

Abstract
The homodimeric umuD gene products play key roles in regulating the cellular response to DNA damage in Escherichia coli. UmuD2 is composed of 139-amino acid subunits and is up-regulated as part of the SOS response. Subsequently, damage-induced RecA·ssDNA nucleoprotein filaments mediate the slow self-cleavage of the N-terminal 24-amino acid arms yielding UmuD′2. UmuD2 and UmuD′2 make a number of distinct protein-protein contacts that both prevent and facilitate mutagenic translesion synthesis. Wild-type UmuD2 and UmuD′2 form exceptionally tight dimers in solution; however, we show that the single amino acid change N41D generates stable, active UmuD and UmuD′ monomers that functionally mimic the dimeric wild-type proteins. The UmuD N41D monomer is proficient for cleavage and interacts physically with DNA polymerase IV (DinB) and the β clamp. Furthermore, the N41D variants facilitate UV-induced mutagenesis and promote overall cell viability. Taken together, these observations show that a monomeric form of UmuD retains substantial function in vivo and in vitro.
Keywords: DNA Damage, DNA Polymerase, Mutagenesis Mechanisms, Protein Conformation, Protein Stability, DinB, SOS Response, UmuD, β Clamp, Translesion DNA Synthesis
Introduction
Organisms are constantly bombarded by harmful DNA damaging agents that can lead to stalling of the replication machinery and cell death (1). The tightly regulated bacterial SOS system is a stress-induced response to DNA damage and is an integral part of UV-induced mutagenesis in Escherichia coli (1). The first stage of this damage response involves relatively accurate DNA repair processes, but as the response progresses, it shifts to a potentially mutagenic damage tolerance mode to ensure cell survival (1, 2). This switch from accurate DNA repair to mutagenic damage tolerance is regulated in part by the umuD gene products. The UmuD2 homodimer is composed of 139-amino acid subunits and is expressed 20–30 min after the induction of the SOS response (1–3). Interaction with the damage-induced RecA·ssDNA nucleoprotein filament facilitates the slow self-cleavage and removal of the N-terminal 24 amino acids of UmuD2, yielding the C-terminal 115 amino acid homodimer, UmuD′2. Together, UmuC and UmuD′2 form the Y family DNA polymerase V (UmuD′2C), a low-fidelity DNA polymerase that has a specialized ability to copy damaged DNA in a process known as translesion DNA synthesis (1, 4, 5).
UmuD2 and UmuD′2 make a number of distinct protein-protein contacts with considerable functional implications. Both UmuD2 and UmuD′2 interact with the RecA·ssDNA nucleoprotein filament, Y family DNA polymerases UmuC and DinB, the α, β, and ϵ subunits of the replicative DNA polymerase III, and proteases Lon and ClpXP (1, 6–12). UmuD2 strongly interacts with the β processivity clamp, whereas UmuD′ preferentially interacts with the α catalytic subunit (12). UmuD2 also prevents DinB-induced −1 frameshift mutations (7), whereas UmuD′2 activates UmuC for translesion DNA synthesis (1, 4, 6). Degradation of UmuD2 is carried out by the Lon protease (10). Also, UmuD delivers either its UmuD or UmuD′ partner to ClpXP for degradation (13). The multiple interactions of UmuD2 and UmuD′2 are critical for regulating mutagenesis in E. coli.
The structural flexibility of UmuD2 and UmuD′2 dimers permits a broad range of interactions (6, 14–18). For full-length UmuD2, the N-terminal 39-amino acid arms are relatively stably bound to the globular C-terminal domain to produce a distinct binding surface (6, 16). Upon cleavage of the N-terminal 24 amino acids, the remaining 15 amino acids of the arm appear unbound from the C-terminal domain and are quite disordered (15, 17). This leaves the globular domain solvent-exposed and available for interaction with a variety of proteins (6, 16). Whereas x-ray (17) and NMR (16) structures (Fig. 1) indicate that UmuD′2 has an overall β-sheet fold, circular dichroism (CD) (18) experiments show that both the UmuD2 and UmuD′2 dimers resemble a random coil under physiological conditions. Additionally, the NMR and x-ray structures of UmuD′2 are substantially different, with the active site of UmuD′2 correctly formed and poised for catalysis only in the x-ray structure (16, 17). These structural differences highlight the plasticity of UmuD′2 and have resulted in the classification of the umuD gene products as intrinsically disordered proteins (IDPs)2 (18). Many IDPs play key roles in regulation despite their lack of a well defined structure (19–21).
FIGURE 1.

Homology models and structures of the UmuD proteins. A, a homology model of dimeric UmuD2 in trans, elbows down (left) (8) is shown. Asn-41 (blue) is located in the interface of the neck region of UmuD. The active site residues Ser-60 (red) and Lys-97 (gray) are depicted. An arrow shows the location of the C-terminal “tails.” Chain A and chain B are shown in orange and light pink, respectively. The box shows detail of the neck region with residue Asn-41 highlighted. B, shown is an energy-minimized homology model of UmuD2 in cis, elbows down (8). The N-terminal arms of each monomer loop down and across the respective active site. C, asparagine residues are conserved at this position across UmuD-like proteins. Multiple sequence alignment of UmuD (PDB code 1I4V) and similar proteins is shown. P, plasmid-borne homologs; Pv, Proteus vulgaris; S, Salmonella typhi; Sm, Serratia marcescens; As, Acinetobacter sp.; Pa, Protochlamydia amoebophila; Pm, Prochlorococcus marinus; Dv, Desulfovibrio vulgaris; St, Salmonella typhimurium; Cv, Chromobacterium violaceum; bacteriophage λ cI repressor. UVPM, UV protection and mutation protein; PUVP, putative UV protection protein; PSOSM, putative SOS mutagenesis protein; SigP, signal peptidase. All proteins were aligned through a combination of BLAST and evolutionary trace searches and ClustalW multiple alignment (38–40). D, shown are NMR (top) (16) and x-ray (bottom) (17) structures of UmuD′2. Chains A and B are shown in green and yellow, respectively. Illustrations in A, B, and D were prepared using Chimera (70).
Although attempts at high resolution structures of UmuD2 have been unsuccessful, four isoenergetic models of full-length UmuD2 have been proposed wherein the N-terminal arms are in the trans (domain-swapped, intermolecular) or cis (intramolecular) conformations, with the elbows up or down (8). In the trans elbows-down version (Fig. 1A), the N-terminal arm of one monomer loops down across the globular C-terminal domain of its partner where it crosses the catalytic site (8). The model of UmuD2 in the trans elbows-up conformation shows each arm bound to the outer edge of the C-terminal domain, potentially allowing the active site region of the protein to be solvent-exposed (8). For the UmuD2 models in cis, the elbows-down (Fig. 1B) or elbows-up conformations suggest that the arms can bind their own globular domains (8). The proposed models are also consistent with cross-linking experiments completed at physiologically relevant concentrations (22, 23).
The N-terminal arms (residues 1–39) of trans-UmuD2 form an extensive interface that spans the homodimer while making widespread contacts with the C-terminal globular domain. Residues 25–40 of the arms of UmuD2 that loop down and across the C-terminal domain not only alter the interacting surface of the protein but are most likely a source of stability for the dimer (6). The flexible regions of UmuD2 also include the “neck” region surrounding peptides 33–43 and 41–52 as seen by high levels of deuterium uptake (24). Cross-linking studies of this region also support the model of solvent accessibility of residues 31–36 and 39–40 (22). Additionally, the relatively efficient cross-linking of mono-cysteine derivatives at position 37 and 38 of UmuD2 implies that these residues are close to the dimer interface (22). However, it is intriguing that the identical residues do not cross-link in the case of UmuD′2, possibly suggesting a greater degree of structural flexibility relative to full-length UmuD (22). The arms of UmuD′2 (residues 25–39), which are extended and disordered, are not engaged in the dimerization of the protein. The C-terminal globular domain of UmuD′2 also displays a great deal of plasticity and flexibility as is evident from the NMR solution structure and hydrogen-deuterium exchange experiments (15–16, 24). The comparative flexibility of wild-type UmuD′2 relative to UmuD is also demonstrated by the robust cross-linking efficiency of UmuD′2 S57C as opposed to the reduced cross-linking efficiency of UmuD2 S57C (6). Taken together, these results suggest that the UmuD proteins may adopt multiple conformations in solution.
In an effort to learn more about the structural dynamics and functions of UmuD proteins, we set out to create variants that were defective in dimerization. Such a variant would not only answer the question as to whether UmuD2 is active in the cis conformation but also address the possibility that UmuD may be functionally active as a monomer. In monomeric UmuD, an intramolecular (cis) conformation is the only conformation possible for the arm binding to the globular domain. Although models of UmuD2 with the arms in the cis conformation have been proposed, evidence that this conformation is physiologically relevant has been lacking to date. We focused on the region near the α-helix composed of residues 39–44, as it has been proposed to be important for UmuD2 dimerization (22, 23, 25). We also considered the importance of α-helices in protein stability as in the case of leucine zipper dimerization, transmembrane helix interactions, and numerous other examples (26, 27). With this in mind, we hypothesized that disrupting the contacts between the α-helical regions of UmuD2 would result in a reduction in dimerization efficiency. We generated a single point mutation, N41D, that is likely to not only alter the hydrogen bonding network but also to produce repulsive interhelical electrostatic interactions. In this work we have found that the single amino acid substitution N41D is sufficient to shift the dimer-monomer equilibrium of UmuD significantly to the monomer form. This single mutation also renders UmuD′ a monomer. We find that this variant of UmuD is a monomer under most conditions, is active in self-cleavage, and is proficient for UV-induced mutagenesis.
EXPERIMENTAL PROCEDURES
Bacteriological Techniques
The E. coli strains and plasmids used for this study are listed in Table 1. The operator sequences of pGY9739 and pGY9738 contain the o1c mutation where a single base substitution leads to modestly increased expression of umuD and umuC (28, 29). Strains were grown in Luria broth at 37 °C supplemented with spectinomycin (60 μg/ml) or ampicillin (100 μg/ml). Competent cells were prepared using the CaCl2 method (30). UmuD and UmuD′ N41D were constructed using a QuikChange kit (Stratagene). Mutations were confirmed by DNA sequence analysis (Massachusetts General Hospital Core Facility, Cambridge, MA). Mutagenic primer sequences are N41D forward (5′-GCGCATCGATCTGGATCAACTGTTGATCC) and N41D reverse (5′-GGATCAACAGTTGATCCAGATCGATGCGC).
TABLE 1.
Strains and plasmids
| Strains and plasmids | Relevant genotype | Source or reference |
|---|---|---|
| Strain | ||
| AB1157 | argE3 | Laboratory stock |
| GW8017 | AB1157 ΔumuDC | 22 |
| PB103 | AB1157 ΔumuDC ΔrecJ | P1 (JW2860) → GW8017 (71) |
| BL21 DE3 | Laboratory stock | |
| Plasmid | ||
| pGY9738 | o1cumuD′C; pSC101-derived, SpecR | 29 |
| pGY9739 | o1c umuDC; pSC101-derived, SpecR | 29 |
| pGB2 | Vector; pSC101-derived, SpecR | 32 |
| pSG4 | umuD′, AmpR | 31 |
| pSG5 | umuD, AmpR | 31 |
Proteins, Strains, and Plasmids
UmuD N41D and UmuD′ N41D expression plasmids were constructed in pSG5 and pSG4. Expression of UmuD and UmuD′ proteins were accomplished as previously described (31). Cells were harvested, and UmuD and UmuD′ proteins were purified according to published methods (28). The β clamp was also purified using the method published for UmuD and UmuD′. DinB and RecA proteins were purified as described (31, 33).
Native PAGE
Purified wild-type UmuD2 and UmuD′2 (10 μm) as well as UmuD N41D and UmuD′ N41D (10 μm) were each incubated in non-denaturing sample buffer (NDS, 62.5 mm Tris-HCl, pH 6.8, 10% glycerol, 0.01% bromphenol blue) for 5 min with or without the addition of 10 mm dithiothreitol (DTT). Electrophoresis was carried out by 10–20% polyacrylamide gel electrophoresis (PAGE) (Lonza) using non-denaturing electrode buffer (NDE, 25 mm Tris-Base, 192 mm glycine) at a constant 120 V at room temperature until proteins were resolved. The gel was then stained with Sypro Ruby (Molecular Probes), which has a detection limit of 0.25–1 ng for most proteins. Fluorescence was detected with a Storm 860 PhosphorImager using excitation wavelength of 635 nm. UmuD monomer and dimer bands were quantified using ImageQuant (GE Healthcare).
Determination of Binding Constant of UmuD N41D
Concentrations of UmuD N41D and UmuD′ N41D ranging from 5 to 200 μm were combined with 10 mm DTT and NDS buffer in preparation for native PAGE. Wild-type UmuD2 and UmuD′2 at concentrations of 10, 100, and 200 μm were included as controls. Protein bands were detected with Sypro Ruby and quantified as described above. The calculations used to determine the KD for homodimerization (including equations and derivations) have been previously described (34). The following equation was applied to the data set to obtain the KD for homodimerization.
In this equation [A]T is the total concentration of A added to the reaction, and [AA] is the concentration of dimers. The KD was obtained by generating a best fit curve for the average of three trials using nonlinear regression curve fitting in GraphPad Prism.
Thermal Shift Assay
An optical 96-well reaction plate (Applied Biosystems) was used to analyze 16-μl reaction volumes. Each well contained UmuD Tm buffer (50 mm Hepes, pH 7.5, and 100 mm NaCl), 45 μm UmuD protein, and 25× Sypro Orange (Molecular Probes). The plates were sealed using optical adhesive film (Applied Biosystems). An iCycler iQ5 real-time PCR detection system (Bio-Rad) was used to heat the plate from 10 to 70 °C in 0.1 °C increments. Changes in the fluorescence were monitored concurrently with a charge-coupled device (CCD) camera. The temperature midpoint for the protein unfolding transition, Tm, was calculated by fitting the fluorescence data to a Boltzmann model using curve-fitting program Microsoft Excel XLfit5 add-on program (ID Business Solutions) (24),
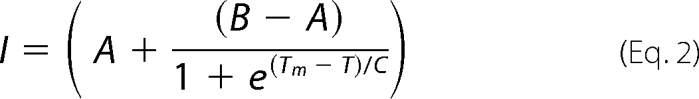 |
In this equation I is the measured fluorescence intensity at temperature T. The pretransitional and posttransitional fluorescence intensities are denoted A and B, respectively, and the slope factor is denoted as C. Fitting did not include data points after the fluorescence intensity maximum.
UmuD in Vitro Cleavage Assays
RecA·ssDNA nucleoprotein filament-dependent cleavage of wild-type UmuD2 and UmuD N41D at concentrations of 0.5, 1, 2, 5, and 10 μm was assayed (22, 31, 35) for 60 min in LG buffer (31) at 37 °C. The addition of sodium dodecyl sulfate (SDS)-PAGE loading buffer (25 mm Tris-HCl, 5% glycerol, 2% SDS, 1.25% β-mercaptoethanol, 0.1% bromphenol blue) was used to quench the reaction before analysis by 4–20% SDS-PAGE (Pierce). Alkaline cleavage was carried out as previously reported (31). Reactions were incubated at 37 °C for 48 h. Cleavage products were analyzed by 4–20% SDS-PAGE (Pierce). Proteins were detected by staining with Sypro Ruby as described above.
To assay the inhibition of UmuD cleavage by DinB, wild-type UmuD2, and UmuD N41D at concentrations of 2 μm were incubated at room temperature for 1 h in the absence of wild-type DinB or in the presence of 2 or 4 μm wild-type DinB in LG buffer (31). In a separate reaction, 3.15 μm RecA, 0.35 μm 24-mer DNA oligo, and 0.68 μm ATPγS were incubated at room temperature for 1 h. The reactions were combined and incubated at 37 °C for 1 h. The 20-μl reaction was then quenched with 5 μl of 4× SDS-PAGE loading buffer before analysis by 4–20% SDS-PAGE (Pierce). Sypro Ruby was used for protein detection as described above.
Immunoblotting
The level of UmuD and UmuD′ expression from the low-copy plasmids pGY9739 and pGY9738, respectively, in GW8017 was determined by Western blotting. Induction of expression with UV irradiation was accomplished as previously described (28). For experiments using denaturing conditions, cell pellets were resuspended in 50 μl of 0.85% saline and 50 μl of SDS-PAGE buffer. Cells were lysed by heating for 20 min at 95 °C before loading 15-μl aliquots onto 4–20% SDS-PAGE gels (Pierce). Electrophoresis was carried out using Tris-Hepes-SDS running buffer (100 mm Tris base, 100 mm Hepes, 1% SDS) at 120 V for 1 h 15 min. These assays were carried out with loading amounts appropriate to give a signal within the linear range of quantification (Fig. 5D).
FIGURE 5.

UmuD expression levels, cleavage products, and heterodimer formation. Protein expression was induced by exposing cultures to 25 J/m2 UV light. A, an immunoblot of gel run under denaturing conditions shows the steady-state expression levels of UmuD from plasmids pGY9739 and pGY9738 in GW8017. Relative UmuD expression levels are shown below the blot, where UmuD N41D is normalized to wild-type UmuD, and UmuD′ N41D is normalized to wild-type UmuD′. Percent cleavage for UmuD2 and UmuD N41D is also indicated. B, an immunoblot of a gel under native conditions shows that UmuD2 and UmuD′2 are expressed as dimers, whereas UmuD N41D and UmuD′ N41D are expressed as monomers in vivo. There is no cleavage product seen under these conditions for wild-type UmuD2; however, the cleavage product for UmuD N41D is evident. C, shown is an immunoblot of identical samples from B under denaturing conditions. In this blot, both full-length and cleaved products are present in almost equivalent amounts, suggesting that a stable wild-type UmuDD′ heterodimer was observed in B. D, Western blots in A, B, and C were carried out to give a signal within the linear range of the assay. The intensity of the signal versus the amount of crude lysate loaded is shown. The point on the y axis of the graph represents the band from gels in A–C with highest intensity, which is the heterodimer in B. E, heterodimer formation is shown under equilibrium conditions using purified UmuD proteins in vitro. Native gel shows preferential heterodimer formation (box) when wild-type UmuD and UmuD′ proteins are combined in equivalent amounts. UmuD N41D and UmuD′ N41D do not form heterodimers as expected. Combining N41D variant and wild-type UmuD or UmuD′ also does not result in heterodimerization.
For experiments carried out under non-denaturing conditions, a gentle lysis BugBuster protein extraction reagent (Novagen) was used per the manufacturer's directions. For native PAGE, 10 μl of supernatant was added to 20 μl of NDS buffer, and half of the reaction was loaded onto 10–20% polyacrylamide gels (Lonza). Electrophoresis was accomplished using NDE buffer at 120 V for 3.5 h. Immunoblotting experiments were completed as previously described (28).
UV Survival and Mutagenesis Assays
Survival and mutagenesis assays were performed as previously described (28, 31). The data represent an average of at least three trials, and error bars represent the S.D.
Heterodimerization of UmuD and UmuD′
The heterodimerization experiment was carried out by incubating a mixture of UmuD and UmuD′ proteins at 10 μm in LG buffer (31) and 10 mm DTT for 15 min. NDS buffer was added directly to the reaction for a total volume of 25 μl. Electrophoresis was carried out by 10–20% native PAGE (Lonza) using NDE buffer until proteins were resolved. Bands were detected and quantified as previously described.
Formaldehyde Cross-linking of UmuD and β Clamp
Formaldehyde cross-linking reactions were completed by incubating 10 μm UmuD and β clamp protein in X-link buffer (20 mm Hepes, pH 7.5, 50 mm NaCl, 0.1 mm EDTA) and 80 mm formaldehyde for a total reaction volume of 12 μl. The reactions were incubated for 45 min at room temperature before the addition of NDS buffer. Proteins were resolved by 10–20% native PAGE (Lonza) for 5 h at 120 V. Sypro Ruby staining was used to detect cross-linked proteins.
RESULTS
Design of UmuD Variants
Previous investigations into the conformation of the N-terminal arms of UmuD2 have concluded that although the arms are likely to be dynamic, the active form of this protein is trans, where the arm of one monomer is bound to the globular C-terminal domain of the adjacent monomer (8, 13, 18, 36, 37). In creating monomeric UmuD proteins, we address two issues, 1) Can UmuD function as a monomer; that is, can it cleave itself to form UmuD′ and facilitate mutagenesis? and 2) If UmuD is active as a monomer, this would suggest that the wild-type UmuD2 dimer can also adopt an active cis conformation where the N-terminal arm of the monomer binds to and is cleaved by its respective active site.
The strategy involved perturbation of the two areas of contact between subunits of the dimer; the N-terminal neck region between Asn-41 and Pro-48 and the C-terminal tail from Val-135 to Arg-139 (Fig. 1A). We identified important candidate residues for mutation by using the evolutionary trace method for identifying active sites and functional interfaces, BLAST searches, and the multiple sequence alignment software ClustalW (38–40). Results of this comprehensive search generated 15 orthologs of E. coli UmuD′2 (PDB code 1I4V) (16) with residues Asn-41 and Pro-48 emerging as highly conserved (Fig. 1C). The matches include homologs of UmuD from a number of bacterial species: signal peptidase, RumA, MucA, ImpA, LexA, bacteriophage λ cI repressor, and putative proteins suggested to be involved in the DNA damage response (Fig. 1C). Like the UmuD2 protein, RumA, MucA, ImpA, and LexA also undergo a RecA-facilitated self-cleavage reaction (41–45). The chemical mechanism by which these proteins cleave is also similar in that the active sites contain a serine-lysine dyad that is located in the globular domain and the cleavage site is the dipeptide sequence (Ala/Cys)-Gly (41, 46, 47). Many of these orthologs are also organized into an operon in which there is a umuD-like gene located upstream of a umuC-like gene. Some examples include mucAB, impAB, and rumAB (41, 43, 48–52).
We hypothesized that the UmuD N41D mutation within the neck region of UmuD2 would disrupt the hydrogen bonding network that provides stability while simultaneously generating destabilizing electrostatic and hydrophobic-hydrophilic interactions with the helix of the opposing monomer (Fig. 1). We carried out the following analysis using the solution NMR structure of UmuD′2 (Fig. 1D, top) (16). The crystal structure of UmuD′2 is shown for comparison (Fig. 1D, bottom) (17). We used the contacts of structural units (CSU) program to analyze the effect of constructing the N41D mutation (53). This software calculates the solvent accessibility of an atom, putative hydrogen bonds, and the various stabilizing and destabilizing interactions that occur between residues (hydrophobic-hydrophobic, aromatic-aromatic, hydrophobic-hydrophilic) (53). Based on this calculation, Asn-41 supports a hydrogen bonding network that involves residues Ile-38, Asp-39, Gln-42, Gln-46, and His-47 within that chain. The substitution N41D supports hydrogen bond formation between Gln-42 and Leu-40 within chain A and Arg-37 of chain B. However, destabilizing interactions were also identified between Asp-41 of chain A and Ile-38 and Asp-41 of chain B.
As a result of these searches, we also constructed P48G, a triple mutant V135S/K136A/R139A, and combinations of mutations from both the N-terminal and C-terminal regions N41D/K136A/R139A and P48G/K136A/R139A. Expression of UmuD2 P48G and V135S/K136A/R139A was substantially lower than that of wild-type UmuD2. For variants N41D/K136A/R139A and P48G/K136A/R139A, expression could not be confirmed even by Western blotting. Therefore, we focused on the UmuD N41D variant, which we obtained in high yield.
UmuD N41D and UmuD′ N41D Are Monomers in Vitro
The dimeric and monomeric conformations of wild-type and N41D UmuD and UmuD′ were determined by native gel electrophoresis (Fig. 2A). UmuD2 and UmuD′2 dimers are clearly resolved. The predominant species of UmuD N41D migrates farther into the gel than either wild-type UmuD2 or UmuD′2 dimers, consistent with UmuD N41D being a monomer. The presence of a small amount of UmuD N41D dimer is apparent. In the presence of DTT this species is eliminated, and only the monomer form is observed. UmuD2 contains one cysteine per monomer, Cys-24, so the addition of reducing agent DTT was necessary to prevent cross-linking of Cys24 on the N-terminal arms of UmuD2. For UmuD′ N41D, no covalent dimers are observed in the presence or absence of DTT as the N-terminal 24-amino acid arms, including the readily cross-linked C24, are not present in UmuD′2.
FIGURE 2.

UmuD N41D and UmuD′ N41D are monomers. A, a native gel shows the resolution of UmuD and UmuD′ monomers (Mono) and dimers (Di) at 10 μm. The plus (+) sign indicates the addition of DTT to the sample. B, a representative native gel shows monomer and dimer formation as a function of increasing concentrations of UmuD N41D (5, 10, 25, 50, 75, 100, 125, 150, 175, 200 μm as indicated below the gel) as well as wild-type UmuD2 (10, 100, 200 μm). C, a representative native gel shows monomer and dimer formation as a function of increasing concentrations of UmuD′ N41D (25, 50, 75, 100, 125, 150, 175, 200 μm as indicated below the gel) as well as wild-type UmuD′2 (10, 100, 200 μm). D, shown are calculated dimer concentrations (fraction bound) as a function of total concentration of UmuD N41D ([A]T). Dimer concentrations were obtained as a function of total concentration of UmuD N41D to give a KD of 52 ± 8.0 μm (S.E.) (solid line).
To determine the extent to which the N41D mutation alters the dimer-monomer equilibrium, the KD for dimerization was determined by analyzing a range of concentrations via native PAGE in the presence of DTT (Fig. 2B). We analyzed UmuD N41D at concentrations of 5–200 μm. At the lowest concentrations used, no dimer or only trace amounts of dimer are formed; at higher concentrations there is an appreciable dimer present, but the monomer is still the major species. The resulting curve was produced by calculating the fraction bound (observed dimer) as a function of the total concentration of UmuD N41D protein added to the reaction. The best fit curve generated a KD for dimerization for UmuD N41D of 52 ± 8.0 μm (S.E.) (Fig. 2D). This KD is more than 6 orders of magnitude greater than the upper limit of the KD for wild-type UmuD2, which is reported to be ≈10−11 m (18). In the case of wild-type UmuD′2, no monomer form was observed in the native gel analysis (Fig. 2C). The KD for dimerization of wild-type UmuD′2 is less than 10 pm (18). In the case of UmuD′ N41D, no dimer could be detected at concentrations up to 200 μm by native PAGE. Therefore, this single amino acid change is sufficient to strongly favor the monomeric form of the protein.
N41D Mutation Significantly Reduces the UmuD′ Melting Temperature
To characterize the stability of UmuD N41D, we analyzed the melting profiles of the wild-type and variant UmuD proteins. Experiments were performed using purified protein and Sypro Orange fluorescent dye. Sypro Orange is weakly fluorescent in aqueous solution but becomes highly fluorescent when in contact with nonpolar environments such as the hydrophobic sites of a protein. Upon the melting of a protein, the hydrophobic regions are exposed, and the dye binds. This results in an increase in fluorescence emission followed by a gradual decrease in the intensity that may be due to precipitation or aggregation of the complex of unfolded protein and the probe (54). It was previously shown that wild-type UmuD2 undergoes two melting transitions, one near 30 °C due to the dissociation of the N-terminal arms and one at 60 °C due to melting of the globular domain (24, 54) (Fig. 3A). No clear transitions were observed for UmuD N41D; this may be because the N-terminal arms are only transiently bound to the globular domain, resulting in exposure of the most hydrophobic regions of the protein (Fig. 3A). A range of concentrations was tested above and below the determined KD for UmuD N41D with no change in denaturation profile. The addition of stabilizing agents, such as polyethylene glycol, sucrose, or glycerol also did not give a clear denaturation transition for UmuD N41D. Therefore, having the N-terminal arms bound to the dimeric globular domain may be a source of stability for the wild-type UmuD2 protein and may explain why this is the only biochemically observed conformation to date (13, 18, 37).
FIGURE 3.

Thermal shift assay of UmuD protein stability. Melting was observed by monitoring Sypro Orange fluorescence as a function of temperature. A, shown is a melting profile of wild-type UmuD2 (24) (green) and UmuD N41D (orange). B, change in calculated melting temperature between wild-type UmuD′2 (24) (navy blue) and UmuD′ N41D (magenta) variant protein at 45 μm is 6.0 °C.
In comparing the melting curves of wild-type UmuD′2 and UmuD′ N41D at 45 μm, we determined a ΔTm of 6.0 °C, with UmuD′ N41D destabilized relative to wild-type UmuD′2 (Fig. 3B). This decrease in melting temperature is likely due to the loss of stability provided through dimerization. Under the conditions of these experiments, UmuD′2 is dimeric, whereas UmuD′ N41D is monomeric. This experiment was carried out using a range of concentrations, all of which gave consistent results.
UmuD N41D Monomer Undergoes Efficient Cleavage
The cleavage of the N-terminal 24 amino acids of UmuD2 to yield the UmuD′2 homodimer is required to activate UmuC for its role in translesion DNA synthesis (1). UmuD binds to the RecA·ssDNA nucleoprotein filament, which brings together the active site residues Ser-60 and Lys-97, facilitating deprotonation of Ser-60 that cleaves the peptide bond between Cys-24 and Gly-25 (16). The RecA·ssDNA-dependent cleavage of UmuD N41D was assayed in vitro to determine whether cleavage of the monomer was possible (Fig. 4A). The concentrations used in this assay were well below the calculated KD for dimerization for UmuD N41D. A range of concentrations was used to rule out the possibility that the cleavage proficiency is concentration-dependent. UmuD N41D not only cleaves under these conditions, but the efficiency of cleavage was found to be independent of concentration and near that of wild-type UmuD2 (Fig. 4A).
FIGURE 4.

Cleavage and DinB-dependent inhibition of cleavage of UmuD N41D in vitro is comparable with wild-type UmuD2. A, cleavage products in the presence (+) and absence (−) of the RecA·ssDNA nucleoprotein filament are indicated. Wild-type UmuD2 and UmuD N41D at concentrations of 0.5, 1, 2, 5, and 10 μm were assayed. B, cleavage under alkaline conditions (pH 10) is shown. Concentrations are as in A. The percentage of cleavage product was determined as the ratio of the density of the UmuD′ band to the total density of UmuD and UmuD′ protein in each lane. C, DinB efficiently inhibits the cleavage of both UmuD N41D variant (dashed line) and wild-type UmuD (solid line) proteins to the same extent in vitro. Error bars represent S.D. for three independent experiments are shown for each point.
To rule out the possibility that UmuD N41D was able to cleave itself because the RecA·ssDNA nucleoprotein filament facilitated dimerization, a cleavage assay under alkaline conditions was carried out (Fig. 4B). This reaction occurs in the absence of additional protein or DNA co-factors. At pH 10, Ser-60 can be activated as a nucleophile without the addition of RecA·ssDNA, although cleavage efficiency is reduced. We found that the UmuD N41D monomer cleaves as efficiently as wild-type UmuD2 under these conditions, with no dependence on concentration. Together, these observations indicate that the UmuD N41D monomer is active for cleavage. Furthermore, these observations provide strong evidence to support the model that wild-type UmuD2 can cleave in the cis conformation.
DinB Inhibits UmuD N41D Cleavage
DinB, UmuD, and RecA proteins form a ternary complex in solution, which suppresses the mutagenic −1 frameshift activity of DinB (7, 55). The physical interaction between UmuD2 and DinB inhibits the RecA-facilitated cleavage of UmuD2 in vitro (7). Therefore, we tested the extent to which DinB inhibits cleavage of UmuD N41D compared with wild-type UmuD2. UmuD protein concentrations used in this experiment were significantly below the KD for dimerization of UmuD N41D. UmuD N41D and DinB were incubated separately from the rest of the components of the cleavage reaction. This ensures that UmuD N41D can bind to DinB without competition from RecA·ssDNA. We found no difference in the capacity of DinB to inhibit RecA·ssDNA-mediated cleavage of wild-type UmuD2 or UmuD N41D (Fig. 4C). The addition of 4 μm DinB to a cleavage reaction containing 2 μm wild-type UmuD2 and UmuD N41D resulted in a >33% reduction in cleavage efficiency for both UmuD proteins. Therefore, the dimerization defect of UmuD N41D does not appear to affect the molecular interactions necessary for proper DinB-UmuD interactions, which suggests that the monomer is functionally active and capable of physical interactions with DinB in this context.
UmuD N41D and UmuD′ N41D Are Expressed as Active Monomers in Vivo
We next determined the proficiency of UmuD and UmuD′ N41D variants for in vivo functions. The expression level and cleavage activity in vivo of wild-type UmuD2 and UmuD N41D expressed from plasmids were determined by immunoblotting under native and denaturing conditions. We UV-irradiated cells harboring low-copy plasmids that expressed UmuD2 or UmuD′2 or the N41D variants and resolved the proteins by electrophoresis under denaturing conditions. The extent of cleavage observed for wild-type UmuD2 and the UmuD N41D monomer was 39 and 41%, respectively (Fig. 5A). The expression levels for wild-type and variant proteins were similar (Fig. 5A). We also used gentle, non-denaturing extraction conditions and analyzed the cellular proteins by native gel electrophoresis. Under native conditions, we found that UmuD N41D and UmuD′ N41D are both resolved as monomers with comparable expression levels as seen under denaturing conditions. Full-length UmuD2 and cleaved UmuD′ N41D can also be seen on the blot, thus, confirming the cleavage proficiency of the monomer in vivo. However, for wild-type UmuD2 under native conditions, only a single band was observed (Fig. 5B). This was intriguing as we would expect both full-length and cleaved UmuD2 proteins to be present as their dimeric forms. Western blots of proteins resolved under denaturing conditions were performed on the identical samples, and the presence of both UmuD and UmuD′ proteins in nearly equivalent amounts was observed (Fig. 5C). From this, we conclude that the UmuDD′ heterodimer is likely the most stable conformation in vivo. Although this phenomenon was previously observed in vitro by using cross-linking (56), we believe this is the first report of heterodimer formation observed in vivo. These findings shed new light on the predominant conformation of wild-type UmuD after UV irradiation and highlight the possibility that the heterodimer may play a larger role in this regard.
UmuD and UmuD′ Rapidly Form Heterodimers in Vitro
It has been known for some time that UmuD2 forms exchangeable dimers and that UmuD2 and UmuD′2 form heterodimers (8, 16, 56). Glutaraldehyde cross-linking of equimolar amounts of UmuD2 and UmuD′2 was previously used to demonstrate the preferential formation of the UmuDD′ heterodimer. In this case, no UmuD2 or UmuD′2 homodimers could be detected after 20 min, leading the authors to suggest that the heterodimer complex was favored (56). Subsequently, it was shown that the arms of the active site variant UmuD2 S60A could be cleaved by incubating with a non-cleavable variant in which the cleavage site is mutated but the active site is intact (36). From this and similar experiments, it was concluded that the dimers must be exchangeable for cleavage to occur and that cleavage occurs in trans (8, 36).
By mixing equal amounts of wild-type UmuD2 and UmuD′2 under equilibrium conditions, we have found that the heterodimer conformation is indeed the most stable conformation (Fig. 5E). This is consistent with our observations in vivo (Fig. 5B). We detect 71% heterodimer formation with 29% UmuD′ present, most likely as a result of cleavage during incubation and/or electrophoresis (Fig. 5E). We do not observe full-length UmuD2 homodimer. Experiments were also performed to determine the ability of monomeric UmuD N41D and UmuD′ N41D to form heterodimers or to form exchangeable dimers with wild-type UmuD and UmuD′ (Fig. 5E). Heterodimerization did not occur with any combination of proteins. Notably, UmuD N41D or UmuD′ N41D are unable to form dimers even with wild-type UmuD or UmuD′ as partners.
UmuD N41D and UmuD′ N41D Facilitate UV Mutagenesis and Survival
UmuD′2C is required for UV-induced mutagenesis in E. coli (1). As part of its mutagenic signature, polymerase V inserts guanine opposite the 3′-thymine of (6-4) T-T photoproducts that are a result of exposure to UV light (57, 58). This activity can be detected via the reversion of the argE3 auxotrophic marker in the arginine biosynthetic pathway (31). To assess the proficiency of UmuD N41D monomers in UV-induced mutagenesis, we compared the mutation frequency of ΔumuDC strains harboring plasmid-borne wild-type UmuD2 and UmuD′2 as well as UmuD N41D and UmuD′ N41D variants. We found that the mutation frequency of cells expressing UmuD N41D and UmuD′ N41D were similar to those expressing wild-type UmuD2 and UmuD′2, respectively (Fig. 6A). This suggests that monomeric UmuD′ is able to interact with the numerous protein partners required for mutagenesis, including UmuC, RecA, and the β clamp (1).
FIGURE 6.

UmuD and UmuD′ N41D are proficient for UV-induced mutagenesis and confer resistance to ultraviolet radiation. A, shown is UV-induced mutation frequency for wild-type UmuD or UmuD′ and UmuD N41D or UmuD′ N41D in plasmids pGY9739 (umuDC) or pGY9738 (umuD′C) and empty vector pGB2 in strain GW8017. Mutation frequencies are reported relative to that of GW8017 pGY9739, which is set to 100%. B, survival assays were carried out with pGY9739 (umuDC) and pGY9738 (umuD′C) plasmids in PB103. pGY9738-N41D (■, umuD′C-N41D; pGY9738 (♦, umuD′C); pGY9739-N41D (▴, umuDC-N41D); pGY9739 (●, umuDC); pGB2 (—, empty vector). Error bars representing S.D. from at least three experiments are shown.
AB1157 ΔumuDC ΔrecJ strains are hypersensitive to UV light (59), a phenotype that can be suppressed by complementation with low-copy plasmids bearing the umuDC genes. We determined that ΔumuDC ΔrecJ strains harboring plasmids expressing UmuD N41D and UmuD′ N41D were not sensitive to UV light (Fig. 6B). Even though UmuD N41D and UmuD′ N41D are defective in dimerization, the level of resistance to UV light for strains harboring these variants surpassed that of strains expressing the wild-type UmuD and UmuD′ proteins. As highlighted above, the expression levels of these variants are similar to those of wild-type UmuD and UmuD′, and the cleavage efficiency of UmuD N41D is also comparable with that of wild-type UmuD2 both in vivo and in vitro (Figs. 4 and 5). Notably, defects in UmuD2 cleavage, as with the UmuD2 S60A active site variant, result in hypersensitivity to killing by UV light, whereas defects in dimerization apparently confer UV resistance (56, 60). These observations confirm that dimeric UmuD2 and UmuD′2 are not essential for UV survival or for overall cell viability.
UmuD N41D Interacts with the β Clamp as a Monomer
Interactions between the UmuD proteins and the homodimeric β clamp have been well characterized using cross-linking methods (61, 62). It was concluded from these experiments that residues on both the N-terminal arms and C-terminal globular domain of UmuD2 interact with the β clamp (62, 63). Truncations constructed in the N-terminal arms of UmuD2 indicate that residues 9–19 are particularly important for binding; therefore, the affinity of UmuD′2 for the β clamp is weaker (62). We utilized formaldehyde cross-linking to assess the relative binding affinity of wild-type UmuD2 versus the UmuD N41D monomer for the β clamp (Fig. 7). Results clearly demonstrate that there is a difference in the stoichiometry of binding, although the resolution of the experiment does not allow for precise quantification. In the case of cross-linked UmuD2 and the β clamp, the multiple bands observed may indicate the binding of one or two UmuD dimers per dimeric β clamp. As this experiment was completed under non-denaturing conditions, wild-type UmuD2 is most likely cross-linked to the β clamp as a dimer. Furthermore, we included the active site variant UmuD2 S60A to confirm that UmuD′2 is not one of the forms detected. Conversely, cross-linking the UmuD N41D monomer to the β clamp produced one clean band. Considering the scenario described above, we conclude that the complex detected is composed of two UmuD N41D monomers in complex with the β clamp, with one UmuD monomer binding to each β clamp monomer.
FIGURE 7.

UmuD N41D cross-links to the β clamp as a monomer. Formaldehyde was used as a cross-linking agent. A native gel shows wild-type UmuD2 or the UmuD N41D monomer cross-linked to the β clamp. The reaction contained equimolar amounts (10 μm) of UmuD proteins and the β clamp.
DISCUSSION
In this report we have used numerous in vitro and in vivo methods to characterize UmuD N41D and UmuD′ N41D as active monomers that functionally mimic the wild-type dimers. It was found that alkaline- and RecA-mediated cleavage for both wild-type UmuD2 dimer and UmuD N41D monomer proteins occurs with similar efficiency. Moreover, cleavage of the N-terminal arms of the UmuD N41D monomer shows that it is a stable and active species and also lends to the conclusion that the wild-type UmuD dimer may cleave in cis. The conformational dynamics of the UmuD and UmuD′ monomer and dimer proteins was further investigated using the thermofluor assay, which clearly shows a difference in overall structural stability and hydrophobicity. It is also evident that the UmuD monomer interacts with partner proteins such as DinB and the β clamp in vitro and is active in facilitating mutagenesis in vivo.
We have estimated the KD for dimerization for monomeric UmuD N41D as ∼52 μm. This is >6 orders of magnitude greater than that determined for wild-type UmuD2 (18). As only a single mutation was made in the N-terminal neck region of the protein, this region appears to be critical for modulating the dimerization stability of wild-type UmuD2 and UmuD′2. Certainly, the charge-charge repulsion is most likely the key contributor to the dimerization defect, as an N-terminal truncation of wild-type UmuD′ through residue 45 reportedly does not affect dimer formation (25).
Intriguingly, LexA is an extremely tight dimer, with a KD of less than 20 pm (64), but unlike wild-type UmuD, the LexA protein sequence contains residue Asp-101 corresponding to UmuD Asn-41 (Fig. 1C). On further inspection, the LexA crystal structure (47) (PDB code 1JHH) shows the carboxyl groups of the Asp-101 residues of each monomer pointing away from the interface and exposed to solvent. Therefore, the destabilizing charge-charge repulsion that is attributed to the dimerization defect of UmuD N41D is averted in LexA.
The dynamic nature of UmuD proteins is also evident in the results of thermofluor assays on the UmuD N41D and UmuD′ N41D monomers. A sharp melting transition was not obtained for full-length UmuD N41D monomer even with the addition of a host of stabilizing agents. However, the melting profile for wild-type UmuD2 has been reported as two distinct unfolding transitions that represent the dissociation of the N-terminal arms at ∼30 °C and the melting of the dimeric globular domain at 60 °C, respectively (24). As previously observed, there is a single melting transition at 60 °C for wild-type UmuD′2 (24). The Tm of 54 °C for the monomeric UmuD′ N41D most likely represents unfolding of the globular domain, and we attribute this reduction in melting temperature as compared with wild-type UmuD′2 to the loss of the stability that is provided through dimerization. Moreover, this study demonstrates the significance of the N-terminal arms and their considerable contributions to stability. Although the contacts within the dimer interface of the C-terminal domains are important, the wrapping of the UmuD2 arms across the globular domain also seems to provide additional structural support that is not present in the UmuD N41D variant.
UmuD2 has been categorized as an IDP (18). These proteins exhibit a high degree of flexibility that allows the accommodation of a large number of protein-protein contacts (65). IDPs defy the traditional structure-function paradigm, as it is not essential to fold into a stable three-dimensional structure for basic cellular function (65). These proteins function via molecular recognition where the structure of the IDP is altered upon binding to a partner protein or as effectors that modify the activity of a single binding partner or a protein complex (66). The dimeric UmuD proteins have been shown to interact with a growing list of partners including translesion DNA synthesis polymerases UmuC and DinB, the α, β, and ϵ subunits of the replicative polymerase III, RecA, and the Lon and ClpXP proteases (1, 6–12). Therefore, UmuD2 is reminiscent of a hub protein that may be stabilized by binding to multiple structured interacting partners that allow for the regulation of protein expression, multiprotein complex formation, and degradation as required (18, 65). Many of these interactions may be transient but highly specific. Regardless, evolutionary selection of a dimeric over monomeric UmuD protein has advantages. Not only is dimeric UmuD2 structurally more stable, but it can also adopt more conformations and potentially bind twice as many interacting partners at any given time.
Monomeric UmuD N41D undergoes RecA·ssDNA-facilitated cleavage of its N-terminal arms as well as cleavage under alkaline conditions (Fig. 4, A and B). Cleavage is independent of protein concentration, indicating that the UmuD N41D monomer cleaves via an intramolecular reaction. Interestingly, the monomeric λ cI repressor protein also undergoes RecA·ssDNA-facilitated intramolecular self-cleavage (67). It was previously shown that wild-type UmuD2 with the N-terminal arms in the trans conformation cleaves to form UmuD′2 by an intermolecular pathway (36). The idea of trans or intermolecular cleavage was first confirmed by introducing plasmids carrying active site mutants (S60A and K97A) and cleavage site mutants (C24D and G25D) into E. coli ΔumuDC strains (36). Cleavage was only observed when both active site and cleavage site mutants were introduced together into cells, suggesting that the reaction is intermolecular (36). The UmuD2 ortholog MucA also undergoes intermolecular cleavage (36). However, isoenergetic models of UmuD2 with the N-terminal arms in the cis conformation led us to consider intramolecular cleavage as a viable mechanism (8). This is in agreement with the cleavage of other UmuD-like proteins such as LexA and λ cI that are primarily intramolecular in nature (46). As monomeric UmuD N41D must cleave itself intramolecularly, it is plausible that wild-type UmuD cleaves itself both intermolecularly and intramolecularly.
Other evidence also suggests that the UmuD N41D monomer may be a competitive substrate for RecA-facilitated cleavage (35). It was determined that cross-linking residues close to the dimer interface of UmuD2 reduced the cleavage efficiency by the RecA nucleoprotein filament substantially (35). The monocysteine derivatives used include V34C, I38C, and L44C, which are in the vicinity of the N-terminal neck region where the key interactions for dimerization occur (35). Because UmuD N41D is proficient for cleavage, the essential interactions with RecA that are required for this process are still intact. Therefore, the RecA nucleoprotein filament may only be needed to activate the catalytic site of UmuD by inducing Ser-60 and Lys-97 to adopt the correct conformation for cleavage (16).
It has been generally accepted that a defect in dimerization of the UmuD proteins would render UmuD inactive in most, if not all, of its functions and make cells non-mutable (25, 28, 68, 69). Indeed, UmuD′ variants A50V, T51I, H82Y, or G129S (68), all suggested to disrupt dimerization, result in reduced levels of spontaneous or induced mutagenesis (28, 68). In contrast, we have shown that the UmuD N41D and UmuD′ N41D monomers are active in facilitating mutagenesis and cell survival after treatment with DNA-damaging UV light. Moreover, the UmuD N41D monomer has little or no defect in cleavage either in vitro or in vivo. Taken together, our observations indicate that although UmuD2 is a tight dimer, dimerization is not required for the cellular functions of UmuD in regulating mutagenesis.
Acknowledgments
We thank April Gu in Civil and Environmental Engineering at Northeastern University for use of the real time PCR instrument. Srinivas Somarowthu and Alexandra Wallace are kindly acknowledged for help and technical assistance. We thank Česlovas Venclovas for careful reading of the manuscript.
This work was supported by a New Faculty Award from the Camille and Henry Dreyfus Foundation (to P. J. B.), the National Science Foundation (CAREER Award, MCB-0845033 to P. J. B.), and the Northeastern University Office of the Provost.
- IDP
- intrinsically disordered proteins
- NDS
- non-denaturing sample buffer
- NDE
- non-denaturing electrode buffer
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. Freidberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., Ellenberger T. (2005) DNA Repair and Mutagenesis, 2nd Ed., American Society for Microbiology, Washington, D. C. Press, Washington, D. C. [Google Scholar]
- 2. Opperman T., Murli S., Smith B. T., Walker G. C. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 9218–9223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gonzalez M., Woodgate R. (2002) Bioessays 24, 141–148 [DOI] [PubMed] [Google Scholar]
- 4. Reuven N. B., Arad G., Maor-Shoshani A., Livneh Z. (1999) J. Biol. Chem. 274, 31763–31766 [DOI] [PubMed] [Google Scholar]
- 5. Tang M., Shen X., Frank E. G., O'Donnell M., Woodgate R., Goodman M. F. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 8919–8924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sutton M. D., Guzzo A., Narumi I., Costanzo M., Altenbach C., Ferentz A. E., Hubbell W. L., Walker G. C. (2002) DNA Repair 1, 77–93 [DOI] [PubMed] [Google Scholar]
- 7. Godoy V. G., Jarosz D. F., Simon S. M., Abyzov A., Ilyin V., Walker G. C. (2007) Mol. Cell 28, 1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beuning P. J., Simon S. M., Zemla A., Barsky D., Walker G. C. (2006) J. Biol. Chem. 281, 9633–9640 [DOI] [PubMed] [Google Scholar]
- 9. Nohmi T., Battista J. R., Dodson L. A., Walker G. C. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 1816–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gonzalez M., Frank E. G., Levine A. S., Woodgate R. (1998) Genes Dev. 12, 3889–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frank E. G., Ennis D. G., Gonzalez M., Levine A. S., Woodgate R. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10291–10296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sutton M. D., Opperman T., Walker G. C. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 12373–12378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neher S. B., Sauer R. T., Baker T. A. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13219–13224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ollivierre J. N., Fang J., Beuning P. J. (2010) J. Nucleic Acids 2010, Article ID 947680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferentz A. E., Opperman T., Walker G. C., Wagner G. (1997) Nat. Struct. Biol. 4, 979–983 [DOI] [PubMed] [Google Scholar]
- 16. Ferentz A. E., Walker G. C., Wagner G. (2001) EMBO J. 20, 4287–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peat T. S., Frank E. G., McDonald J. P., Levine A. S., Woodgate R., Hendrickson W. A. (1996) Nature 380, 727–730 [DOI] [PubMed] [Google Scholar]
- 18. Simon S. M., Sousa F. J., Mohana-Borges R., Walker G. C. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1152–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dyson H. J., Wright P. E. (2005) Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 20. Radivojac P., Iakoucheva L. M., Oldfield C. J., Obradovic Z., Uversky V. N., Dunker A. K. (2007) Biophys. J. 92, 1439–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uversky V. N., Oldfield C. J., Dunker A. K. (2005) J. Mol. Recognit. 18, 343–384 [DOI] [PubMed] [Google Scholar]
- 22. Guzzo A., Lee M. H., Oda K., Walker G. C. (1996) J. Bacteriol. 178, 7295–7303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohta T., Sutton M. D., Guzzo A., Cole S., Ferentz A. E., Walker G. C. (1999) J. Bacteriol. 181, 177–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fang J., Rand K. D., Silva M. C., Wales T. E., Engen J. R., Beuning P. J. (2010) J. Mol. Biol. 398, 40–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peat T. S., Frank E. G., McDonald J. P., Levine A. S., Woodgate R., Hendrickson W. A. (1996) Structure 4, 1401–1412 [DOI] [PubMed] [Google Scholar]
- 26. Finger C., Volkmer T., Prodöhl A., Otzen D. E., Engelman D. M., Schneider D. (2006) J. Mol. Biol. 358, 1221–1228 [DOI] [PubMed] [Google Scholar]
- 27. Vinson C., Myakishev M., Acharya A., Mir A. A., Moll J. R., Bonovich M. (2002) Mol. Cell. Biol. 22, 6321–6335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beuning P. J., Chan S., Waters L. S., Addepalli H., Ollivierre J. N., Walker G. C. (2009) J. Bacteriol. 191, 5910–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sommer S., Knezevic J., Bailone A., Devoret R. (1993) Mol. Gen. Genet. 239, 137–144 [DOI] [PubMed] [Google Scholar]
- 30. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 31. Beuning P. J., Simon S. M., Godoy V. G., Jarosz D. F., Walker G. C. (2006) Methods Enzymol. 408, 318–340 [DOI] [PubMed] [Google Scholar]
- 32. Churchward G., Belin D., Nagamine Y. (1984) Gene 31, 165–171 [DOI] [PubMed] [Google Scholar]
- 33. Singleton S. F., Simonette R. A., Sharma N. C., Roca A. I. (2002) Protein Expr. Purif. 26, 476–488 [DOI] [PubMed] [Google Scholar]
- 34. Goodrich J. A., Kugel J. F. (2007) Binding and Kinetics for Molecular Biologists, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 35. Lee M. H., Guzzo A., Walker G. C. (1996) J. Bacteriol. 178, 7304–7307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McDonald J. P., Frank E. G., Levine A. S., Woodgate R. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 1478–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schlacher K., Goodman M. F. (2007) Nat. Rev. Mol. Cell Biol. 8, 587–594 [DOI] [PubMed] [Google Scholar]
- 38. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thompson J. D., Higgins D. G., Gibson T. J. (1994) Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mihalek I., Res I., Lichtarge O. (2004) J. Mol. Biol. 336, 1265–1282 [DOI] [PubMed] [Google Scholar]
- 41. Schumann W. (2006) Dynamics of the Bacterial Chromosome: Structure and Function, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 42. Shiba T., Iwasaki H., Nakata A., Shinagawa H. (1990) Mol. Gen. Genet. 224, 169–176 [DOI] [PubMed] [Google Scholar]
- 43. Perry K. L., Elledge S. J., Mitchell B. B., Marsh L., Walker G. C. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 4331–4335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Slilaty S. N., Little J. W. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 3987–3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sauer R. T., Yocum R. R., Doolittle R. F., Lewis M., Pabo C. O. (1982) Nature 298, 447–451 [DOI] [PubMed] [Google Scholar]
- 46. Little J. W. (1993) J. Bacteriol. 175, 4943–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luo Y., Pfuetzner R. A., Mosimann S., Paetzel M., Frey E. A., Cherney M., Kim B., Little J. W., Strynadka N. C. (2001) Cell 106, 585–594 [DOI] [PubMed] [Google Scholar]
- 48. Perry K. L., Walker G. C. (1982) Nature 300, 278–281 [DOI] [PubMed] [Google Scholar]
- 49. Glazebrook J. A., Grewal K. K., Strike P. (1986) J. Bacteriol. 168, 251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lodwick D., Owen D., Strike P. (1990) Nucleic Acids Res. 18, 5045–5050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ho C., Kulaeva O. I., Levine A. S., Woodgate R. (1993) J. Bacteriol. 175, 5411–5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kulaeva O. I., Wootton J. C., Levine A. S., Woodgate R. (1995) J. Bacteriol. 177, 2737–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sobolev V., Sorokine A., Prilusky J., Abola E. E., Edelman M. (1999) Bioinformatics 15, 327–332 [DOI] [PubMed] [Google Scholar]
- 54. Ericsson U. B., Hallberg B. M., Detitta G. T., Dekker N., Nordlund P. (2006) Anal. Biochem. 357, 289–298 [DOI] [PubMed] [Google Scholar]
- 55. Foti J. J., Delucia A. M., Joyce C. M., Walker G. C. (2010) J. Biol. Chem. 285, 23086–23095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Battista J. R., Ohta T., Nohmi T., Sun W., Walker G. C. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 7190–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tang M., Pham P., Shen X., Taylor J. S., O'Donnell M., Woodgate R., Goodman M. F. (2000) Nature 404, 1014–1018 [DOI] [PubMed] [Google Scholar]
- 58. Becherel O. J., Fuchs R. P. (1999) J. Mol. Biol. 294, 299–306 [DOI] [PubMed] [Google Scholar]
- 59. Courcelle C. T., Chow K. H., Casey A., Courcelle J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 9154–9159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Opperman T., Murli S., Walker G. C. (1996) J. Bacteriol. 178, 4400–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Duzen J. M., Walker G. C., Sutton M. D. (2004) DNA Repair 3, 301–312 [DOI] [PubMed] [Google Scholar]
- 62. Sutton M. D., Narumi I., Walker G. C. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 5307–5312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dalrymple B. P., Kongsuwan K., Wijffels G., Dixon N. E., Jennings P. A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 11627–11632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mohana-Borges R., Pacheco A. B., Sousa F. J., Foguel D., Almeida D. F., Silva J. L. (2000) J. Biol. Chem. 275, 4708–4712 [DOI] [PubMed] [Google Scholar]
- 65. Dunker A. K., Cortese M. S., Romero P., Iakoucheva L. M., Uversky V. N. (2005) FEBS J. 272, 5129–5148 [DOI] [PubMed] [Google Scholar]
- 66. Tompa P. (2002) Trends Biochem. Sci. 27, 527–533 [DOI] [PubMed] [Google Scholar]
- 67. Gimble F. S., Sauer R. T. (1989) J. Mol. Biol. 206, 29–39 [DOI] [PubMed] [Google Scholar]
- 68. McLenigan M., Peat T. S., Frank E. G., McDonald J. P., Gonzalez M., Levine A. S., Hendrickson W. A., Woodgate R. (1998) J. Bacteriol. 180, 4658–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fujii S., Gasser V., Fuchs R. P. (2004) J. Mol. Biol. 341, 405–417 [DOI] [PubMed] [Google Scholar]
- 70. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 71. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H. (2006) Mol. Syst. Biol. 2, 2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]