer, even at relatively high protein concentrations (here between 5 and 8.6 mg/ml).
TABLE 2.
SEC-MALS data
Indicated in the last column (oligomeric state) is the stoichiometry determined.
| Protein | Concentration | Elution volume | Measured mass | Theoretical mass | Oligomeric state |
|---|---|---|---|---|---|
| μm | ml | Da | Da | ||
| PDZ12 | 233 | 17.2 | 21,640 ± 0.8% | 21,307 | Monomer |
| PDZ23 | 233 | 16.3 | 26,970 ± 0.6% | 27,052 | Monomer |
| PDZ123 | 233 | 15.5 | 36,800 ± 0.3% | 36,922 | Monomer |
| E6 | 500–1340 | Variable | 99,000–271,000 | 8,759 | Higher order oligomers |
| E6 | 890 | 15.5 | 13,260 ± 35% | 8,759 | Monomer:dimer equilibrium |
| PDZ123:E6 (1:2.25) | 117:263 | 15.7 | 55,050 ± 0.4% | 54,439 (1PDZ:2E6) | 1 PDZ:2E6 |
| PDZ123:E6 (1:3.0) | 120:360 | 15.7 | 61,960 ± 2% | 63,198 (1PDZ:3E6) | 1 PDZ:3E6 |
The situation for E6L151V was, however, more complicated. Small monomeric proteins, for example the size of an E6 monomer, inherently scatter light quite weakly, thus requiring the use of higher protein concentrations to obtain measurable scattering signals. Thus, it is important to bear in mind that the use of high protein concentrations for SEC-MALS may affect the oligomeric state of the protein under study. Recombinant E6L151V preparations, although pure and unmodified according to SDS-PAGE and mass spectrometry analyses, gave rise to SEC peaks with varying elution volumes and with sustained “tails” (data not shown). This behavior contrasts with that of the concatenated PDZ domains, which gave rise to Gaussian peak shapes with highly consistent elution volumes. Analysis of five different E6L151V preparations gave qualitatively similar results. Consistent with the variable elution volume for different E6 preparations, the measured molar masses for the peak centers varied between 99 and 271 kDa (Table 2, where the expected mass of the monomer is 8.76 kDa). The addition of high concentrations of reductants (e.g. 50 mm dithiothreitol) had no significant effect on the measured masses, demonstrating that the higher order oligomerization of E6L151V was unlikely to be a consequence of intermolecular disulfide bond formation. Thus, it appears that, at the concentrations required for SEC-MALS measurements, E6L151V tends to form noncovalently linked high molecular weight aggregates (here 11-mers to 30-mers were observed). This observation fits well with recent cell assays, where E6 was observed to oligomerize to form high molecular weight aggregates (21, 29, 30). Whereas the oligomeric state of E6 was not consistent at the protein concentrations required for SEC-MALS, we did observe, with one preparation, a single clear E6L151V peak eluting from the SEC column that had a molecular mass consistent with a 50–50% mixture of E6 monomers and dimers (Table 2).
Having demonstrated that each PDZ construct was monomeric and that E6L151V tends to form higher order oligomers, we then used SEC-MALS to try and independently determine the apparent binding stoichiometry. Given the complex solution behavior of E6L151V, the measured molecular masses for PDZ123 complexes were surprisingly straightforward. At a molar excess of just over 2.25 molecules of E6L151V to one PDZ123, we found no evidence for any complex larger than a 2:1 complex (Table 2). Similarly, at a molar excess of three molecules of E6 to one PDZ123, there was no species larger than a 3:1 complex (Table 2 and Fig. 1, d and e). Thus, it appears that PDZ123, which contains the three PDZ domains of SAP97, can bind three molecules of E6L151V. We cannot exclude the possibility that if E6L151V can form stable dimers (as per the singular observation for apo-E6L151V), what appears to be a 3:1 complex comprises only two occupied binding sites, one of which contains a bound dimer. However, the fact that we measured a 3:1 complex from ITC and, as discussed below, from kinetics, corroborates that PDZ123 has three binding sites for the E6 protein. Thus, the 3:1 complex observed from SEC-MALS appears to be an accurate observation and not an artifact from high protein concentrations. Interestingly, the elution volumes for this E6L151V-PDZ123 3:1 complex were extremely similar to that of the apo-PDZ123 (15.65 versus 15.5 ml, respectively). This similar elution volume suggests that the hydrodynamic shapes of apo- and saturated PDZ123 are very similar despite their large difference in molecular mass (36,922 versus 63,198 Da). One likely interpretation of this observation is that the complex has undergone a conformational change upon binding of E6. The data in Fig. 1 (d and e) were interpreted with the utmost caution. In reproducible 3:1 SEC-MALS experiments, we obtained three main “peaks”: (i) the front end of the first peak; (ii) the tail of the first peak; and (iii) unbound E6, presumably in a mixed oligomeric state, as per the apo-form of E6. Despite repeated attempts with the highest resolution silica SEC columns, these peaks could not be further resolved. Thus, any mass determination across the peak is a weighted average of the masses for all species present. For this reason, we fitted the mass for the peak (i), the front end of the main peak that was minimally “contaminated” by other species. The fact that the mass of this species was virtually identical to that expected for the saturated complex strongly suggests that it corresponds to the 3:1 complex. Peak (ii) is likely a mixture of E6 oligomers and perhaps partially saturated PDZ123, whereas the third peak is unbound E6.
The Dissociation Rate from SAP97 Distinguishes the Binding of HPV16 and HPV18 E6 Proteins
Having established that all three PDZ domains of SAP97 bind E6 and that PDZ123 can form a 1:3 complex with the E6, we wanted to learn more about the mechanism of the SAP97 and E6 interaction, by studying binding kinetics. Sorting out the mechanism of interaction of ligands, such as E6, that bind to multiple binding sites on the same protein is a complex task. However, a useful approach is to investigate binding at the respective binding site individually and then in concert. Initially we measured the on and off rate constants for the PDZ-E6WT and PDZ-E6L151V interactions by rapidly mixing the proteins in a stopped flow spectrometer and monitoring the change in intrinsic fluorescence of the E6 protein upon binding. The on-off rate constants (Table 3) were estimated from a plot of the observed rate constants versus E6 and/or PDZ concentration by fitting the data to Equation 3 (Fig. 2 and “Materials and Methods”). The on rate constants for the E6WT-PDZ interaction were very similar to those of the E6L151V-PDZ interaction, 12/6.9 μm−1 s−1 for PDZ1, 8.4/6.7 μm−1 s−1 for PDZ2, and 7.7/9.7 μm−1 s−1 for PDZ3, for E6WT and E6L151V, respectively. The off rate constants, however, were 3–16-fold higher for E6WT compared with E6L151V for the three PDZ domains (Table 3). Therefore, the higher off rate constants of HPV16 E6, as compared with those of the HPV18 E6 mimic E6L151V, account for its lower affinity toward the SAP97 PDZ domains, in particular for PDZ2 and PDZ3.
TABLE 3.
Rate and equilibrium constants for the reaction between different PDZ domains and E6WT and E6ML151V, respectively
| Protein | kon | koffa | koff1b | koff2b | Kd (koff/kon) |
|---|---|---|---|---|---|
| μm−1s−1 | s−1 | s−1 | s−1 | μm | |
| E6WT-PDZ | |||||
| PDZ1 | 12 ± 1.8 | 12.0 ± 5.0 | 1.0 ± 0.5a | ||
| PDZ2 | 8.4 ± 1.0 | 34.0 ± 6.0 | 4.0 ± 0.9a | ||
| PDZ3 | 7.7 ± 1.0 | 16.0 ± 1.5 | 2.0 ± 0.3a | ||
| E6L151V-PDZ | |||||
| PDZ1 | 6.9 ± 0.3 | 4.7 ± 0.5 | 6.2 ± 0.5 | 0.9 ± 0.08b | |
| PDZ2 | 6.7 ± 0.5 | 5.2 ± 0.4 | 2.5 ± 0.3 | 0.4 ± 0.06b | |
| PDZ3 | 9.7 ± 0.2 | 2.7 ± 0.2 | 1.0 ± 0.2 | 0.1 ± 0.02b | |
| PDZ12 | 8.7 ± 0.2 | 7.5 ± 0.4 | 4.6 ± 0.4 | 1.3 ± 0.4 | |
| PDZ23 | 25 ± 0.4 | 4.3 ± 0.5 | 2.0 ± 0.4 | 0.5 ± 0.2 | |
| PDZ123 | 27 ± 1.2 | 9.3 ± 1.4 | 1.9 ± 0.5 | 0.7 ± 0.2 | |
a koff determined from Equation 3.
b koff determined from displacement experiments.
FIGURE 2.

Stopped flow binding and displacement experiments for PDZ and E6L151V. a and b, observed rate constants for PDZ1, PDZ2, and PDZ3 binding (a) and PDZ12, PDZ23, and PDZ123 binding (b). c and d, observed rate constants for PDZ3/E6L151V (c) and PDZ123/E6L151V displacement (d) plotted against increasing concentration of the peptide used to trap the PDZ proteins subsequent to the dissociation from E6L151V (SRTRRETQV, corresponding to the C terminus of E6L151V). e, experiment to estimate the number of binding sites in PDZ12. The concentration of E6 was increased at a constant concentration of PDZ12 (6 μm), and Equation 3 was fitted to the data to obtain n (Table 1). f, the observed rate constant kobs as a function of E6L151V at a constant concentration of either PDZ2 or PDZ3 (1 μm). The solid line is the expected dependence of kobs for E6L151V under pseudo-first order conditions according to the parameters in Table 3, determined by varying PDZ2. The kink in the data between 5 and 10 μm may reflect dissociation of E6 oligomers prior to the association with PDZ2 or PDZ3. See Table 3 for fitted parameters.
Concatenation of PDZ Domains Modulates Binding
To investigate whether the E6/PDZ interaction in SAP97 is influenced by neighboring PDZ domains, we performed stopped flow binding experiments using the concatenated PDZ constructs PDZ12, PDZ23, and PDZ123. The binding reaction was monophasic for E6-PDZ12 (Equation 1). Interestingly, E6-PDZ23 and E6-PDZ123 displayed biphasic binding kinetics (Equation 2) including a slow phase, which appeared to decrease (within error) with increasing concentrations of PDZ23 or PDZ123 (Fig. 2b). Such a phase is fully consistent with an initial binding to both domains followed by a slow equilibration governed by the respective off rate constants of the two domains. The on-off rate constants were estimated from the slopes and intercept of the fast phase (Fig. 2b). The on rate constant for the PDZ12 construct was similar or slightly higher as compared with PDZ1 or PDZ2 alone (8.7 μm−1 s−1 compared with 6.9 and 6.7 μm−1 s−1, respectively), whereas the on rate constants for PDZ23 and PDZ123 were significantly higher (25 and 27 μm−1 s−1, respectively) compared with those of the individual domains. The kinetics for multivalent binding is complex and has been discussed in more detail elsewhere (34). In theory, any of the individual on rate constants or their sums may appear as a phase. Here, it is clear that the kon of PDZ12 is different from the sum of those for PDZ1 and PDZ2. For PDZ23, the kon appears to be higher than the sum of PDZ2 and PDZ3, whereas the observed kon for PDZ123 is close to the expected or slightly higher. In fact, the on rate constants of PDZ23 and PDZ123 appear to be similar, which probably is a reflection of the low kon of the PDZ12 tandem, which results in a small difference between PDZ23 and PDZ123. We conclude, based on the nonadditive on rate constants for PDZ12 and on the similar on rate constants for PDZ23 and PDZ123, that some adverse steric effect modulates the binding of E6 to PDZ12, which decreases the on rate constant to one or both of the domains.
To get an independent measure of the off rate constants, we performed displacement experiments as described previously (24). Two observed off rate constants were obtained for each of PDZ12, PDZ23, and PDZ123 (Fig. 2 and Table 3). For PDZ123 one might expect three observed off rate constants, but because all three koff values for the single domains are within 1 order of magnitude, a triple exponential is difficult to resolve and may well appear as a double exponential in the experiment. The off rate constants of PDZ12 and PDZ23 agreed well with those measured for the respective single domains in similar experiments (Table 3), suggesting that the tandem PDZ domains bind E6 simultaneously and thus further corroborating the stoichiometries determined by SEC-MALS and ITC. However, because the precision and accuracy is very high in displacement experiments, the 2-fold lower off rate constants of these two tandems, as compared with the single domains, indicate additional interactions that are not present in the single PDZ-E6 complexes. Such additional interactions may include a conformational change upon binding.
To further confirm the number of E6 interaction sites in the PDZ constructs, the E6 binding was assessed by stopped flow in the region of second order kinetics (Equation 3, Fig. 2e, and Table 1). These data agreed well with the stoichiometry observed in ITC and SEC-MALS.
Dissociation of E6 Oligomers Appears Rate-limiting for Binding at High Micromolar Concentration of E6
The fact that full-length E6 forms oligomers has been demonstrated previously (21, 29). Here, we showed by SEC-MALS that this oligomerization occurs also for the E6 C-terminal domain. This behavior would explain the observed rate constant for binding of E6 to PDZ domains, when the E6 concentration was increased (Fig. 2f). With ∼10 μm of E6 (i.e. 20 μm before the 1:1 mixing in the stopped flow), the linear increase in kobs was abruptly stopped, and the dependence was apparently “saturated.” However, this rather distinct kink in the dependence of kobs on E6 concentration is likely the result of an aggregation/oligomerization event rather than of a change in rate-limiting step in the binding reaction. The observed behavior is consistent with a mechanism where the E6 forms oligomeric species that need to separate before associating with the PDZ domain. At lower concentrations of E6, the kinetic data were consistent with E6 being a monomer, and its C terminus was thus accessible for binding. First, dilution of E6 into buffer (going from 6 to 3 μm) resulted in a trace, which was a perfect straight line. Second, when PDZ1, PDZ2, and PDZ3 concentrations were varied (Fig. 2a), there was no evidence for a rate-limiting dissociation of E6. Instead, the traces were perfectly monophasic (Equation 1), and kobs was increasing linearly with PDZ concentration, showing that E6 did not form (rate-limiting) oligomers at the initial concentration of the experiment (6 μm). It is also worth noting that the apparent rate of the proposed dissociation at higher E6 concentrations appears to differ between those of PDZ2 and PDZ3 in Fig. 2f (∼20 s−1) and PDZ12 in Fig. 2e (40–50 s−1). The discrepancy in rate constants is, however, consistent with the heterogeneity of the oligomeric states that E6 may populate, as suggested by the SEC-MALS experiments. In conclusion, the C-terminal domain of E6 appears to oligomerize at ∼10–20 μm, and the rate of dissociation is 20–50 s−1. Note that the on-off rate constants and equilibrium constants reported in Table 3 are not affected by this E6 oligomerization, because the E6 concentration was below 10 μm in these experiments. Further, the on rate constant for the association of a short peptide corresponding to the C terminus of HPV18 E6 and SAP97 PDZ2 (24) is virtually identical to that for the E6L151V-PDZ2 in the present study. In the ITC experiments, the data points at higher E6 concentrations might be affected by the equilibrium between free and oligomerized E6 molecules, but the general agreement between Kd from kinetics and ITC suggests that there is a minor effect on the fitted parameters.
Noncanonical Interactions Are Involved in the Binding between E6 and PDZ2
To investigate the changes that occur upon binding of E6 to a PDZ domain at the molecular level on a residue per residue basis, we performed NMR titration experiments on the interactions between the single PDZ2 domain and E6WT and E6L151V, respectively. PDZ2 was chosen for the NMR studies because extensive biophysical characterization of the interaction between PDZ2 and peptides corresponding to the C terminus of the E6 protein has been performed (24, 33, 35). 15N-1H correlation maps were recorded for the free and bound forms of the PDZ2 protein (Fig. 3), thus allowing for subtle effects in the E6 protein-PDZ interaction to be sorted out. The combined chemical shift changes of amides were calculated using Equation 4. Structural changes were mapped based on the extent each peak moved in the bound state relative to the free state and were divided into four different classes: large, medium, small, and no change (see legend to Fig. 3). Similar structural changes were seen for the E6WT-PDZ2 and E6L151V-PDZ2 interactions (Fig. 3 and supplemental materials). Those residues that underwent large and medium chemical shift changes were divided into two groups based on their proximity to the binding site. Group 1 comprised those residues close to the canonical peptide-binding site of the PDZ domain (36), which are expected to make a direct or indirect interaction with the E6 (Lys-324, Gly-328, Gly-330, Gly-335, His-341, and Asn-393), whereas group 2 comprised those distant from the ligand binding site (Thr-351, Ile-354, Glu-355, Glu-356, Glu-357, His-360, Leu-371, Glu-380, Glu-385, and Phe-397) (Fig. 3). Interestingly, residues Glu-355, Gly-357, Leu-371, and Phe-397 (distance to side-chains of the bound peptide in an NMR structure was 8.7–13.5 Å; Fig. 3, b and c) that underwent large or medium change in the E6-PDZ2 interaction exhibited small or no change when the C-terminal peptide of E6 was used (4, 6–8, 33). Therefore, residues Glu-355, Gly-357, Leu-371, and Phe-397 constitute a subset of residues that participate in noncanonical interactions either via intradomain allostery or directly with residues on the E6 protein.
FIGURE 3.

NMR data of the E6-PDZ interaction. a, an overlay of HSQC spectra of free PDZ2 (red) and E6L151V-PDZ2 complex (blue). The arrows indicate residues that moved substantially and are discussed in the text. b and c, residues in PDZ2 undergoing structural change upon binding to the E6 protein. Color codes for chemical shift changes of residues in PDZ2 (Protein Data Bank code 2IOL) are: red, large change, Δ ≥ 0.15 ppm; orange, medium change, 0.10 ≤ Δ < 0.15 ppm; yellow, small change, 0.05 ≤ Δ < 0.10 ppm; and gray, no change, 0.00 ≤ Δ < 0.05 ppm. The PDZ2 domain was titrated with E6WT (b) and E6L151V (c). The four residues that display distinct chemical shift changes as compared with binding of a C-terminal peptide (33) are labeled. The canonical binding pocket is indicated by the solid line. This picture was drawn with PyMol (32).
DISCUSSION
Cervical cancer is caused by certain strains of HPV through expression of the oncogenic proteins E6 and E7. The E6 protein has evolved to bind to several important host regulatory proteins, such as the tumor suppressor proteins p53 and SAP97, and thus mark these proteins for destruction to keep HPV-infected cells alive. The exact mechanisms by which host proteins are targeted by the viral proteins are very complex (4, 6–8), and also it is not known how the viral proteins E6 and E7 can be expressed for many years without being detected and destroyed by the immune system (37). Here we have looked at mechanistic aspects of the complex formation between E6 and the PDZ domains of SAP97 and revealed several novel facets of this interaction that are recapitulated in Fig. 4.
FIGURE 4.

Scheme for the interaction between HPV E6 and the PDZ domains of SAP97. E6 forms oligomers, which dissociate upon binding to SAP97 (k = ∼20–50 s−1). All three PDZ domain of SAP97 may bind one E6 molecule each, and a conformational change of the quaternary complex gives a similar hydrodynamic radius to that of apo PDZ123. Dissociation rates govern the affinities for HPV16 and HPV18 E6 proteins for the PDZ domains of SAP97. Residues outside the canonical binding site of the second PDZ domain are affected by the E6 protein in the binding reaction.
One major finding is that the E6 proteins of both HPV16 and HPV18 bind to all three PDZ domains of SAP97 in vitro. By using a cell pulldown assay, it was previously suggested that only the PDZ2 domain of SAP97 could bind the E6 protein of HPV16 (13), whereas E6 from HPV18 was shown to bind to all three PDZ domains from SAP97 (15). The basis of this difference is not clear. Furthermore, a swap of the last amino acids (Leu and Val) completely reversed the immortalization and binding affinities of the two E6 proteins (19, 38). In this study we have measured the affinities of the respective PDZ domains with the E6 from HPV16 and a “pseudo” HPV18 E6 protein (E6L151V). All three PDZ domains have similar association or on rate constants for the two different E6 constructs. However, the dissociation (off rate) constants differed and may explain the observed difference in virulence between the two HPV types (19, 38). Although Val and Leu residues both have a hydrophobic side chain, the Val residue of HPV18 E6 is smaller than the Leu of HPV16 E6, which may allow for a more snug fit in the hydrophobic binding pockets of the PDZ domains.
Contrary to the previous results, we find that PDZ1 displays the highest affinity for HPV16 E6, whereas PDZ3 binds strongest to E6L151V (i.e. to HPV18 E6). However, the affinities are within the same order of magnitude (Tables 1 and 3), suggesting a high degree of promiscuity in E6-PDZ interactions. Furthermore, SEC-MALS, ITC, and kinetic studies show that all three putative binding sites in SAP97 are occupied by E6 proteins in a quaternary complex. Previous experiments using electron microscopy on full-length SAP97 showed that the molecule is present in a monomer-dimer equilibrium and that the dimerization occurs via its N-terminal L27 domain (39). Furthermore, it was demonstrated that monomeric SAP97 is a relatively dynamic protein that exists either in an extended conformation or as a more compact ring-like structure. In another study on the PDZ region of SAP97, it was demonstrated by small angle x-ray scattering that PDZ123 is indeed flexible, in particular the region between PDZ2 and PDZ3 (40). It was further shown that the PDZ12 part was more conformationally restricted and displayed a dumbbell-like shape, in agreement with our kinetic experiments where the on rate constant for PDZ12 is not the sum of those for PDZ1 and PDZ2. The SEC-MALS experiments presented here are consistent with the PDZ region of SAP97 undergoing a structural change, perhaps a collapse or a compaction, upon interaction with the E6 proteins. These data are supported by rate constants of dissociation from stopped flow fluorescence, in particular the slower dissociations from PDZ12, PDZ23, and PDZ123, as compared with those of the single domains (Fig. 2, c and d, and Table 3). Whether the quaternary complex occurs in vivo depends on the expression levels of the E6 protein and of SAP97. Likewise, the reported oligomerization of E6 (21, 29, 30), which is also observed here, would also be dependent on E6 concentration, i.e. the expression level. Immunoblots suggest that the expression of E6 in vivo is low (41, 42), but actual concentrations have not been reported and are very difficult to estimate. Expression of E6 may also vary both temporally and spatially, and the effects of oligomerization and dissociation of E6 in the infected cell remain to be investigated.
PDZ domains bind ligands like the E6 protein through the C terminus of the ligand, which becomes a strand in an extended β-sheet (43). Nevertheless, the potential of allosteric interactions in PDZ domains has been discussed and experimentally substantiated (36, 44–47). Here, by NMR experiments, we found residues in PDZ2 that are not part of the peptide-binding pocket but nevertheless experience chemical shift changes upon binding of E6. Importantly, four of these residues (Fig. 3) were unaffected by binding of an E6 C-terminal peptide (33). This subset of four residues, distal from the binding pocket, may change chemical shifts either through an intradomain allosteric effect (45) or by direct interactions between other parts of the E6 than its C terminus. In either case these noncanonical interactions may be employed by the E6 protein to increase affinity or even modulate binding to a third partner. Low affinity nonspecific interactions between E6 and PDZ2 are less likely to cause these changes in chemical shifts because these four residues saturate at similar concentration as residues in the canonical binding pocket, upon titration with E6.
In conclusion, we report affinities, stoichiometries, rate constants, and conformational change(s) for the complex between E6 and SAP97. As a step toward better understanding of E6-mediated oncogenesis, our results highlight the dynamic nature of the E6-SAP97 binding and reveal mechanistic and molecular details of the interaction.
This work was supported by funds from the Swedish Research Council (to P. J. and P. L.), Jeansson's Foundation (to P. J.), Clas Groschinskys Minnesfond (to P. J.), Science Foundation Ireland Awards 07/SK/B1224and 09/YI/B1682 (to N. F.), and a start-up award from University College Dublin (to N. F.).
This article was selected as a Paper of the Week.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Fig. S1.
- HPV
- human papillomavirus
- ITC
- isothermal titration calorimetry
- SEC-MALS
- size exclusion chromatography-multi-angle laser light scattering
- HSQC
- heteronuclear single quantum coherence
- SAP97
- synapse-associated protein 97.
REFERENCES
- 1. de Villiers E. M., Wagner D., Schneider A., Wesch H., Miklaw H., Wahrendorf J., Papendick U., zur Hausen H. (1987) Lancet 330, 703–706 [DOI] [PubMed] [Google Scholar]
- 2. zur Hausen H. (1991) Virology 184, 9–13 [DOI] [PubMed] [Google Scholar]
- 3. zur Hausen H. (2002) Nat. Rev. Cancer 2, 342–350 [DOI] [PubMed] [Google Scholar]
- 4. Mammas I. N., Sourvinos G., Giannoudis A., Spandidos D. A. (2008) Pathol. Oncol. Res. 14, 345–354 [DOI] [PubMed] [Google Scholar]
- 5. Bosch F. X., Manos M. M., Muñoz N., Sherman M., Jansen A. M., Peto J., Schiffman M. H., Moreno V., Kurman R., Shah K. V. (1995) J. Natl. Cancer Inst. 87, 796–802 [DOI] [PubMed] [Google Scholar]
- 6. Boulet G., Horvath C., Vanden Broeck D., Sahebali S., Bogers J. (2007) Int. J. Biochem. Cell Biol. 39, 2006–2011 [DOI] [PubMed] [Google Scholar]
- 7. Tungteakkhun S. S., Duerksen-Hughes P. J. (2008) Arch. Virol. 153, 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wise-Draper T. M., Wells S. I. (2008) Front. Biosci. 13, 1003–1017 [DOI] [PubMed] [Google Scholar]
- 9. Fehrmann F., Laimins L. A. (2003) Oncogene 22, 5201–5207 [DOI] [PubMed] [Google Scholar]
- 10. Tong X., Howley P. M. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 4412–4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thomas M., Banks L. (1998) Oncogene 17, 2943–2954 [DOI] [PubMed] [Google Scholar]
- 12. Chen J. J., Reid C. E., Band V., Androphy E. J. (1995) Science 269, 529–531 [DOI] [PubMed] [Google Scholar]
- 13. Kiyono T., Hiraiwa A., Fujita M., Hayashi Y., Akiyama T., Ishibashi M. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 11612–11616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee S. S., Weiss R. S., Javier R. T. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 6670–6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gardiol D., Kühne C., Glaunsinger B., Lee S. S., Javier R., Banks L. (1999) Oncogene 18, 5487–5496 [DOI] [PubMed] [Google Scholar]
- 16. Glaunsinger B. A., Lee S. S., Thomas M., Banks L., Javier R. (2000) Oncogene 19, 5270–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee S. S., Glaunsinger B., Mantovani F., Banks L., Javier R. T. (2000) J. Virol. 74, 9680–9693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakagawa S., Huibregtse J. M. (2000) Mol. Cell. Biol. 20, 8244–8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas M., Glaunsinger B., Pim D., Javier R., Banks L. (2001) Oncogene 20, 5431–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng W., Zhang M. (2009) Nat. Rev. Neurosci. 10, 87–99 [DOI] [PubMed] [Google Scholar]
- 21. Liu Y., Cherry J. J., Dineen J. V., Androphy E. J., Baleja J. D. (2009) J. Mol. Biol. 386, 1123–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zanier K., Charbonnier S., Baltzinger M., Nominé Y., Altschuh D., Travé G. (2005) J. Mol. Biol. 349, 401–412 [DOI] [PubMed] [Google Scholar]
- 23. Nominé Y., Ristriani T., Laurent C., Lefèvre J. F., Weiss E., Travé G. (2001) Protein Eng. 14, 297–305 [DOI] [PubMed] [Google Scholar]
- 24. Chi C. N., Bach A., Engström A., Wang H., Strømgaard K., Gianni S., Jemth P. (2009) Biochemistry 48, 7089–7097 [DOI] [PubMed] [Google Scholar]
- 25. Chi C. N., Engström A., Gianni S., Larsson M., Jemth P. (2006) J. Biol. Chem. 281, 36811–36818 [DOI] [PubMed] [Google Scholar]
- 26. Nominé Y., Masson M., Charbonnier S., Zanier K., Ristriani T., Deryckère F., Sibler A. P., Desplancq D., Atkinson R. A., Weiss E., Orfanoudakis G., Kieffer B., Travé G. (2006) Mol. Cell 21, 665–678 [DOI] [PubMed] [Google Scholar]
- 27. Gianni S., Engström A., Larsson M., Calosci N., Malatesta F., Eklund L., Ngang C. C., Travaglini-Allocatelli C., Jemth P. (2005) J. Biol. Chem. 280, 34805–34812 [DOI] [PubMed] [Google Scholar]
- 28. Malatesta F. (2005) Biophys. Chem. 116, 251–256 [DOI] [PubMed] [Google Scholar]
- 29. García-Alai M. M., Dantur K. I., Smal C., Pietrasanta L., de Prat-Gay G. (2007) Biochemistry 46, 341–349 [DOI] [PubMed] [Google Scholar]
- 30. Nomine Y., Ristriani T., Laurent C., Lefevre J. F., Weiss E., Trave G. (2001) Protein Expression Purif. 23, 22–32 [DOI] [PubMed] [Google Scholar]
- 31. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 32. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 33. Liu Y., Baleja J. D. (2008) Front. Biosci. 13, 121–134 [DOI] [PubMed] [Google Scholar]
- 34. Chi C. N., Bach A., Gottschalk M., Kristensen A. S., Strømgaard K., Jemth P. (2010) J. Biol. Chem. 285, 28252–28260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y., Dasgupta J., Ma R. Z., Banks L., Thomas M., Chen X. S. (2007) J. Virol. 81, 3618–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jemth P., Gianni S. (2007) Biochemistry 46, 8701–8708 [DOI] [PubMed] [Google Scholar]
- 37. Thomas M., Narayan N., Pim D., Tomaić V., Massimi P., Nagasaka K., Kranjec C., Gammoh N., Banks L. (2008) Oncogene 27, 7018–7030 [DOI] [PubMed] [Google Scholar]
- 38. Thomas M., Dasgupta J., Zhang Y., Chen X., Banks L. (2008) Virology 376, 371–378 [DOI] [PubMed] [Google Scholar]
- 39. Nakagawa T., Futai K., Lashuel H. A., Lo I., Okamoto K., Walz T., Hayashi Y., Sheng M. (2004) Neuron 44, 453–467 [DOI] [PubMed] [Google Scholar]
- 40. Goult B. T., Rapley J. D., Dart C., Kitmitto A., Grossmann J. G., Leyland M. L., Lian L. Y. (2007) Biochemistry 46, 14117–14128 [DOI] [PubMed] [Google Scholar]
- 41. Androphy E. J., Hubbert N. L., Schiller J. T., Lowy D. R. (1987) EMBO J. 6, 989–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Banks L., Spence P., Androphy E., Hubbert N., Matlashewski G., Murray A., Crawford L. (1987) J. Gen. Virol. 68, 1351–1359 [DOI] [PubMed] [Google Scholar]
- 43. Doyle D. A., Lee A., Lewis J., Kim E., Sheng M., MacKinnon R. (1996) Cell 85, 1067–1076 [DOI] [PubMed] [Google Scholar]
- 44. Peterson F. C., Penkert R. R., Volkman B. F., Prehoda K. E. (2004) Mol. Cell 13, 665–676 [DOI] [PubMed] [Google Scholar]
- 45. Fuentes E. J., Der C. J., Lee A. L. (2004) J. Mol. Biol. 335, 1105–1115 [DOI] [PubMed] [Google Scholar]
- 46. Gianni S., Walma T., Arcovito A., Calosci N., Bellelli A., Engström A., Travaglini-Allocatelli C., Brunori M., Jemth P., Vuister G. W. (2006) Structure 14, 1801–1809 [DOI] [PubMed] [Google Scholar]
- 47. Niu X., Chen Q., Zhang J., Shen W., Shi Y., Wu J. (2007) Biochemistry 46, 15042–15053 [DOI] [PubMed] [Google Scholar]