

Abstract
DNA polymerases catalyze the incorporation of deoxynucleoside triphosphates into a growing DNA chain using a pair of Mg2+ ions, coordinated at the active site by two invariant aspartates, whose removal by mutation typically reduces the polymerase activity to barely detectable levels. Using two stopped-flow fluorescence assays that we developed previously, we have investigated the role of the carboxylate ligands, Asp705 and Asp882, of DNA polymerase I (Klenow fragment) in the early prechemistry steps that prepare the active site for catalysis. We find that neither carboxylate is required for an early conformational transition, reported by a 2-aminopurine probe, that takes place in the open ternary complex after binding of the complementary dNTP. However, the subsequent fingers-closing step requires Asp882; this step converts the open ternary complex into the closed conformation, creating the active-site geometry required for catalysis. Crystal structures indicate that the Asp882 position changes very little during fingers-closing; this side chain may therefore serve as an anchor point to receive the dNTP-associated metal ion as the nucleotide is delivered into the active site. The Asp705 carboxylate is not required until after the fingers-closing step, and we suggest that its role is to facilitate the entry of the second Mg2+ into the active site. The two early prechemistry steps that we have studied take place normally at very low Mg2+ concentrations, although higher concentrations are needed for covalent nucleotide addition, consistent with the second metal ion entering the ternary complex after fingers-closing.
Keywords: DNA Polymerase, Enzyme Mechanisms, Enzyme Mutation, Fluorescence, Kinetics, Protein Metal Ion Interaction, Conformational Change
Introduction
DNA polymerases catalyze phosphodiester bond formation between the 3′-OH of a DNA primer strand and the α-phosphorus of an incoming deoxynucleoside triphosphate. Before covalent bond formation, the enzyme-substrate complex undergoes a series of noncovalent changes that prepare the active site for the chemical step and also serve as kinetic checkpoints to ensure selection of the correct nucleotide (1). In the Klenow fragment of DNA polymerase I (Pol I(KF))2, the subject of the present study, there is evidence for at least three prechemistry steps following the binding of nucleotide, as shown in the reaction pathway of Fig. 1.
FIGURE 1.

Reaction pathway for Pol I(KF) from DNA binding up to phosphoryl transfer. DNA* (step 2.1) represents a DNA rearrangement detected with a 2-AP probe 5′ to the templating position. EO and EC (step 2.2) represent the open and closed conformations of the polymerase. EC to EC‡ (step 3) represents a further transformation, whose identity is not known, corresponding to the rate-limiting step in single nucleotide incorporation. The existence of additional, as yet undetected, steps is not ruled out.
Two prechemistry steps in Pol I(KF) have been observed directly by the use of fluorescent probes. The first of these conformational transitions (step 2.1) is a DNA rearrangement, detected by a 2-aminopurine (2-AP) reporter placed 5′ to the templating base (2). The following step (step 2.2), detected using a FRET-based assay (3), is the closing of the fingers subdomain, which had been inferred from the comparison of DNA polymerase binary (Pol-DNA) and ternary (Pol-DNA-dNTP) cocrystal structures (Fig. 2) (4–8). The order of these two conformational transitions in the reaction pathway was deduced from the behavior of ribonucleotide substrates. With an incoming rNTP complementary to the templating base, the DNA rearrangement takes place normally, but fingers-closing is strongly inhibited (3). It follows, therefore, that the intermediate detected by the 2-AP probe is a Pol-DNA-dNTP complex with the fingers subdomain in an open conformation. Significantly, neither of the prechemistry steps 2.1 or 2.2 is observed when the incoming dNTP is mispaired with the templating base (2, 3), indicating the existence of a very early checkpoint that detects mispairs.
FIGURE 2.

Experimental design and the position of the mutants. The α-carbons of the binary complex (Protein Data Bank entry 1L3U) and of the ternary complex (Protein Data Bank entry 1LV5) of Bst DNA polymerase (6) were aligned using PyMOL (DeLano Scientific LLC). Both complexes were essentially identical except for the mobile portion of the fingers subdomain (residues 680–714, equivalent to 732–766 of Pol I(KF), shown in blue in the binary and gray in the ternary complex; the non-mobile portion of the protein (from 1L3U) is shown in cyan. The residues Asp705 and Asp882 of Pol I(KF), used in our study, correspond to the residues Asp653 and Asp830 of Bst pol, colored in yellow and green, respectively. The red spheres indicate the attachment point for the donor fluorophore on the protein (residue 692 of Bst pol, equivalent to residue 744 of Pol I(KF)). The DNA primer-template is shown in orange, with the attachment position of the dabcyl quencher on the template strand shown as a black sphere.
In Pol I(KF), there is evidence for at least one additional noncovalent step after the fingers-closing transition. Comparison of product yields in pulse-chase and pulse-quench experiments provided evidence for a noncovalent rate-limiting step preceding phosphoryl transfer (9), and rate considerations placed this slow prechemistry step (step 3) after fingers-closing (3). Currently, the precise nature of this step remains a mystery.
All polymerases share a two-metal ion catalytic mechanism, which is clearly visualized in polymerase ternary complex cocrystal structures (reviewed in Ref. 8). The catalytic metal is normally Mg2+, but Mn2+ can substitute. One metal ion (metal A) coordinates the 3′-OH of the primer and facilitates its nucleophilic attack on the α-phosphate of the incoming nucleotide. The other metal ion (metal B) coordinates the β- and γ-phosphate oxygens of the nucleotide and thus stabilizes the leaving group in the phosphoryl transfer reaction. Additionally, both metal ions can stabilize negative charge in the pentacovalent intermediate or transition state. It is reasonable to assume that metal B enters the polymerase complex along with the dNTP; however, it is currently unclear when metal A becomes bound. Experiments with DNA polymerase β and RB69 DNA polymerase, using exchange-inert dNTP complexes with Cr(III) or Rh(III), show that some early steps of the polymerase reaction can indeed take place when the only metal ion present is in the metal B position, coordinated to the dNTP phosphates (10–13). The subsequent addition of a second metal ion to fill the metal A site is required before phosphoryl transfer can take place.
Crystal structures of a large number of nucleic acid polymerases complexed with DNA and nucleotide show the two catalytic metal ions bound at the polymerase active site by two carboxylate side chains that are invariant throughout the entire polymerase superfamily (8, 14, 15). In Pol I(KF), an A-family DNA polymerase, the carboxylate ligands, located in the palm subdomain, are Asp705 and Asp882 (Fig. 2). Substitution of either of these side chains by alanine reduced polymerase activity to almost undetectable levels (16, 17). Although the mutational studies clearly established that Asp705 and Asp882 are essential for nucleotide incorporation, they did not indicate which step(s) of the reaction pathway is affected. In this study, we have investigated the D705A and D882A Pol I(KF) mutants using the two fluorescence assays described above, which report directly on early conformational transitions in the polymerase reaction. The results indicated which prechemistry steps are dependent on the aspartates and, by implication, on their coordinated metal ions, and suggested a sequence of events that assembles the polymerase active site for catalysis of phosphoryl transfer.
EXPERIMENTAL PROCEDURES
Materials
DNA oligonucleotides were synthesized by the Keck Biotechnology Resource Laboratory at Yale Medical School. Oligonucleotides for fluorescence and kinetics experiments were purified by denaturating gel electrophoresis. Ultrapure deoxynucleotides and [γ-32P]ATP were purchased from GE Healthcare. 5-(((2-iodoacetyl)amino)ethyl)aminonaphthalene-1-sulfonic acid (IAEDANS) and tris-(2-carboxyethyl)phosphine, were purchased from Molecular Probes (Eugene, OR).
Expression and Purification of Pol I(KF) and Mutant Derivatives
The Pol I(KF) construct used for fluorophore labeling has been described previously (3). It has an N-terminal hexahistidine tag for purification, the D424A mutation to inactivate the 3′-5′ exonuclease, the C907S mutation to remove the single native cysteine, and the L744C mutation to provide a unique labeling site on the fingers subdomain. For simplicity, the N-His6, D424A, C907S, L744C genotype will be referred to as wild type (WT) throughout. The D705A and D882A mutations were introduced into the Pol I(KF) construct described above, using the QuikChange site-directed mutagenesis kit (Stratagene), according to the manufacturer's instructions. Pol I(KF) derivatives were expressed and purified to homogeneity as described previously (3, 18).
Labeling of Pol I(KF) Derivatives with IAEDANS
The proteins carrying a single cysteine at position 744 were labeled with a 2–3-fold molar excess of IAEDANS as described previously (3). Excess fluorophore was removed by gel filtration on a Bio-Spin 30 column. Labeled proteins were stored at −20 °C in 50 mm Tris-HCl, pH 7.5, 1 mm DTT, 40% (v/v) glycerol. The extent of labeling, calculated from the UV spectrum, was typically ≥70%.
Chemical Quench Experiments
Single-turnover measurements of nucleotide incorporation were carried out at room temperature (22 °C) in a rapid quench-flow instrument (KinTek Corp., model RQF-3) for fast reactions or with manual quenching when the reaction was slow. The DNA substrate was the linear duplex L:unmod:3′OH (Fig. 3A), consisting of a 13-mer primer, 5′-labeled with 32P, annealed to a 1.5-fold molar excess of the complementary 19-mer, with A as the templating base. The enzyme-DNA solution contained 0.5 μm polymerase and 0.1 or 0.05 μm primer-template duplex. The reaction was initiated by mixing the enzyme-DNA solution with an equal volume of a dNTP solution. The standard reaction buffer was 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, and 10 mm MgCl2. In some experiments, the concentration of MgCl2 or MnCl2 was varied, as indicated. Reactions were quenched at appropriate time intervals using excess EDTA and were fractionated on denaturating polyacrylamide-urea gels and quantitated on a Fuji FLA 5100 scanner.
FIGURE 3.

Oligonucleotides used in this study. A, DNA substrate used in chemical quench experiments. The 13-mer primer, 5′-labeled with 32P (indicated as an asterisk), was annealed to the 19-mer template. B, DNA duplex oligonucleotide used for 2-AP fluorescence measurements. The 2-AP fluorophore (X) was placed 5′ to the templating base, designated as the T(+1) position (relative to the templating position as 0 in our numbering system). C, hairpin DNA substrate used in FRET-based fluorescence measurements of fingers-closing. The dabcyl-dT quencher was placed at the T(−8) position. D, as in C but without the dabcyl quencher, used as a control for the FRET-based measurements. Except for A, the primer strand was dideoxy-terminated (3′H) so as to prevent the reaction from proceeding beyond the ternary complex. In some experiments, extendable (3′OH) versions of B–D were also used. All DNA substrates employed essentially the same duplex DNA sequence. The observed rates of fluorescence changes or of dNTP incorporation in our assays are altered very little by changing the templating base (2, 3).
Fluorescence Emission Spectra
Steady-state fluorescence spectra were recorded at 22 °C using a Photon Technology International scanning spectrofluorometer. The solutions for 2-AP fluorescence measurements contained 1 μm L:T(+1)2-AP:3′H duplex DNA (Fig. 3B; formed by annealing the T(+1)2-AP template strand with a 1.5-fold molar excess of the dideoxy-terminated complementary primer strand) and 2 μm wild-type or 4 μm mutant Pol I(KF) in 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 10 mm MgCl2, and 0.1 mg/ml BSA. The polymerase/DNA ratio was selected as that which gave the maximum increase in the fluorescence of the 2-AP DNA duplex. The complementary nucleotide, dGTP, was added to a final concentration of 200 μm dGTP for wild-type Pol I(KF) or 1 mm for the mutants. Samples were excited at 310 nm, and emission spectra were scanned from 330 to 460 nm. Spectra were corrected by subtraction of the fluorescence emission contributed by the identical concentration of Pol I(KF) in buffer and were adjusted to account for the dilution due to the addition of the dNTP solution.
Emission spectra of AEDANS-labeled Pol I(KF) complexes with the dabcyl-modified H:T(−8)D:3′H DNA (Fig. 3C) were recorded using solutions containing 1 μm labeled polymerase and 2 μm DNA (for wild type) or 4 μm DNA (for the mutants) in 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 10 mm MgCl2, and 0.1 mg/ml BSA. The polymerase/DNA ratio was chosen so as to obtain the maximum change in the fluorescence of the AEDANS-labeled protein. Ternary complexes were formed by adding the complementary nucleotide, dTTP, to a final concentration of 200 μm for wild-type Pol I(KF) or 1 mm for the mutants. AEDANS-labeled samples were excited at 350 nm, and emission spectra were collected from 360 to 650 nm. All spectra were corrected by subtraction of the buffer background and for the dilution factor.
Stopped-flow Fluorescence
Stopped-flow experiments were performed at 22 °C using an Applied Photophysics SX.18MV spectrofluorometer. For measurements using the T(+1) 2-AP reporter, one drive syringe contained a solution of L:T(+1)2-AP:3′H (Fig. 3B) duplex DNA with wild-type or mutant Pol I(KF), and the other contained dGTP, complementary to the templating base. The buffer in both syringes was 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 10 mm MgCl2. Rapid mixing of the two solutions gave final concentrations of 1 μm DNA and 2 μm wild-type or 4 μm mutant Pol I(KF), with varied dGTP concentrations (indicated in the figures). Some experiments were carried out using Mn2+, at 5 mm MnCl2 (for wild-type) or 10 mm MnCl2 (for D705A). In the experiments carried out at low MgCl2 concentrations with an extendable DNA substrate, one drive syringe contained 0.4 μm L:T(+1)2-AP:3′OH duplex DNA (Fig. 3B) and 1.8 μm wild-type Pol I(KF) in 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, and no MgCl2. The other syringe contained 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 2 mm dGTP, and various concentrations of MgCl2. The MgCl2 concentrations were chosen so that, after mixing with the E-DNA solution, the final total Mg2+ concentrations ranged from 0.07 to 0.7 mm (as indicated in Fig. 7), giving 0.25–5.8 μm free Mg2+ and 3.1–67 μm Mg-dGTP, as described by Bakhtina et al. (12). The excitation wavelength for the 2-AP probe was 313 nm, and fluorescence emission was detected using a 345-nm long pass filter.
FIGURE 7.

A single metal ion is sufficient for open and closed ternary complex formation; the second metal ion is needed for nucleotide incorporation. A, stopped-flow fluorescence assay for step 2.1. The binary complex of Pol I(KF) and L:T(+1)2-AP:3′OH (Fig. 3B) was mixed with 2 mm dGTP. B, stopped-flow fluorescence assay for fingers-closing (step 2.2). The binary complex of 744-AEDANS Pol I(KF) and H:T(−8)D:3′OH DNA duplex (Fig. 3C) was mixed with 2 mm dTTP. In both experiments, MgCl2 was omitted from the Pol-DNA drive syringe and was present only in the syringe containing the complementary nucleotide. The reaction buffer in both syringes contained 1 mm EDTA, which served to limit the free Mg2+ concentration. As described by Bakhtina et al. (12), the total MgCl2 concentrations in the reaction were 0.07, 0.2, 0.4, and 0.7 mm (indicated in the figure), corresponding to free Mg2+ concentrations of 0.25, 0.81, 2, and 5.8 μm, and Mg-dNTP concentrations of 3.1, 9.8, 25, and 67 μm. Control reactions in the presence of 3 mm MgCl2 indicate the signal obtained when covalent dNTP incorporation takes place. The fluorescence changes observed between 1 and 10 s in the presence of 0.7 mm Mg2+ correspond to the slow addition of dNTP; these changes were absent in an otherwise identical reaction using the corresponding dideoxy-terminated DNA substrate.
For FRET-based stopped-flow experiments to investigate the fingers-closing transition, one drive syringe contained AEDANS-labeled polymerase and the dabcyl-modified H:T(−8)D:3′H DNA (Fig. 3C), whereas the other contained the complementary nucleotide, dTTP. Rapid mixing of these two solutions gave final concentrations of 0.5 μm labeled Pol I(KF), 1 μm DNA (for wild type), or 2 μm DNA (for the mutants) and varied dTTP concentrations (as indicated in the figures). The buffer in both syringes was 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, and 5 mm MgCl2 (for wild type) or 10 mm MgCl2 (for the mutants). In some experiments, Mn2+ replaced Mg2+, at 2 mm MnCl2 (for wild type) or 10 mm MnCl2 (for the mutants). Experiments at low MgCl2 concentrations were carried out as described above, using the extendable H:T(−8)D:3′OH DNA and adding MgCl2 only to the drive syringe containing dTTP (at 2 mm).
To measure DNA dissociation from a binary complex with D882A Pol I(KF), one drive syringe contained 0.1 μm AEDANS-labeled polymerase with 0.2 μm of dabcyl-modified H:T(−8)D:3′OH DNA (Fig. 3C), and the other contained a 2 μm concentration of an unmodified DNA duplex. The buffer in both syringes was 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, and 10 mm MgCl2. Upon mixing the two solutions, the excess unmodified DNA acts as a trap preventing the free labeled polymerase from rebinding to dabcyl-modified DNA molecules. The resulting increase in protein fluorescence reports DNA dissociation. To measure DNA dissociation from the Pol-DNA-dNTP ternary complex, the DNA trap solution contained 2 mm dNTP.
In all stopped-flow experiments using the AEDANS reporter, the excitation wavelength was 350 nm, and fluorescence emission was detected with a 400-nm long pass filter. Stopped-flow data were collected for 10 s using a logarithmic time base, and averages were typically taken from four or more traces. Reaction rates were derived from curve fitting to exponential equations using Kaleidagraph (Synergy Software, Reading, PA).
RESULTS
Consistent with earlier data on the D705A and D882A Pol I(KF) mutants (16, 17), single-turnover rate measurements of dNTP incorporation (Table 1) demonstrate the importance of the invariant carboxylates, Asp705 and Asp882, in the polymerase reaction. Ala substitution of either side chain caused a decrease of at least 6 orders of magnitude in the rate of dNTP addition. Because Asp705 and Asp882 are ligands to divalent metal ions at the polymerase active site, we examined the effect of metal ion concentration on the polymerase activity of these mutants (supplemental Fig. S1). Wild-type Pol I(KF) had a broad Mg2+ optimum from 5 to 10 mm. The very low activity of the D705A and D882A Pol I(KF) mutants, measured at 10 mm Mg2+, was not improved by further increasing the metal ion concentration. In wild-type Pol I(KF) and the D882A mutant, substitution of Mn2+ for Mg2+ had little effect, causing only a small decrease in the reaction rate. By contrast, the use of Mn2+ instead of Mg2+ increased the activity of the D705A mutant by ∼10-fold at 20 mm metal ion and by up to 20-fold at metal ion concentrations above 50 mm.
TABLE 1.
Single-turnover rates of dNTP incorporation by wild-type Pol I(KF) and the D705A and D882A mutants
Values reported as mean ± S.D. were from two independent experiments; the others were from single determinations. The reactions were carried out under single-turnover conditions (see “Experimental Procedures”) in the presence of a 10 mm concentration of the indicated divalent metal ion for WT and D882A and 20 mm for D705A. The DNA substrate, L:unmod:3′OH (Fig. 3A), has A at the templating position.
| Proteina | kobs (s−1) in Mg2+ | kobs (s−1) in Mn2+ |
|---|---|---|
| WTb | 43 | 35 |
| D705Ac | (0.21 ± 0.07) × 10−5 | (2.5 ± 0.1) × 10−5 |
| D882Ac | (2.4 ± 0.5) × 10−5 | (1.1 ± 0.2) × 10−5 |
a In addition to the listed mutations, all proteins in this study were N-His6, D424A, L744C, and C907S.
b The reaction with WT Pol I(KF) contained 40 μm dTTP.
c Reactions with the mutant Pol I(KF) derivatives contained 500 μm dTTP. Because the reactions were extremely slow, the rates were calculated from the yield of product DNA after 4 h.
In light of the crucial role of Asp705 and Asp882 in the overall process of dNTP incorporation, further experiments in this study investigated their participation in the early steps of the reaction. We used two fluorescence assays that we had previously developed to study prechemistry conformational transitions (2, 3). A 2-AP probe on the DNA substrate reports step 2.1, and a FRET-based assay reports step 2.2, the fingers-closing transition (Fig. 1).
Asp705 and Asp882 Are Not Required for the Prechemistry Step 2.1
The DNA duplex, L:T(+1)2-AP:3′H (Fig. 3B), which has a 2-AP reporter as the 5′ neighbor of the templating base, provides a sensitive fluorescence assay for step 2.1. The primer strand was dideoxy-terminated so as to block covalent addition of a dNTP while allowing observation of the prechemistry steps. The fluorescence emission spectra of complexes of the 2-AP DNA with the D705A and D882A mutants were similar to that previously reported for wild-type Pol I(KF) (2); relative to unbound 2-AP DNA, there was a ∼1.5-fold increase in 2-AP fluorescence upon forming the Pol-DNA binary complex and a ∼2.5-fold increase upon forming the Pol-DNA-dNTP ternary complex with the complementary dGTP (supplemental Fig. S2). We assume that the increase in 2-AP fluorescence represents a conformational rearrangement that decreases the stacking of the T(+1) base with adjacent bases and may resemble the dislocation of the T(+1) base seen in ternary complex cocrystal structures (4–6).
We used the stopped-flow instrument to monitor the changes in 2-AP fluorescence, corresponding to step 2.1, as a function of time. The results for wild-type Pol I(KF) and both mutants were similar (Fig. 4). Upon the addition of dGTP to the Pol-DNA binary complex, there was an initial very rapid fluorescence increase within the instrument dead time (∼3 ms), indicated by the increasing start points of the traces, followed by a slower second phase. Fitting the observable slow fluorescence increase to a single exponential gave an approximate rate for the second phase of the reaction. To refine this determination and to estimate the rate of the initial fast phase, we fitted each trace to a double exponential, taking account of the amplitude missing due to the rapid fluorescence changes in the first 3 ms (for details of the fitting procedure, see supplemental Fig. S3). The results of this analysis (Table 2) indicate a rate of at least 400 s−1 for the initial phase of the fluorescence change. The differences between the behavior of the two carboxylate mutants and wild-type Pol I(KF) are relatively subtle: a faster Rate1 for D705A, a slower Rate2 for both mutants, and differences in the relative amplitudes of the two phases.
FIGURE 4.

Asp705 and Asp882 are not required for the rapid template strand conformational transition detected by 2-AP. The plots show the stopped-flow fluorescence signal upon the addition of dGTP, at the indicated concentrations, to the 2-AP-containing DNA duplex L:T(+1)2-AP:3′H (Fig. 3B) in complex with WT (A), D705A (B), and D882A Pol I(KF) (C). In the absence of protein, the addition of dGTP (1, 10, and 100 μm) to the 2-AP-DNA gave no fluorescence change (data not shown). Only the first 0.1 s of data are plotted in order to show rapid changes. The black lines superimposed on the data traces show fitting to a double exponential, taking account of the fluorescence changes that occur in the instrument dead time (see supplemental Fig. S3 and Table S2).
TABLE 2.
Kinetic parameters of wild-type and mutant Pol I(KF) from stopped-flow fluorescence data with a 2-AP(+1) reporter
Data are the average values from two experiments and are reported as mean ± S.D.
| Protein | Rate1a | Rate2 | Amp1/Amp2b | Kdoverallc |
|---|---|---|---|---|
| s−1 | s−1 | μm | ||
| WT | ⩾400 | 65 ± 13 | 75:25 | 2.8 ± 0.6 |
| D705A | ⩾900 | 13 ± 4 | 90:10 | 8.6 ± 3.1 |
| D882A | ⩾400 | 22 ± 4 | 55:45 | 8.1 ± 1.2 |
a Rate1, Rate2, Amp1, and Amp2 were calculated taking account of the amplitude that is lost in the instrument dead time (supplemental Fig. S3 and Table S2). Rate2 showed very little variation with dNTP concentration, and therefore we report the average of Rate2 values at all nucleotide concentrations.
b The ratio of the maximum values of the amplitudes of the fast and slow phases, each determined by plotting amplitude against dGTP concentration and fitting the data to a hyperbolic equation.
c Kd overall was determined by plotting the end point of each fluorescence trace against dGTP concentration and fitting to a hyperbolic equation (supplemental Fig. S3).
We interpret the stopped-flow kinetics within the framework of Scheme 1.
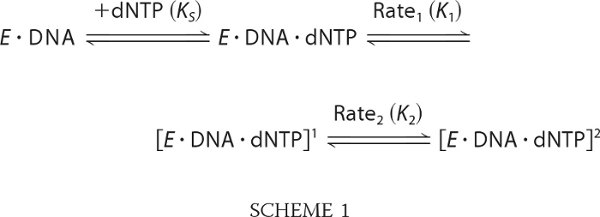 |
In this scheme, KS, K1, and K2 are the equilibrium constants for the initial binding of nucleotide and the two subsequent transitions. For simplicity, we assume that the 2-AP fluorescence increase results from the Rate1 conformational transition, and the slower (Rate2) transition shifts the equilibrium in favor of products without further changes to the 2-AP fluorescence. Because of the substantial difference in Rate1 and Rate2, we can make the following inferences from our analysis (Table 2). The ratio of the amplitudes of the two phases should approximate the equilibrium across the first conformational transition; relative to wild type, this transition appears more favorable in D705A and less favorable in D882A.3 The overall Kd for dNTP binding was determined from the hyperbolic dependence on dNTP concentration of the end point of the fluorescence traces (supplemental Fig. S3) and is equal to 1/(KSK1K2).
Asp705 Is Not Required for the Fingers-closing Transition (Step 2.2)
Our assay for the fingers-closing transition (3) uses FRET between an AEDANS fluorophore at position 744, on the mobile portion of the fingers subdomain, and a dabcyl quencher at position T(−8) on the DNA template strand (Fig. 3C). Fig. 2 illustrates the decrease in donor-to-quencher distance, resulting in a decrease in AEDANS emission, associated with fingers-closing and also shows the positions of the Asp705 and Asp882 side chains in the present study. As in the 2-AP experiments, the DNA primer terminus was dideoxy-terminated to limit observations to the prechemistry steps. Emission spectra of 744-AEDANS Pol I(KF) were consistent with our previous study (3); formation of the binary Pol-DNA complex with the dabcyl-DNA decreased the AEDANS fluorescence by ∼30%, and the addition of the complementary dTTP caused a further ∼20% decrease. The D705A and D882A mutations did not change the response upon forming the binary complex; however, the fluorescence decrease upon the addition of dTTP was smaller for D705A (∼10% decrease), and there was no change (within experimental error) for D882A (supplemental Fig. S4).
Using the stopped-flow instrument, we analyzed the kinetics of the fluorescence decrease associated with closed complex formation. As described previously (3), mixing of 744-AEDANS Pol I(KF), in complex with the dabcyl-DNA duplex, with the complementary dTTP resulted in a biphasic fluorescence decrease (Fig. 5A and Table 3). The rates obtained from the data are consistent with our previous study. Our interpretation of the biphasic kinetics is analogous to the scheme described above for the 2-AP stopped-flow fluorescence experiments, namely that the fingers-closing transition (causing the fluorescence decrease) corresponds to the faster rate, and the second phase is a subsequent non-fluorescent transition that displaces the overall equilibrium toward products.
FIGURE 5.

Asp705 is not required for fingers-closing activity. Stopped-flow fluorescence used 744-AEDANS Pol I(KF) and the dabcyl-containing DNA duplex, H:T(−8)D:3′H (Fig. 3C) in Mg2+ or Mn2+ buffer. The binary complex of labeled WT Pol I(KF) and DNA in 5 mm Mg2+(A) or 2 mm Mn2+ buffer (C) was mixed with dTTP to give the indicated final concentrations. B and D, analogous plots obtained with D705A Pol I(KF) in 10 mm Mg2+ or 10 mm Mn2+ buffer, respectively. Time is plotted on a logarithmic scale in order to illustrate clearly all phases of the reaction. The black lines superimposed on the data traces show fitting to a double exponential for the wild-type reactions or to a single exponential for the D705A reactions (see supplemental Fig. S5 and Table S3 for curve fitting and residuals), yielding the reaction rates and amplitudes reported in Table 3. In the absence of the dabcyl quencher on DNA, essentially no change in AEDANS fluorescence was observed (supplemental Fig. S6A).
TABLE 3.
Kinetic parameters of wild-type and D705A Pol I(KF) in the stopped-flow FRET-based assay of the fingers-closing conformational change
| Protein | Rate1a | Rate2a | Amp1:Amp2b | kmaxc | Kd(app)c | Kdoveralld |
|---|---|---|---|---|---|---|
| s−1 | s−1 | s−1 | μm | μm | ||
| Mg2+ as metal cofactor | ||||||
| WT | 75 ± 18 | 6.3 ± 1.8 | 5.1 | 110 | 19 | 8.6 |
| D705A | ⩾500e | 110 ± 4 | ∼6 | 120 ± 40 | ||
| Mn2+ as metal cofactor | ||||||
| WT | 91 ± 6 | 6.3 ± 2.3 | 130 | 19 | <1 | |
| D705A | 50 ± 12 | 60 ± 6 | 18 ± 7 | 6.9 ± 0.2 | ||
a The individual rates reported for WT Pol I(KF) were measured at 50 μm dTTP, and those for the D705A mutant were measured at 1 mm dTTP (in Mg2+) and 100 μm dTTP (in Mn2+). Values reported as mean ± S.D were from at least two independent experiments; the others were from single determinations. Examples of curve fitting and residuals for single and double exponentials are shown in supplemental Fig. S5.
b The ratio of the maximum values of the amplitudes of the fast and slow phases, determined by plotting the individual amplitudes against dTTP concentration and fitting the data to a hyperbolic equation.
c The parameters kmax and Kd(app) were derived from rate measurements at a series of dTTP concentrations by plotting Rate1 against dTTP concentration and fitting the data to a hyperbolic equation.
d Kd overall was determined from the hyperbolic dependence on dTTP concentration of either the total amplitude or the end point of the fluorescence traces.
e The rate of this very fast process was estimated assuming that 80% of the process was accomplished within 3 ms.
The D705A mutant also showed biphasic kinetics, but both rates were substantially faster than those of wild-type Pol I(KF) (Fig. 5B and Table 3). The first phase of the reaction was essentially complete within 3 ms (corresponding to a rate of ≥500 s−1) and therefore was observed as a decrease in the start points of the fluorescence traces. No fluorescence change was observed when using a control DNA duplex that lacked the dabcyl quencher, ruling out the possibility that the start point decrease in Fig. 5B might be an instrument artifact (supplemental Fig. S6A). The D705A mutation also caused a decrease in the dNTP binding affinity, as shown by the 15-fold increase, relative to wild type, in the overall Kd(dTTP) for the fingers-closing process (Table 3). Essentially identical fluorescence changes were observed when a deoxy-terminated primer was used instead of the dideoxy-terminated DNA (supplemental Fig. S6B and Table S1).
Asp882 Is Required for the Fingers-closing Transition
No fluorescence decrease corresponding to fingers-closing was observed with the D882A mutant, regardless of whether the DNA substrate had a 3′-H (Fig. 6A) or a 3′-OH (supplemental Fig. S6D). Instead, we observed a dNTP-dependent fluorescence increase with a rate of 3.1 s−1. Because dissociation of the dabcyl-DNA from the ternary complex could account for this fluorescence increase, we measured the DNA dissociation rate directly using a DNA trap. The complex of labeled D882A Pol I(KF) with dabcyl-DNA was mixed in the stopped-flow instrument with a 10-fold excess of an unmodified DNA duplex to serve as a trap and prevent reassociation of free labeled protein with the dabcyl-DNA (Fig. 6B). The rate of DNA dissociation measured in the presence of dTTP, so as to form a correctly paired ternary complex, was similar to the rate of the fluorescence increase observed for D882A in the fingers-closing assay (Table 4), suggesting that the latter is indeed caused by DNA dissociation. Moreover, the DNA dissociation rate was faster in the presence of nucleotide (both the complementary dTTP and the mispaired dGTP) than in its absence, consistent with the traces in Fig. 6A showing the fluorescence increase to be dNTP-dependent.
FIGURE 6.

D882A inhibits fingers-closing, and dNTPs promote DNA dissociation from the open complex. A, stopped-flow fluorescence used 744-AEDANS D882A Pol I(KF) and the H:T(−8)D:3′H DNA duplex (Fig. 3C). The binary complex of labeled protein and DNA was mixed with the complementary nucleotide, dTTP, to give the indicated final concentrations. The buffer in both syringes included 10 mm MgCl2. B, the rate of dissociation of 744-AEDANS D882A Pol I(KF) from H:T(−8)D:3′OH DNA (Fig. 3C) was measured using a DNA trap as described under “Experimental Procedures.” The red trace is the dissociation of the Pol-DNA binary complex when mixed with an excess of an unmodified DNA duplex (DNA trap). The blue trace shows the dissociation of the correctly paired ternary complex, measured by including a 2 mm concentration of the complementary nucleotide, dTTP, in the DNA trap solution. The green trace shows the dissociation of a mismatched ternary complex, when the DNA trap solution contained a 2 mm concentration of a noncomplementary nucleotide, dGTP. C, as in A, except that the reaction buffer contained 10 mm MnCl2 instead of MgCl2. The black lines superimposed on the data traces in A and B show fitting of the fluorescence increases to single exponential equations, with rates and amplitudes reported in supplemental Table S3.
TABLE 4.
Rates of dissociation of D882A Pol I(KF) complex with DNA
The DNA oligonucleotide was H:T(−8)D:3′OH (Fig. 3C). Values reported as mean ± S.D were from two independent experiments; the others were from single determinations.
| Reagenta | Complex | koffb |
|---|---|---|
| s−1 | ||
| DNA trap, no dNTP | Binary | 0.7 ± 0.1 |
| DNA trap, 2 mm dTTP | Ternary (A-dTTP) | 2.4 ± 0.2 |
| DNA trap, 2 mm dGTP | Ternary (A-dGTP) | 5.3 |
| 2 mm dTTPc | Ternary (A-dTTP) | 3.3 ± 0.2 |
a The indicated reagent was mixed with an equal volume of the Pol-DNA binary complex in the stopped-flow instrument. The buffer in both syringes contained 50 mm Tris-HCl, 1 mm EDTA, and 10 mm MgCl2.
b Dissociation rates were determined by fitting the fluorescence increase to a single exponential. In a similar experiment, wild-type Pol I(KF) gave DNA dissociation rates of 2.4 s−1 from the binary complex, 0.3 s−1 from the correctly paired ternary complex, and 20 s−1 from the mispaired ternary complex (3).
c From Fig. 6A.
A Single Metal Ion Is Sufficient for the Prechemistry Steps 2.1 and 2.2
To investigate the requirements for the two Mg2+ ions during the prechemistry steps, we limited the amount of free Mg2+ in the reaction buffer using the approach developed by Bakhtina et al. (12). The rationale of this experiment is that Mg2+ will bind to dNTP and thus to the polymerase at the metal B site at micromolar concentrations, whereas the metal A site has a much lower affinity for Mg2+, indicated by a Mg2+ optimum for dNTP incorporation in the millimolar range. Maintaining the concentration of free Mg2+ in the micromolar range therefore has an effect on the polymerase reaction similar to that of an exchange-inert metal-dNTP complex, without introducing an unnatural metal ion. We carried out stopped-flow fluorescence assays for steps 2.1 and 2.2 in the presence of very low concentrations of free Mg2+ and Mg-dNTP, controlled by the inclusion of EDTA in the nucleotide solution, as described (12).
Fig. 7A shows the 2-AP fluorescence assay for step 2.1 using the extendable DNA duplex, L:T(+1)2-AP:3′OH DNA. In the presence of 3 mm Mg2+, the initial rapid fluorescence increase was followed by a fluorescence decrease corresponding to the incorporation of dGTP, as reported previously (2). However, at the lower Mg2+ concentrations indicated in Fig. 7A, the fluorescence signal was almost indistinguishable from that observed with a nonextendable DNA (compare Fig. 4A), with only a slight fluorescence decrease seen at long time points with 0.7 mm total Mg2+.
Analogous results were obtained in the fingers-closing FRET assay using 744-AEDANS Pol I(KF) and the extendable dabcyl substrate, H:T(−8)D:3′OH (Fig. 7B). In 3 mm Mg2+, the addition of the complementary dTTP resulted in the expected fingers-closing fluorescence decrease followed by a fluorescence increase due to the covalent addition of the nucleotide (3). As in the 2-AP experiment, the lower Mg2+ concentrations gave traces very similar to those obtained with the corresponding nonextendable DNA (Fig. 5A), with the fluorescence increase detectable only at long times with 0.4 and 0.7 mm total Mg2+. Together, these experiments show that binding of dNTP-Mg2+ alone is sufficient for the polymerase reaction to progress as far as formation of the closed ternary complex, but further addition of Mg2+ is required to fill the second metal ion site and support dNTP incorporation.
Effects of Mn2+ on the Early Stages of the Reaction
When the stopped-flow assays for steps 2.1 and 2.2 were carried out with Mn2+ as the metal cofactor, the results were significantly different from those obtained with Mg2+. With the T(+1)2-AP probe, reporting step 2.1, the characteristic fluorescence increase was much slower, ∼12 s−1 for wild-type Pol I(KF) and 1 s−1 for D705A (supplemental Fig. S7), compared with ≥400 s−1 in the Mg2+ reaction.
In the fingers-closing assay, wild-type Pol I(KF) gave similar reaction rates in Mn2+ and Mg2+ (Fig. 5C and Table 3). However, Kd overall, obtained from the dTTP concentration dependence of the amplitudes or the end points of the fluorescence traces, was much lower in Mn2+ than in Mg2+. As a result, the fingers-closing transition in Mn2+ was essentially irreversible, with all of the fluorescence traces reaching the identical end point, even at dTTP concentrations as low as 1 μm. With the D705A mutant, the reaction was much slower in Mn2+ compared with Mg2+ and was no longer detectably biphasic (Fig. 5D and Table 3). The dTTP affinity of D705A, measured by Kd overall, was almost 20-fold greater in Mn2+. Because the T(+1)2-AP fluorescence increase in Mn2+ is slower than the fingers-closing transition, we infer that the DNA rearrangement reported by the 2-AP probe must take place after fingers-closing in Mn2+ and is not necessarily identical to the equivalent step observed in Mg2+. Intriguingly, the 2-AP fluorescence increase seen with the D705A mutant in Mn2+ is preceded by a fluorescence decrease at ∼60 s−1, similar to the fingers-closing rate (supplemental Fig. S7 and Table 3).
In the case of the D882A mutant, a low amplitude fluorescence decrease was observed when Mn2+ was used as the metal cofactor (Fig. 6C), suggesting that Mn2+, unlike Mg2+, allows the fingers-closing transition to proceed to a small extent. As with Mg2+, a fluorescence increase, most probably due to DNA dissociation, was also observed. Because of the low amplitudes, it was difficult to get accurate determinations of the rates of these two fluorescence changes; we estimate the fluorescence decrease to be ∼60 s−1, similar to the wild-type fingers-closing rate, and the increase to be ∼2 s−1. The behavior of the D705A and D882A mutants in the presence of Mn2+ was unchanged when an extendable (deoxy-terminated) DNA substrate was used instead of the nonextendable DNA in the experiments described above (supplemental Fig. S6 and Table S1).
DISCUSSION
Residues Asp705 and Asp882 of Pol I(KF) each make bifurcated interactions with the two catalytically essential metal ions (A and B) at the polymerase active site in the closed ternary complex. Therefore, it is no surprise that mutation of either carboxylate results in an enzyme that is essentially inactive in terms of incorporation of nucleotides into DNA (16, 17). The current study examines the role of Asp705 and Asp882 and their associated metal ions in the prechemistry steps of the DNA polymerase reaction that assemble the active site in readiness for the phosphoryl transfer reaction. Despite the dramatic effect of the D705A and D882A mutations on the chemical incorporation rate, the carboxylate side chains have much less influence on some of the early steps of the reaction. The behavior of the D705A and D882A mutants in fluorescence-based assays that probe prechemistry conformational transitions sheds light on the sequence of events in the early steps of the DNA polymerase reaction pathway. Interpreting our results in the context of available crystal structures of A-family DNA polymerases in complexes with DNA and nucleotide substrates, we present a structural description of the assembly of the catalytically competent polymerase active site (Fig. 8), with the caveat that such a structural description is limited by the lack of cocrystal data for the early (and probably unstable) intermediates that we are most interested in.
FIGURE 8.

Proposed sequence of events for the prechemistry steps involving Asp882 and Asp705. All protein structures are based on crystal structures of Bst pol, but side chains are labeled with Pol I(KF) residue numbers; the protein backbone is shown in gray, except for the O-helix (black). A, the initial, open, ternary complex was modeled from the binary complex (Protein Data Bank entry 1L5U) (6) with the position of the nucleotide (green) taken from the structure of Klentaq bound to dCTP in the absence of DNA (Protein Data Bank entry 5KTQ) (19). The nucleotide's triphosphate tail is bound by the conserved side chains Arg754 and Lys758 (beige) on the O-helix. The Asp882 and Asp705 side chains are shown in yellow; Asp882 is hydrogen-bonded (dashed line) to the 3′-OH of the primer strand, whereas Asp705 points away from the active site. The primer strand of the DNA is shown in pink, and the template strand is shown in blue. A suggested position for metal B (purple), chelated via the dNTP β- and γ-phosphates, is indicated (no bound metal is present in the 5KTQ coordinates). B, the ternary complex (taken from Protein Data Bank entry 1LV5) (6) immediately after the fingers have closed but before metal A binds, showing the change in position of the Asp705 and Asp882 side chains, upon going from the open (yellow, as in A) to closed (cyan) conformation. Arg754 and Lys758 remain in contact with the incoming nucleotide in the closed conformation but are omitted for clarity. Since the primer is dideoxy-terminated in this structure, the interaction between the 3′-OH group and the carboxylate of Asp882 is absent. C, the closed complex is illustrated (as in B), showing the entry of metal A (red, from 1LV5). This structure represents the enzyme poised for catalysis.
Nucleotide Binding
The sequence of prechemistry steps in the DNA polymerase reaction is initiated by the binding of nucleotide to a Pol-DNA binary complex, whose structure we have taken as the starting point in our mechanism. We propose that the initial contacts made by an incoming dNTP resemble those seen in the DNA-free polymerase-dNTP binary complex cocrystals of Pol I(KF) and Klentaq (19, 20), specifically interactions between the dNTP phosphates and conserved positively charged side chains on or close to the O-helix of the fingers subdomain (Fig. 8A). Kinetic studies have demonstrated the importance for dNTP binding of residues such as Arg754 of Pol I(KF), which is predicted to interact with the γ-phosphate (21). Moreover, the interactions between Klentaq and dCTP, observed in the Pol-dNTP binary complex, are preserved in the closed Pol-DNA-dNTP ternary complex, implying that movement of the O-helix simply delivers the bound dNTP into the active site (5).
Our experiments using very low concentrations of Mg2+ support a model in which the dNTP binds to the Pol-DNA complex as dNTP-Mg2+, whereas the second Mg2+, required for the completion of catalysis, does not bind until later. The fluorescence changes associated with steps 2.1 and 2.2 were not observed in the absence of divalent metal ions; however, these conformational transitions, but not the subsequent phosphoryl transfer, could take place with Mg2+ at only micromolar levels (Fig. 7). These observations are consistent with studies of Pol β and RB69 DNA polymerase showing that exchange-inert metal-dNTP complexes, containing Cr(III) or Rh(III), promote early conformational change steps in the absence of exogenous divalent metal ions, the latter being required for covalent incorporation of the nucleotide into DNA (10–13). The idea that metal B enters the active site as dNTP-Mg2+ is supported by NMR studies of Pol I(KF) showing that binding of dNTP to the polymerase domain creates a high affinity binding site for a divalent metal ion (22). Moreover, cocrystals of A-family Pol-DNA binary complexes do not have a bound metal ion in a position equivalent to the metal A site (6, 23). (Some binary complex cocrystals have a divalent metal ion coordinated to the two active-site carboxylates, in a location close to the metal B site, but its mechanistic significance is unclear.)
DNA Rearrangement, Step 2.1
The fluorescence change of the T(+1)2-AP reporter is affected very little by the D705A or D882A mutations, indicating that the active-site carboxylates are not important either for the initial binding of dNTP and its bound Mg2+ or for this subsequent conformational transition. If indeed the incoming dNTP is bound as we have suggested in Fig. 8A, the Asp705 and Asp882 side chains would be too distant to interact at this early stage with the chelated metal B, although both aspartates are ligands to metal B in the closed ternary complex. We know very little about the structural change associated with step 2.1, except that the polymerase must remain in an open conformation because fingers-closing is a subsequent step. The fluorescence increase associated with step 2.1 implies that the T(+1) base becomes less stacked; however, the binary complex crystal structures do not provide much information on the interactions responsible for the observed change. In the Pol-DNA binary complex, the templating base is bound in the preinsertion binding pocket formed by the O and O1 helices and consequently is not able to stack with the T(+1) base, its 5′ neighbor. Therefore, the relevant interactions with the T(+1) base probably involve either the protein or the T(+2) base (absent in the majority of binary complex structures), but our earlier attempts to identify the contacts proved inconclusive (2). Currently, we visualize step 2.1 as a local rearrangement of the unpaired template DNA that decreases the stacking interactions made by the T(+1) base. An intriguing possibility is that a change in the environment of the T(+1) base could be associated with movement of the templating base into a position suitable for testing the complementarity of the incoming dNTP.
The fluorescence increase of step 2.1 is not observed if the incoming dNTP is not complementary to the templating base (2, 3),4 consistent with the idea of an early fidelity checkpoint that discriminates against mispairs. It is possible that the discriminator step precedes step 2.1 so that only correctly paired ternary complexes initiate step 2.1. Alternatively, step 2.1 itself could be the discriminator, with the equilibrium favoring the forward reaction for a correctly paired dNTP and favoring the reverse reaction for a mispair.
Fingers-closing and the Role of Asp882
Step 2.2 (fingers-closing) takes place essentially normally with the D705A mutant but is strongly impaired in D882A Pol I(KF). No fingers-closing fluorescence signal was observed with D882A in the presence of Mg2+; in Mn2+, there was a limited extent of reaction at a rate similar to the wild-type fingers-closing rate. This suggests that D882A has not lost the ability to form the closed complex, but instead, the mutation has changed the open-closed equilibrium of the ternary complex so that it is biased in favor of the open complex (more so in Mg2+ than in Mn2+). Single-molecule FRET studies of Pol I(KF) demonstrate a finely balanced equilibrium between open and closed conformations, with the closed conformation favored by ∼1 kcal/mol in a correctly paired ternary complex (24), so it is not unreasonable to imagine that the loss of a key side chain could shift the equilibrium so that the ternary complex is predominantly in the open conformation. In this scenario, we assume that the low level of catalysis observed with D882A occurs via the sparsely populated closed ternary complex.
What is the role of Asp882 in stabilizing the closed conformation? Alignment of open binary and closed ternary complexes of Bst pol and Klentaq shows that the position of the side chain equivalent to Asp882 changes very little. Thus, Asp882 may serve as an anchor point during the fingers-closing transition, making an interaction with metal B as the movement of the O-helix delivers the dNTP-Mg2+ complex into the active site (Fig. 8B). The Bst pol binary complex illustrated here (as well as other binary complexes in which the primer is deoxy-terminated) has a hydrogen bond between the homologue of Asp882 and the primer 3′-OH (Fig. 8A) (6, 23). In the closed ternary complex crystal structure, this hydrogen bond is absent because the primer is dideoxy-terminated; however, the similar positioning of Asp882 in open and closed conformations suggests that, with the natural substrates, Asp882 bridges the active site, linking the 3′-OH and the dNTP-Mg2+, and this could be the basis for its role as an anchor point during fingers-closing.
There is an interesting contrast between wild-type Pol I(KF), where formation of the closed ternary complex stabilizes DNA binding (3), and D882A, where the ternary complex is predominantly in the open conformation and the addition of nucleotide, even when it is complementary to the templating base, destabilizes DNA binding relative to the binary complex (Fig. 6 and Table 4; compare with wild-type data listed in Table 4 Footnote b). We suggest that weaker DNA binding may be a property inherent to the open ternary complex rather than being a direct consequence of the absent Asp882 side chain. The behavior of D882A resembles the destabilization of DNA binding observed for mismatched ternary complexes of wild-type Pol I(KF), which also remain in an open conformation (3). As we have suggested previously (3, 24), the open ternary complex may represent an important checkpoint in which the incoming dNTP previews the template base, and the eventual outcome is determined by whether the nascent base pair is correct or mismatched. The D882A mutant could therefore be a valuable tool, allowing us to arrest the reaction at the open ternary complex and to study this intermediate using a variety of physical techniques.
The Role of Asp705
In our stopped-flow experiments, the D705A mutant is fully capable of carrying out the fingers-closing transition and actually does so at a faster rate than wild-type Pol I(KF). Therefore, Asp705 does not assume its important role in catalysis until after the closed complex has formed. The Klentaq and Bst pol binary (Pol-DNA) complex structures show that, at the start of the reaction, Asp705, unlike Asp882, is not positioned appropriately to be a metal ligand; instead, it faces away from the active site region (Fig. 8A). In the closed ternary complex structures (with both metals A and B) the side chain of Asp705 has rotated so that one of the carboxylate oxygens is a ligand to metal B (Fig. 8B). Two possible scenarios explain the faster rate of fingers-closing observed with the D705A mutant: 1) the repositioning of the Asp705 side chain could take place, at least in part, during the fingers-closing conformational change and might involve a significant energy barrier; alternatively, 2) the position of the Asp705 side chain itself might impede the fingers-closing process. In either case, the presence of Asp705 (in the wild-type enzyme) requires the input of energy, relative to D705A, giving a higher energy transition state, which could enhance the catalysis of subsequent steps. Structural studies of RB69 DNA polymerase, HIV-1 reverse transcriptase, and Pol β also show alternative conformations of active-site side chains in the prechemistry steps of the reaction pathway, suggesting that the mobility of key side chains may be a common feature of the polymerase mechanism (11, 25–27). In Pol β, both direct measurements and computational studies indicate that some side chain motions are energy-requiring and can potentially be rate-limiting for the reaction kinetics (28).
The repositioning of Asp705 during or subsequent to fingers-closing contributes a ligand to the metal A binding site, and we suggest that the important function of Asp705 is to ensure the proper coordination of metal A in preparation for phosphoryl transfer. The participation of Asp705 in a step after fingers-closing should further stabilize the closed complex, accounting for the tighter binding of dNTP to wild-type Pol I(KF), compared with D705A. Our earlier data on the fingers-closing process in the presence of Ca2+ had suggested a metal-dependent step after fingers-closing but before chemistry (3), and the current study provides additional lines of evidence that coordination of metal A occurs after fingers-closing. First, both of the early fluorescently detectable steps take place normally in the presence of very low Mg2+ concentrations, but higher metal ion concentrations are required to complete phosphoryl transfer, implying that an additional metal site must be filled after fingers-closing. Second, the behavior of the D705A and D882A mutant proteins in the fingers-closing assay is not appreciably influenced by the presence of the primer 3′-OH, one of the ligands to metal A, suggesting that the metal A site is irrelevant until after the fingers-closing step. The participation of Asp705 in a late step involving metal coordination is also consistent with the 10-fold increase in the rate of dNTP incorporation by D705A when Mg2+ is replaced by Mn2+, a metal ion that is better able to bind to sites with imperfect geometry or missing ligands (29, 30). Mn2+ had the opposite effect, a slight decrease in the rate of dNTP incorporation, on wild-type Pol I(KF), which should have a normal metal A site, and on D882A, which does not proceed efficiently beyond the open ternary complex.
Steps following Fingers-closing
As described above, our data suggest that Asp705 plays a role in the binding of metal A after the fingers-closing conformational change, a process that may correspond to the unidentified rate-limiting prechemistry step 3. Plausible candidates for this rate-limiting step are the movement of Asp705 into its position as a metal A ligand or the entry of metal A into its coordination site. Studies of other DNA polymerases using exchange-inert dNTP-metal complexes indicate that one or more early conformational transitions can take place in the absence of the second active-site metal ion, just as we have demonstrated here, although there is a lack of consensus on when the second metal ion enters the ternary complex. In the case of the X-family DNA polymerase, Pol β, structural studies have shown that the fingers close in the absence of the second metal ion (10, 11), and stopped-flow fluorescence indicated that binding of the second metal and catalysis occurs without kinetic consequences (12); however, computational studies suggested that fingers-closing in Pol β requires both active-site metal ions (28, 31). With the B-family DNA polymerase from phage RB69, a conformational transition (detected by a template 2-AP probe but assumed to be fingers-closing) is induced by the exchange-inert dNTP-Rh(III), but subsequent binding of the second (catalytically essential) metal ion is extremely slow, suggesting that it normally binds in advance of the nucleotide and cannot gain entry to the ternary complex without reversing the conformational transition (13, 32).
Our data do not rule out an additional role for Asp705 in the chemical step itself; nor do they rule out significant participation by Asp882 in the steps that follow fingers-closing. (The latter question cannot be addressed because D882A does not proceed beyond the open ternary complex.) Indeed, a computational study of T7 DNA polymerase, a homologue of Pol I(KF), has suggested that the residue equivalent to Asp882 may act as a general base to receive the proton from the primer 3′-OH, so as to generate the attacking nucleophile for the phosphoryl transfer reaction (33). This idea fits well with the consistent observation of a hydrogen bond between the Asp882 homologue and the primer 3′-OH in A-family Pol-DNA binary complex cocrystals in which the primer strand has a 3′-OH (6, 23).
Effects of Mn2+
Our preliminary observations when replacing the natural cofactor, Mg2+, with Mn2+ are intriguing, and further studies are ongoing in order to understand their full implications. The largest effects were the decrease in rate, by ∼2 orders of magnitude, of the fluorescence increase reported by the T(+1)2-AP probe and the increase in overall dNTP binding affinity measured by the end point of the fingers-closing fluorescence change. An obvious inference from the slower rate seen with the 2-AP probe is that, in Mn2+, the putative DNA rearrangement must take place after fingers-closing and may not necessarily be identical to the step 2.1 process seen in the Mg2+ reaction. This makes it difficult to interpret the metal-dependent differences in the fingers-closing assay because the open ternary complex structure at the start of the fingers-closing is different in Mg2+ and Mn2+.
There is little definitive information that establishes why Mn2+ should have such a profound effect on the early stages of the DNA polymerase reaction. In a crystallographic study of Pol β, Pelletier et al. (34) reported that Mn2+ is coordinated to the dNTP at the active site as an α-γ bidentate, instead of the β-γ bidentate observed with Mg2+. They also reported tighter nucleotide binding in the presence of Mn2+, which is similar to our observations. These results raise the possibility that dNTP-Mn2+ may find an aberrant, perhaps lower energy, binding site on the DNA polymerase and that this may alter the early steps of the reaction pathway. Changes to the pathway that alter or eliminate an early fidelity checkpoint might explain the ability of Mn2+ to decrease polymerase fidelity (30, 35–37).
Conclusions
From our studies of the effects of active-site carboxylate mutations on the prechemistry steps in the Pol I(KF) reaction pathway, a picture is starting to emerge of the sequence of events in the pathway and the stages at which the carboxylates and their associated metal ions are involved. Our results suggest an exquisitely choreographed series of conformational transitions that sequentially assemble the active site into a reaction-competent configuration. Similar conclusions were reached in computational studies of Pol β (28, 31), an X-family polymerase unrelated to Pol I, suggesting that the stepwise assembly of the correct active-site geometry may be a common feature of DNA polymerases, providing multiple fidelity checkpoints at which inappropriate substrates can be diverted from the productive reaction pathway.
Supplementary Material
Acknowledgments
We thank Olga Potapova for excellent technical support and Anna M. Pyle for use of the stopped-flow instrument.
This work was supported, in whole or in part, by National Institutes of Health Grant GM-28550.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7 and Tables S1–S3.
As described below, the subsequent fingers-closing step in D705A is extremely rapid (≥500 s−1) and therefore may account for the larger amplitude (compared with WT) of the first phase of the 2-AP(+1) fluorescence change. Analogously, the absence of the fingers-closing step in D882A may contribute to the altered kinetics of the 2-AP(+1) fluorescence change in the D882A mutant.
O. Bermek, unpublished observations.
- Pol I(KF)
- Klenow fragment of E. coli DNA polymerase I
- Klentaq
- the portion of Thermus aquaticus DNA polymerase equivalent to Klenow fragment
- Bst pol
- the portion of Bacillus stearothermophilus DNA polymerase equivalent to Klenow fragment
- Pol
- (DNA) polymerase
- IAEDANS
- 5-(((2-iodoacetyl)amino)ethyl)aminonaphthalene-1-sulfonic acid
- 2-AP
- 2-aminopurine.
REFERENCES
- 1. Joyce C. M., Benkovic S. J. (2004) Biochemistry 43, 14317–14324 [DOI] [PubMed] [Google Scholar]
- 2. Purohit V., Grindley N. D. F., Joyce C. M. (2003) Biochemistry 42, 10200–10211 [DOI] [PubMed] [Google Scholar]
- 3. Joyce C. M., Potapova O., Delucia A. M., Huang X., Basu V. P., Grindley N. D. F. (2008) Biochemistry 47, 6103–6116 [DOI] [PubMed] [Google Scholar]
- 4. Doublié S., Tabor S., Long A. M., Richardson C. C., Ellenberger T. (1998) Nature 391, 251–258 [DOI] [PubMed] [Google Scholar]
- 5. Li Y., Korolev S., Waksman G. (1998) EMBO J. 17, 7514–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnson S. J., Taylor J. S., Beese L. S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 3895–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doublié S., Sawaya M. R., Ellenberger T. (1999) Structure 7, R31-R35 [DOI] [PubMed] [Google Scholar]
- 8. Brautigam C. A., Steitz T. A. (1998) Curr. Opin. Struct. Biol. 8, 54–63 [DOI] [PubMed] [Google Scholar]
- 9. Dahlberg M. E., Benkovic S. J. (1991) Biochemistry 30, 4835–4843 [DOI] [PubMed] [Google Scholar]
- 10. Zhong X., Patel S. S., Tsai M.-D. (1998) J. Am. Chem. Soc. 120, 235–236 [Google Scholar]
- 11. Arndt J. W., Gong W., Zhong X., Showalter A. K., Liu J., Dunlap C. A., Lin Z., Paxson C., Tsai M.-D., Chan M. K. (2001) Biochemistry 40, 5368–5375 [DOI] [PubMed] [Google Scholar]
- 12. Bakhtina M., Lee S., Wang Y., Dunlap C., Lamarche B., Tsai M.-D. (2005) Biochemistry 44, 5177–5187 [DOI] [PubMed] [Google Scholar]
- 13. Wang M., Lee H. R., Konigsberg W. (2009) Biochemistry 48, 2075–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Delarue M., Poch O., Tordo N., Moras D., Argos P. (1990) Protein Eng. 3, 461–467 [DOI] [PubMed] [Google Scholar]
- 15. Steitz T. A., Smerdon S. J., Jäger J., Joyce C. M. (1994) Science 266, 2022–2025 [DOI] [PubMed] [Google Scholar]
- 16. Polesky A. H., Steitz T. A., Grindley N. D. F., Joyce C. M. (1990) J. Biol. Chem. 265, 14579–14591 [PubMed] [Google Scholar]
- 17. Polesky A. H., Dahlberg M. E., Benkovic S. J., Grindley N. D. F., Joyce C. M. (1992) J. Biol. Chem. 267, 8417–8428 [PubMed] [Google Scholar]
- 18. Joyce C. M., Derbyshire V. (1995) Methods Enzymol. 262, 3–13 [DOI] [PubMed] [Google Scholar]
- 19. Li Y., Kong Y., Korolev S., Waksman G. (1998) Protein Sci. 7, 1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beese L. S., Friedman J. M., Steitz T. A. (1993) Biochemistry 32, 14095–14101 [DOI] [PubMed] [Google Scholar]
- 21. Astatke M., Grindley N. D. F., Joyce C. M. (1995) J. Biol. Chem. 270, 1945–1954 [DOI] [PubMed] [Google Scholar]
- 22. Mullen G. P., Serpersu E. H., Ferrin L. J., Loeb L. A., Mildvan A. S. (1990) J. Biol. Chem. 265, 14327–14334 [PubMed] [Google Scholar]
- 23. Kiefer J. R., Mao C., Braman J. C., Beese L. S. (1998) Nature 391, 304–307 [DOI] [PubMed] [Google Scholar]
- 24. Santoso Y., Joyce C. M., Potapova O., Le Reste L., Hohlbein J., Torella J. P., Grindley N. D. F., Kapanidis A. N. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zakharova E., Wang J., Konigsberg W. (2004) Biochemistry 43, 6587–6595 [DOI] [PubMed] [Google Scholar]
- 26. Sarafianos S. G., Clark A. D., Jr., Das K., Tuske S., Birktoft J. J., Ilankumaran P., Ramesha A. R., Sayer J. M., Jerina D. M., Boyer P. L., Hughes S. H., Arnold E. (2002) EMBO J. 21, 6614–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Batra V. K., Beard W. A., Shock D. D., Krahn J. M., Pedersen L. C., Wilson S. H. (2006) Structure 14, 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Radhakrishnan R., Arora K., Wang Y., Beard W. A., Wilson S. H., Schlick T. (2006) Biochemistry 45, 15142–15156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang W., Lee J. Y., Nowotny M. (2006) Mol. Cell 22, 5–13 [DOI] [PubMed] [Google Scholar]
- 30. Cowan J. A. (1998) Chem. Rev. 98, 1067–1088 [DOI] [PubMed] [Google Scholar]
- 31. Yang L., Arora K., Beard W. A., Wilson S. H., Schlick T. (2004) J. Am. Chem. Soc. 126, 8441–8453 [DOI] [PubMed] [Google Scholar]
- 32. Lee H. R., Wang M., Konigsberg W. (2009) Biochemistry 48, 2087–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Florián J., Goodman M. F., Warshel A. (2003) J. Am. Chem. Soc. 125, 8163–8177 [DOI] [PubMed] [Google Scholar]
- 34. Pelletier H., Sawaya M. R., Wolfle W., Wilson S. H., Kraut J. (1996) Biochemistry 35, 12762–12777 [DOI] [PubMed] [Google Scholar]
- 35. Kunkel T. A., Loeb L. A. (1979) J. Biol. Chem. 254, 5718–5725 [PubMed] [Google Scholar]
- 36. El-Deiry W. S., Downey K. M., So A. G. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 7378–7382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goodman M. F., Keener S., Guidotti S., Branscomb E. W. (1983) J. Biol. Chem. 258, 3469–3475 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.