

Abstract
Genome-wide mapping analyses are now commonplace in many species and several networks of interacting loci have been reported. However, relatively few details regarding epistatic interactions and their contribution to complex trait variation in multicellular organisms are available and the identification of positional candidate loci for epistatic QTL (epiQTL) is hampered, especially in mammals, by the limited genetic resolution inherent in most study designs. Here we further investigate the genetic architecture of reproductive fatpad weight in mice using the F10 generation of the LG,SM advanced intercross (AI) line. We apply multiple mapping techniques including a single-locus model, locus-specific composite interval mapping (CIM), and tests for multiple QTL per chromosome to the 12 chromosomes known to harbor single-locus QTL (slQTL) affecting obesity in this cross. We also perform a genome-wide scan for pairwise epistasis. Using this combination of approaches we detect 199 peaks spread over all 19 autosomes, which potentially contribute to trait variation including all eight original F2 loci (Adip1-8), novel slQTL peaks on chromosomes 7 and 9, and several novel epistatic loci. Extensive epistasis is confirmed involving both slQTL confidence intervals (C.I.) as well as regions that show no significant additive or dominance effects. These results provide important new insights into mapping complex genetic architectures and the role of epistasis in complex trait variation.
THE development and elaboration of techniques such as interval mapping (Lander and Botstein 1989), composite interval mapping (CIM) (Zeng 1994), and methods based on complex pedigree structures (Jannink et al. 2001) have produced an extensive repertoire for the statistical exploration of genotype–phenotype relationships, especially for single loci. Using these approaches, genome-wide analyses have identified single-locus QTL (slQTL) underlying variance in characters as varied as agronomic traits and pest resistance in corn (Papst et al. 2004), life span in fruit flies (Wilson et al. 2006), alkylator-induced cancer susceptibility in mice (Fenske et al. 2006), murine skeletal morphology (Kenney-Hunt et al. 2008), and an ever-expanding list of human diseases and disorders including age-related macular degeneration (e.g., Klein et al. 2005), type 2 diabetes (e.g., Sladek et al. 2007; Zeggini et al. 2008), and Crohn's disase (e.g., Duerr et al. 2006). In addition, several studies have successfully employed epistatic QTL (epiQTL) mapping strategies to describe multilocus networks (e.g., Cheverud et al. 2001; Stylianou et al. 2006; Wentzell et al. 2007; Fawcett et al. 2008, 2010).
However, most mapping studies in model systems involve either F2 intercross populations or recombinant inbred (RI) strain panels (see also Hanlon et al. 2006). These populations harbor limited recombination and so tend to identify relatively large confidence intervals, complicating the physiological investigation of statistical results. Furthermore, while RI strain sets represent a fourfold expansion of the F2 recombination-based map, they require a minimum of 20 generations of brother–sister mating (Silver 1995) and the number of strains per set is usually low, especially in mammals. Conversely, the production of advanced intercross (AI) lines involves many generations of outbreeding in a relatively large population. This preserves heterozygosity, retains many more recombinant gametes in the gene pool, decreases the average size of segregating linkage blocks, and increases mapping resolution (Haldane and Waddington 1931; Bartlett and Haldane 1935; Hanson 1959a,b,c,d; Darvasi and Soller 1995; Rockman and Kruglyak 2008). Specifically, the F10 generation of a murine AI line represents an approximately fivefold expansion of the F2 map and thus an improvement in resolution over both F2 intercross and RI line study designs.
Obesity and related phenotypes are among the most intensively studied complex traits in mice and the LG,SM AI has proven particularly useful in the identification of adiposity QTL. Previous work in this cross has characterized over 70 loci contributing to variance in fatpad weight, body weight, and relevant organ weights (Cheverud et al. 1999, 2001, 2004a,b; Fawcett et al. 2008). In addition, a recent study used the combined F9 and F10 generations (Fawcett et al. 2010) to fine map loci for a suite of obesity-related characters and achieved an average confidence interval (C.I.) for fatpad loci of 4.14 Mb. These C.I.s were subsequently tested for epistasis and extensive interaction was confirmed, though several direct-effect loci identified in the F2 and F2/3 generations failed to replicate and were thus not included. However, in a full genome-wide scan for pairwise epistasis in the F2 generation of this cross (Jarvis and Cheverud 2009) 38 fatpad loci, which were not identified using a single-locus mapping model, show significant epistatic interactions. Consistent with results from other experimental systems (reviewed in Phillips 2008) this suggests that many biologically relevant loci are invisible to single-locus scans. Thus, combining the increased genetic resolution of an F10 AI line study, with the full range of single-locus and epistatic mapping strategies promises to produce novel insights into the contribution of epistatic interactions to variation in reproductive fatpad weight in mice. Furthermore, the accumulating data on positional candidate genes (e.g., Chehab 2008; Gat-Yablonski and Phillip 2008; Ichihara and Yamada 2008; Cheverud et al. 2010) provides the opportunity to explore functional hypotheses for identified loci and their interactions.
Utilizing the F10 generation of the LG,SM AI line (Cheverud et al. 2001), we further characterized the complex genetic architecture underlying murine reproductive fatpad weight. We first performed a slQTL scan on the original eight chromosomes harboring direct effect loci in the F2 generation (1, 6–9, 12, 13, and 18) as well as the four shown to harbor slQTL in the combined F9–F10 population (3, 4, 10, and 16) (Fawcett et al. 2010). Composite interval mapping and two QTL tests were subsequently performed, the latter when multiple loci on a single chromosome were suspected. Finally, we carried out a full genome-wide scan for pairwise epistasis. To identify the most meaningful set of loci to screen for candidate genes, marker genotypes representing slQTL and epiQTL that exceeded their appropriate thresholds were combined in linear models, first for each chromosome separately and ultimately the entire genetic system. Confidence intervals for peaks that remained significant in the full model were screened for positional candidate loci and potential physiological interactions via both the Mouse Genome Informatics (MGI) database (www.informatics.jax.org/) and a literature search.
MATERIALS AND METHODS
Data:
The population analyzed is the F10 generation (N = 1298; 85 full-sib families; average litter size 8.45) of an AI line, generated from an F2 intercross of the inbred mouse strains SM/J and LG/J (Chai 1956a,b; Cheverud et al. 1996, 2001; Kramer et al. 1998; Vaughn et al. 1999). The animal facility is maintained at a constant temperature of 21° with 12-hour light/dark cycles. Animals were fed a standard rodent chow (PicoLab Rodent Chow 20 (no. 5053) with 12% of its energy from fat, 23% from protein, and 65% from carbohydrate) ad libitum and were weaned at 3 weeks of age. After weaning, animals were housed in single-sex cages containing no more than five individuals.
Between the F2 and F10 generations, the population was maintained at an effective size of ∼300 with 75 mating pairs and no variance in family size. Mating between littermates was actively avoided. At greater than 13 weeks of age, animals were killed and necropsies performed. The reproductive fatpads of each animal were removed, combined, and weighed on a digital scale to the nearest 100th of a gram. Phenotypes were statistically corrected for age at necropsy, sex, litter size, and parity status (whether or not they were mated to produce the F11), using multiple regression, and the residuals used for further analysis. Genotypes for each individual were obtained at 1470 polymorphic SNPs across the genome by GoldenGate Assay (Illumina; San Diego) using DNA extracted from liver tissue obtained at necropsy. Intermarker genotypes were imputed at 1-cM intervals using the equations of Haley and Knott (1992).
Mapping analyses:
A single-locus QTL (slQTL) scan at all measured and imputed loci was first conducted on chromosomes 1, 3, 4, 6–10, 12, 13, 16, and 18 using the regression model
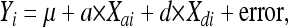 |
(1) |
where Yi is the vector of corrected phenotypes, μ is a constant, and Xai and Xdi are the vectors of genotype scores; a and d are the fitted additive and dominance regression coefficients, respectively. The sums of squares for both model terms were pooled for significance testing. The results of the full genome-wide slQTL mapping in the combined F9–F10 generations were previously reported (Fawcett et al. 2010).
CIM (Zeng 1994) was applied to the identified, preliminary confidence intervals using the following model:
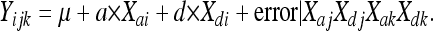 |
(2) |
In this case, Xaj, Xdj, Xak, and Xdk represent vectors of genotype scores at loci >20 F10 cM up- and downstream of the confidence interval on whose effects the within-interval regressions were conditioned. This eliminates the effects of proximal and distal QTL on the same chromosome from being confounded with the target QTL. When multiple peaks on the same chromosome were suggested, the fit of all pairwise two-locus models were compared to the appropriate single-locus case using a χ2 test with 2 degrees of freedom (d.f.) (χcrit2 = 2 * abs[ln(1/pone) − ln(1/ptwo)], where pone and ptwo are P-values from the one- and two-locus models, respectively (Sokal and Rohlf 1995).
Finally all genome-wide, between-chromosome, pairwise combinations of measured and imputed autosomal loci were tested using the following epistatic mapping model:
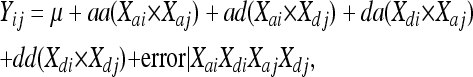 |
(3) |
where aa, ad, da, and dd are the additive-by-additive, additive-by-dominance, dominance-by-additive, and dominance-by-dominance epistasis regression coefficients, and Xai Xdi Xaj Xdj represent vectors of the corresponding additive and dominance genotypes at the two loci involved. The sums of squares and degrees of freedom for all four epistatic components were pooled for initial significance testing. The raw probability associated with each multiple regression for all mapping analyses above was transformed to a linear scale using the base 10 logarithm of the inverse of the probability of no epistasis (LPR = log10(1/p)), producing values comparable to LOD scores obtained through maximum likelihood analysis (Lander and Botstein 1989).
Thresholds:
Interpretation of these analyses is complicated both by the large number of comparisons involved as well as the family structure present in the population. To account for these two issues simultaneously, simulations were performed using the known pedigree of all individuals between the F2 and F10 generations to generate a null distribution of expected effects from which the appropriate single-locus LPR threshold was determined (Fawcett et al. 2008, Norgard et al. 2009). Given a heritability of reproductive fatpad weight in the F10 of 0.47 (from sib correlations) chromosome-specific thresholds for identifying novel slQTL ranged from 6.15 (chromosome 8) to 6.6 (chromosome 1). The experiment-wide threshold for novel slQTL detection was 7.34. For the purposes of replication, a corrected pointwise threshold (equivalent to P = 0.05) of 3.32 was applied for slQTL peaks within previously identified confidence intervals.
Following the method described in Fawcett et al. (2010), the analysis-wide epistasis threshold for the identification of novel interactions was calculated to be 8.33. The threshold for tests between a given slQTL and all other unlinked markers in the analysis was 6.06 and the analogous chromosome-specific thresholds ranged from 4.73 (chromosome 8) to 5.25 (chromosome 1). The corrected pointwise threshold for epistatic tests between two slQTL was 3.44. Tests involving slQTL are partially protected from multiple comparisons as they were identified with independent information.
Confidence intervals:
Due to the complexity of our mapping strategy, the conventional 1 LPR drop criterion was applied to define all reported confidence intervals. When multiple peaks, either slQTL, epiQTL, or both occurred in the same region, the most proximal and most distal 1 LPR drop was used to determine C.I. endpoints. C.I.s for slQTL peaks were also calculated for each location individually using the standard deviation of the simulated distribution of 1000 mapping iterations involving known effects on simulated chromosomes (Norgard et al. 2009). The two techniques yielded very similar C.I. for all slQTL, although the simulation-based intervals were slightly smaller.
Linear models:
We constructed and evaluated separate chromosome-specific models using the linear model function in R (R Development Core Team 2009) before combining their results into a full model of the genetic system. This process began with terms representing each significant effect at all slQTL peaks identified by the single-locus model (Equation 1) and composite interval mapping (Equation 2). For example, the chromosome 1 model (see Figure 1A) began with five slQTL terms representing the additive (P = 0.00726) and dominance (P = 0.0007) effects at 20.15 Mb, the additive (P = 0.000268) and dominance (P = 0.0383) effects at 70.77 Mb and the dominance effect (P = 1.06 × 10−06) at 134.82 Mb. The additive effect at 134.82 Mb was nonsignificant in the slQTL mapping model (P = 0.868) and so was not included. Likewise, the chromosome 13 model (see Figure 1B) included two terms representing the additive effects at 53.54 Mb (P = 3.05 × 10−06) and 90.61 Mb (P = 4.88 × 10−05), respectively. In this case, neither dominance effect was significant in the slQTL mapping model (P = 0.798 and P = 0.634) and so both were excluded. When considered jointly, some individual terms (e.g., the dominance effect only at 70.77 Mb on chromosome 1) no longer remained significant (P < 0.05) in type I ANOVA tables (using the “anova” function). Such terms were removed. For those chromosomes not found to harbor slQTL, a similar process was performed beginning with all significant interactions.
Figure 1.—

Mapping results of significant terms from the full model of reproductive fatpad weight in the LG,SM AI line for chromosomes 1 (A) and 13 (B). Results from the single-locus model are given as connected shaded dots, composite interval mapping as smooth solid lines, and epistatic interactions by other connected shapes. Confidence intervals from previous analyses are represented by horizontal bars below the QTL plot.
Next, individual coefficients from the epistatic mapping model (aa, ad, da, dd; Equation 3) at all peaks that exceeded their appropriate thresholds in the epiQTL scan were similarly examined to determine the type or types of interactions occurring. Terms representing all significant interactions were then added stepwise to each appropriate chromosome-specific model. Only epistatic terms that remained significant (P < 0.05) in both type I and type II ANOVA tables, using the R functions “anova” and “Anova” (the latter from the package “car”), respectively, and did not cause any established additive or dominance effects to become nonsignificant (P < 0.05) were retained to define each final chromosome-specific model. These stringent criteria were established to obtain a tractable number of high-confidence C.I. to screen for positional candidates and physiological interactions.
Next, additive and dominance terms from all chromosome-specific models were combined and terms that became nonsignificant in either type I or type II ANOVA tables (or both) were culled to define the “slQTL system.” This model included 20 terms at 18 loci (15 additive and 5 dominance; boldface type in supporting information, Table S1). Epistasis terms significant in the chromosome-specific models were then added stepwise to the slQTL system as above to define the “full model.” In addition to the 20 marginal-effect terms, this model includes 23 interactions involving 26 different epiQTL confidence intervals. Finally, since many epiQTL peaks occur at locations not represented in the slQTL system, the appropriate additive and dominance terms for each interaction were added to the full model to ensure that the identified epistatic contributions were not unduly biased upward by variance attributable to single-locus effects. This had relatively little effect and resulted in the elimination of only 3 interactions, all of which are significant in type I tests. The results from the full model are reported with these nominally significant terms noted in boldface type (Table 1, see below).
TABLE 1.
Results from the full linear model of the epistatic network underlying murine reproductive fatpad weight in the LG,SM AI line
| Chr 1 | C.I. 1 begin (Mb) | C.I. 1 end (Mb) | Peak 1 (Mb) | Chr 2 | C.I. 2 begin (Mb) | C.I. 2 end (Mb) | Peak 2 (Mb) | slQTL LPR | Peak SNP 1 | Peak SNP 2 | Epistatic LPR | Effect(s) | Threshold type | Threshold | Reported Adipose QTL in CI(s) | QTL reference(s) | Candidates (C.I. 1) | Candidates (C.I. 2) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 16.40 | 21.28 | 20.15 | NA | NA | NA | NA | 4.26 | rs6334092 | NA | NA | A,D | Pointwise | 3.32 | Adip1; Obq2 | Cheverud et al. 2001; Fawcett et al. 2008; Taylor and Phillips 1996 | Pkhd1 | NA |
| 1 | 65.79 | 74.08 | 70.77 | NA | NA | NA | NA | 4.76 | rs6323094 | NA | NA | A | Pointwise | 6.60 | Obq7 | Taylor et al. 2001 | Vwc2l; Fn1 | NA |
| 1 | 118.37 | 138.01 | 134.82 | NA | NA | NA | NA | 9.17 | gnf01.132.831 | NA | NA | A | Pointwise | 3.32 | Obsty1; Gwth1; Obq17 | Cheverudet al. 2004; Yi et al. 2006; Ishimori et al. 2004 | Pik3c2b | NA |
| 3 | 20.54 | 27.82 | 22.51 | NA | NA | NA | NA | 5.56 | rs13477017 | NA | NA | A | Pointwise | 3.32 | None | None | Nlgn1; Ghsr | NA |
| 4 | 9.71 | 11.92 | 10.83 | NA | NA | NA | NA | 4.78 | rs13477558 | NA | NA | D | Pointwise | 3.32 | Unnamed RI QTL | Cheverudet al. 2004 | Plekhf2 | NA |
| 4 | 78.28 | 90.30 | 79.46 | NA | NA | NA | NA | 11.87 | CEL-4-78089985 | NA | NA | A | Pointwise | 3.32 | Adip11; Adip24; Adip11a | Fawcett et al. 2008; Fawcett et al. 2010; Stylianou et al. 2006 | Tyrp1 | NA |
| 6 | 114.73 | 121.97 | 117.73 | NA | NA | NA | NA | 5.01 | mCV23042866 | NA | NA | D | Pointwise | 3.32 | Adip2; Igf1sl1 | Cheverud et al. 2001; Rosen et al. 2005 | Adipor2; Ankrd26; Pparg | NA |
| 6 | 133.92 | 142.67 | 134.20 | NA | NA | NA | NA | 8.89 | rs13479053 | NA | NA | A | Pointwise | 3.32 | Adip2 | Cheverud et al. 2001 | Lrp6; Grin2b; Cdkn1b | NA |
| 7 | 30.18 | 44.44 | 37.21 | NA | NA | NA | NA | 4.08 | rs6217275 | NA | NA | D | Pointwise | 3.32 | Adip3; Adip3A; Adip3Ab | Cheverud et al. 2001; Fawcett et al. 2008; Fawcett et al. 2010 | Tshz3; Plekhf1 | NA |
| 7 | 59.83 | 77.73 | 63.51 | NA | NA | NA | NA | 6.85 | rs3717293 | NA | NA | A,D | Pointwise | 3.32 | Tabw; Adip3Ad; Adip25; Obq1 | Kim et al. 2001; Fawcett et al. 2010; Taylor and Phillips 1996 | Nipa1; Nipa2; Gabrg3; Gabra5; Gabrb3 | NA |
| 7 | 132.03 | 143.20 | 135.24 | NA | NA | NA | NA | 6.38 | CEL-7-116160192 | NA | NA | A | New slQTL chr7 | 6.36 | Bsbob2 | Yi et al. 2004 | Trim72 | NA |
| 8 | 64.98 | 90.95 | 84.79 | NA | NA | NA | NA | 4.76 | rs13479860 | NA | NA | A | Pointwise | 3.32 | Adip4 | Cheverud et al. 2001; Fawcett et al. 2008 | Il15 | NA |
| 9 | 61.70 | 67.72 | 65.39 | NA | NA | NA | NA | 6.98 | rs13480247 | NA | NA | A | New slQTL chr9 | 6.38 | None | None | Mtfmt | NA |
| 9 | 118.30 | 125.00 | 118.88 | NA | NA | NA | NA | 9.64 | rs6316481 | NA | NA | A | Pointwise | 3.32 | Adip5; Adip5a; Adip5b; Adip5c; Obq18 | Cheverud et al. 2001; Fawcett et al. 2008; Fawcett et al. 2010; Ishimori et al. 2004 | Acvr2b | NA |
| 12 | 60.62 | 67.43 | 64.06 | NA | NA | NA | NA | 5.24 | mCV24690992 | NA | NA | A | Pointwise | 3.32 | Adip6; Adip16; Fob2 | Fawcett et al. 2008; Stylianou et al. 2006; Horvat et al. 2000 | Lrfn5 | NA |
| 13 | 40.74 | 55.35 | 53.54 | NA | NA | NA | NA | 4.90 | rs3699522 | NA | NA | A | Pointwise | 3.32 | Adip7; Adip18; Adip18a; Pfat3 | Cheverud et al. 2001; Fawcett et al. 2008; Fawcett et al. 2010; Keightley et al. 1998 | Cplx2; Drd1a | NA |
| 18 | 24.19 | 56.21 | 48.82 | NA | NA | NA | NA | 4.83 | rs3684561 | NA | NA | A | Pointwise | 3.32 | Adip8; Adip8a; Adip8b; Kcal1; Mnif2 | Cheverud et al. 2001; Fawcett et al. 2010; Smith Richards et al. 2002 | Sema6a; Hsd17b4 | NA |
| 18 | 58.77 | 80.76 | 63.84 | NA | NA | NA | NA | 12.31 | rs13483398 | NA | NA | A | Pointwise | 3.32 | Adip8; Adip8c; Adip8d; Obsty4 | Cheverud et al. 2001; Fawcett et al. 2008; Fawcett et al. 2010; Cheverudet al. 2004 | Adrb2; Htr4 | NA |
| 1 | 42.41 | 52.71 | 51.38 | 9 | 68.10 | 95.10 | 77.25 | NA | rs13475863 | rs13480288 | 5.52 | DD | QTL × QTL epi | 3.44 | Adip1; Obq7; Adip5; Mob8 | Cheverud et al. 2001; Taylor et al. 2001; Mehrabian et al. 1998 | Gls | Gclc |
| 1 | 118.37 | 138.01 | 128.52 | 6 | 133.92 | 142.67 | 141.48 | NA | rs6228473 | rs8268650 | 4.95 | AD | QTL × QTL epi | 3.44 | Obsty1; Gwth1; Obq17; Adip2 | Cheverud et al. 2001; Cheverudet al. 2004; Yi et al. 2006; Ishimori et al. 2004 | Gpr39 | Pde3a |
| 1 | 118.37 | 138.01 | 128.84 | 12 | 73.42 | 89.12 | 75.11 | NA | rs13476100 | rs3687032 | 4.64 | DD | QTL × QTL epi | 3.44 | Obsty1; Gwth1; Obq17; Adip6 | Cheverud et al. 2001; Cheverudet al. 2004; Yi et al. 2006; Ishimori et al. 2004 | Gpr39 | Hif1a |
| 1 | 174.21 | 189.05 | 186.63 | 13 | 0.00 | 24.24 | 23.48 | NA | mCV24555989 | gnf13.020.621 | 10.27 | AA | QTL × chr1 epi | 5.25 | Obq9 | Taylor et al. 2001 | Hlx | Abt1 |
| 4 | 30.53 | 39.16 | 36.58 | 9 | 118.30 | 125.00 | 123.70 | NA | rs13477649 | rs8241505 | 6.03 | DD | QTL × QTL epi | 3.44 | Unnamed RI QTL; Dob2; Obq18 | Cheverudet al. 2004; Ishimori et al. 2004; West et al. 1994 | Cga | Slc6a20a; Slc6a20b |
| 4 | 125.68 | 139.92 | 130.91 | 7 | 132.03 | 143.20 | 139.70 | NA | rs3673061 | rs8236684 | 4.93 | AD | QTL × QTL epi | 3.44 | Adip12; Qbis1; Afpq2; Adip3 | Cheverud et al. 2001; Stylianou et al. 2006; Togawa et al. 2006; Brockmann et al. 2000 | Ptpru | Oat |
| 4 | 143.52 | 154.77 | 152.94 | 7 | 132.03 | 143.20 | 141.88 | NA | rs6378384 | rs3719258 | 4.69 | AD | QTL × QTL epi | 3.44 | Adip12; Adip3 | Cheverud et al. 2001; Stylianou et al. 2006 | Ajap1 | Adam12 |
| 6 | 33.46 | 46.84 | 37.64 | 9 | 118.30 | 125.00 | 123.70 | NA | rs13478717 | rs8241505 | 5.11 | DA | QTL × chr6 epi | 5.09 | Dob2; Obq18 | Ishimori et al. 2004;West et al. 1994 | Trim24 | Ccr9 |
| 6 | 53.92 | 71.82 | 54.18 | 7 | 102.32 | 108.47 | 105.10 | NA | rs13478762 | UT-7-90.803899 | 5.13 | AA | QTL × chr7 epi | 4.96 | Adip2; Obq13 | Cheverud et al. 2001; Taylor et al. 2001 | Crhr2; Ghrhr | Capn5 |
| 7 | 132.03 | 143.20 | 137.17 | 8 | 42.26 | 57.10 | 50.65 | NA | rs8236684 | rs13479769 | 5.08 | AA | QTL × chr8 epi | 4.73 | Bsbob2 | Yi et al. 2004 | Fgfr2 | Ing2 |
| 8 | 124.83 | 129.12 | 127.97 | 9 | 20.24 | 39.76 | 23.57 | NA | rs6300613 | rs13480112 | 5.57 | AD | QTL × chr9 epi | 4.99 | Obsty2 | Cheverudet al. 2004 | Disc1 | Npsr1 |
| 9 | 20.24 | 39.76 | 31.31 | 12 | 108.99 | 120.28 | 111.04 | NA | CEL-9-29909656 | CEL-12-104545022 | 5.36 | AA,DD | QTL × chr9 epi | 4.99 | Carfhg2 | Corva et al. 2001 | Kcnj5 | Dlk1; Meg3; Rtl1 |
| 9 | 104.05 | 118.18 | 109.62 | 1 | 191.98 | NA | 193.61 | NA | rs3723953 | rs13476308 | 7.78 | AA | QTL × chr1 epi | 5.99 | Adip5;Dob2 | Cheverud et al. 2001; West et al. 1994 | Fbxw cluster | Nek2 |
| 12 | 108.99 | 120.28 | 113.11 | 1 | 191.98 | NA | 195.79 | NA | rs13481651 | rs13476312 | 6.06 | DA | QTL × chr1 epi | 5.25 | Adip6; Bsbob4; Mob3 | Cheverud et al. 2001; Fawcett et al. 2008; Yi et al. 2004; Warden et al. 1995 | Traf3 | Hsd11b1 |
| 13 | 0.00 | 24.24 | 14.85 | 1 | 118.37 | 138.01 | 119.02 | NA | rs13481702 | rs3694226 | 5.96 | AA | QTL × chr1 epi | 5.25 | Adip7 | Cheverud et al. 2001 | Inhba | Inhbb |
| 13 | 0.00 | 24.24 | 17.38 | 9 | 68.10 | 95.10 | 82.84 | NA | rs3678616 | rs13480312 | 4.54 | AA | QTL × QTL epi | 3.44 | Adip7; Adip5 | Cheverud et al. 2001 | Inhba | Htr1b |
| 13 | 0.00 | 24.24 | 20.21 | 12 | 73.42 | 89.12 | 82.08 | NA | rs6314295 | rs3654718 | 4.78 | AA | QTL × QTL epi | 3.44 | Adip7; Adip6 | Cheverud et al. 2001 | Olfactory receptor cluster | Slc8a3 |
| 13 | 40.74 | 55.35 | 43.69 | 6 | 80.99 | 92.88 | 89.62 | NA | rs13481789 | rs13479099 | 4.90 | AA | QTL × QTL epi | 3.44 | Adip7; Adip18; Adip18a; Pfat3; Adip2 | Cheverud et al. 2001; Fawcett et al. 2008; Fawcett et al. 2010; Keightley et al. 1998 | Ranbp9 | Alms1 |
| 13 | 40.74 | 55.35 | 45.45 | 4 | 143.52 | 154.77 | 152.94 | NA | rs3688207 | rs6378384 | 4.48 | AD | QTL × QTL epi | 3.44 | Adip7; Adip18; Adip18a; Pfat3; Adip12 | Cheverud et al. 2001; Fawcett et al. 2008; Fawcett et al. 2010; Stylianou et al. 2006; Keightley et al. 1998 | Atxn1 | Kcnab2 |
| 18 | 24.19 | 56.21 | 37.51 | 12 | 60.62 | 67.43 | 64.06 | NA | gnf18.033.953 | mCV24690992 | 5.88 | AD | QTL × QTL epi | 3.44 | Adip8; Adip8a; Adip8b; Kcal1; Mnif2; Adip6 | Cheverud et al. 2001; Fawcett et al. 2010; Smith Richards et al. 2002 | Pcdhb cluster | Lrfn5 |
| 18 | 24.19 | 56.21 | 37.93 | 13 | 0.00 | 24.24 | 15.11 | NA | gnf18.033.953 | rs13481702 | 5.87 | DA | QTL × QTL epi | 3.44 | Adip8; Adip8a; Adip8b; Kcal1; Mnif2; Adip7 | Cheverud et al. 2001;Fawcett et al. 2010; Smith Richards et al. 2002 | Pcdhb cluster | Gli3 |
| 18 | 24.19 | 56.21 | 50.47 | 7 | 30.18 | 44.44 | 30.56 | NA | rs13483356 | rs13479174 | 5.76 | AD | QTL × QTL epi | 3.44 | Adip8; Adip8a; Adip8b; Kcal1; Mnif2; Adip3 | Cheverud et al. 2001; Fawcett et al. 2010; Smith Richards et al. 2002 | Hsd17b4 | Lrfn3 |
Chromosome, confidence intervals (Mb), peak locations (Mb), peak LPR scores, nearest SNP to the peak, effect-type threshold, and threshold value are all given for each term. The appropriate references for any a priori hypotheses are listed along with positional candidate loci for both slQTL and epiQTL. Terms in boldface type are nominally significant (P > 0.05) when additive and dominance effects for all interactions are included in the model.
Candidate genes:
All C.I.s for peaks identified in the full model were screened for plausible positional candidate genes and known interactions. This involved both queries of the MGI database for functional variants affecting adiposity as well as a broad literature search and was intended to generate meaningful and testable physiological hypotheses regarding the observed statistical associations.
RESULTS
Replication and identification:
Significant marginal effects, epistatic effects, or both are observed in the F10 population on all eight chromosomes harboring the original Adip loci and three of the four additional chromosomes implicated in the combined F9–F10 slQTL scan (Figure S1). In the F10 alone, there were no significant slQTL on chromosome 16. Similar to the results of Fawcett et al. (2010), peak LPR scores from either the single-locus scan or composite interval mapping at or near the confidence intervals of five Adip loci exceeded the experiment-wide threshold (7.34) for novel QTL detection (Adip1, LPR = 9.2; Adip2, LPR = 8.9; Adip3, LPR = 8.3; Adip5, LPR = 9.6; and Adip8, LPR = 12.3). All three remaining F2 loci exceed the pointwise threshold (3.32) required for tests within previously defined confidence intervals (Adip4, LPR = 5.6; Adip6, LPR = 5.24; and Adip7, LPR = 4.8). Additional slQTL on chromosomes 3, 4, and 10 also replicated. Interestingly, the chromosome 4 locus (Adip24) (Fawcett et al. 2010; LPR = 12.65) roughly corresponds to two loci previously reported in the literature as Adip11 and Adip12 in a cross between C57BL/6J and DBA/2J (Keightley et al. 1996; Brockmann et al. 1998; Stylianou et al. 2006). Finally, composite interval mapping revealed novel loci on chromosomes 7 and 9 that both exceed their appropriate chromosome-specific thresholds of 6.36 and 6.38, respectively. A total of 22 potential marginal effect peaks were identified (Table S1).
epiQTL mapping:
In the genome-wide scan for epistasis, 177 peaks involving 217 interactions exceeded their appropriate significance thresholds and physically cluster into ∼51 potential epiQTL (Table S1). Additive-by-additive interactions were the most common (98), additive-by-dominance or dominance-by-additive were the next most common (97), and dominance-by-dominance interactions were the most rare (22). Consistent with the results of Jarvis and Cheverud (2009) and several other studies (see Phillips 2008), many of these occurred at locations showing no significant marginal effects in this cross, though some occurred at locations significant in slQTL scans in other crosses (Table 1; Figure 1; Table S1; Figure S2; Figure S3; Figure S4; Figure S5; Figure S6; Figure S7; Figure S8; Figure S9; Figure S10; Figure S11; Figure S12; Figure S13; Figure S14; Figure S15; Figure S16; Figure S17; Figure S18; Figure S19; Figure S20).
Linear models:
In total, we identified 199 slQTL and epiQTL peaks that potentially contribute to trait variation. These cluster into roughly 73 confidence intervals showing a variety of combinations of additive, dominance, and epistatic effects (Table S1). To identify the most robust signals, we systematically added vectors of genotype scores representing each into linear models and determined the set that is simultaneously significant in both type I and type II tests. We began by establishing a single-locus model that contained all slQTL peaks that remain significant together. This slQTL system includes 20 marginal-effect terms (15 additive and 5 dominance) and shows an adjusted R2 value of 0.2254 (F statistic = 18.64 on 20 and 1281 d.f.). We next added epistatic peaks stepwise to generate a full model of the genetic system. This full model (Table 1) includes 23 additional interaction terms (9 aa, 10 ad/da, and 4 dd) involving 26 different epiQTL confidence intervals and shows an adjusted R2 value of 0.3322 (F statistic = 15.71 on 43 and 1257 d.f.). Using a χ2 goodness-of-fit test with 23 (43–20) d.f. this represents a highly significant improvement in fit over the base slQTL model (P < 10−25). Following the addition of all marginal terms involved in epistasis, three interaction terms become nonsignificant at the P < 0.05 level in either type I or type II tables or both (boldface terms in Table 1). Removing these interactions from the full model, its adjusted R2 value is 0.3220 (F statistic = 16.07 on 40 and 1260 d.f.), which also represents a highly significant improvement in model fit (P < 10−20).
Positional candidates:
While in-depth functional assays and other detailed molecular studies are required to sort out the biological basis of QTL and their interactions, examination of positional candidate genes in slQTL confidence intervals suggests testable physiological hypotheses for several observed statistical effects. In general, confidence intervals contain a variety of candidate loci including transcription factors, components of various signaling cascades (e.g., the Wnt, Insulin, and Igf signaling networks), neuroendocrine hormones and their receptors, as well as genes directly implicated in glucose processing and metabolism. For example, the C.I. found at 6:133.92–142.67 Mb contains the promising candidate Lrp6, a low-density lipoprotein receptor-related protein that is thought to contribute to variation in a variety of metabolic risk factors in humans (Kahn et al. 2007; Mani et al. 2007) and Cdkn1b, a cyclin-dependent kinase inhibitor with known effects on pancreatic islet mass in diabetic mice (Uchida et al. 2005). Both Lrp6 and Cdkn1b have differences in expression level in white fat (P = 3.82 × 10−12 and 0.013, respectively) and in the liver (P = 1.62 × 10−13 and 7.48 × 10−8, respectively) between the two parental lines in this cross (J. M. Cheverud, unpublished results). The C.I. 18:58.77–80.76 Mb shows potential functional links to mammalian neurotransmitter signaling via Htr4 (Gardner et al. 2008), as do 13:40.74–55.35 Mb via Cplx2 (Brachya et al. 2006) and Drd1a (de Leeuw van Weenen et al. 2009). In addition, the region 6:114.73–121.97 Mb contains neuroendocrine candidates Adipor2 (Yamauchi et al. 2007; Ziemke and Mantzoros 2010) and Ankrd26 (Bera et al. 2008), which also shows a significant difference in expression in liver between LG/J and SM/J (P = 0.0002) (J. M. Cheverud, unpublished results). Together, these loci suggest a functionally similar genetic architecture to the emerging picture of type 2 diabetes in humans (Doria et al. 2008).
There are also a number of strong candidate loci for observed epistatic interactions. The most striking involves the C.I.s 13:0–24.24 Mb and 1:118.37–138.01 Mb, which contain Inhba and Inhbb, respectively. The proteins encoded by these loci are components of the Activin and Inhibin complexes, which have wide-ranging effects on a variety of physiologic, homeostatic, and metabolic processes including mammalian reproduction, inflammation, and adipocyte differentiation (Woodruff and Mather 1995; Hirai et al. 2005; Werner and Alzheimer 2006). Interestingly, 13:0–24.24 Mb participates in five separate interactions that are significant in the full model (Table 1) and appears to interact with a region (9:68.10–95.10 Mb) containing an important receptor for serotonin (Htr1b).
Glutamate signaling and metabolism are also likely to underlie a portion of fatpad variation due to epistasis in this cross. The interacting epiQTL CI 1:42.41–52.71 Mb and 9:68.10–95.10 Mb contain the enzyme that catalyses the first reaction in the primary pathway for the renal catabolism of glutamine (Gls) and the first rate-limiting enzyme in glutathione synthesis (Gclc), respectively. Gls also shows differential expression in white fat cells between the parental lines (P = 0.00097). Ghrelin and its associated pathways also appear as likely candidates. For example, 1:118.37–138.01 Mb contains Gpr39, a member of the ghrelin receptor family. This C.I. interacts with 6:133.92–142.67 Mb, which harbors Pde3a, a locus known to be downstream of ghrelin signaling in platelets (Elbatarny et al. 2007) and which shows significant differences in gene expression in white fat between SM/J and LG/J (P = 0.00018) and 12:73.42–89.12 Mb, which contains Hif1a, whose protein product increases the expression of Vegf (Hoffmann et al. 2008). Interestingly, Vegfc shows a significant difference in expression in white fat between the parental lines (P = 0.001) and Vegfb shows differences in liver (P = 0.009). Ghrelin is also known to increase the expression of Vegf in human luteal cells (Tropea et al. 2007) and Vegf, in turn, is thought to be an important regulator of adipogenesis and obesity (Cao 2007). A final interesting epiQTL CI is 12:108.99–120.28 Mb. It contains Dlk1, Meg3, and Rtl1, all three of which appear to participate in an interacting (and imprinted) network affecting growth in mice (Gabory et al. 2009).
DISCUSSION
While the family structure of an outbred population complicates some aspects of the mapping process, the F10 (and later) generations of advanced intercross lines hold an intrinsic advantage in mapping resolution over more conventional study designs. Here this advantage translated into a variety of results with important implications for mapping complex trait variation and new insights into the genetic architecture of murine fatpad weight.
The first and most striking result of this analysis from a mapping perspective is the relatively low level of overlap in the physical positions of slQTL and epiQTL peaks despite the analytical bias toward finding epistasis involving slQTL due to their protected status with respect to multiple comparisons. Though slight discrepancies may be expected due to subtle patterns of linkage, larger map distances between peaks likely indicate that multiple functional variants are present. Indeed, when both types are observed in close proximity, epistatic peaks tend not to line up well with their single-locus counterparts and epiQTL are frequently observed in regions showing no significant marginal effects at all (Figure 1; Table 1; Table S1; Figure S1; Figure S2; Figure S3; Figure S4; Figure S5; Figure S6; Figure S7; Figure S8; Figure S9; Figure S10; Figure S11; Figure S12; Figure S13; Figure S14; Figure S15; Figure S16; Figure S17; Figure S18; Figure S19; Figure S20). This supports the notion that a relatively large number of variable, functionally relevant loci exert their influence on complex trait variation primarily via epistatic interactions rather than through conventional additive and dominance effects. It is also interesting to note that some regions interact with multiple locations in the genome. For example, proximal chromosome 13 (13:0–24.24 Mb) shows five significant interactions in the full model, including two with separate locations on chromosome 1. Identifying such repeated signals may be useful in developing significance thresholds that help ameliorate the penalties incurred by performing multiple comparisons. Such consistency may also help distinguish epiQTL at the center vs. the edges of functional networks.
Next, in keeping with observations in congenic lines (e.g., Christians et al. 2006) as well as other recent slQTL mapping studies (Fawcett et al. 2010), F2 confidence intervals were frequently observed to divide into multiple significant slQTL (Figure 1; Figure S1). Interestingly, we observe similar splitting of single-locus and epistatic signals. For example, at the proximal end of chromosome 1 (Figure 1A) marginal-effect peaks observed in the F2, combined F2–3, and in an intercross between SM and NZO (obq7; Taylor et al. 2001) appear to resolve in our mapping population into three distinct peaks with two marginal effect loci flanking an epiQTL. This suggests that the original F2 and the subsequent F2–3 signals in this cross were composites of both single-locus and epistatic effects and that the boundaries of previously reported C.I. may have been influenced by epistatic contributions to single-locus values. Thus, current estimates of the number of loci underlying trait variation are likely to be overly conservative and reported effect-size estimates are potentially biased by the presence of multiple, closely linked functional elements. Interestingly, it also suggests that confidence intervals identified in other intercross experiments, especially those that share a parental strain, can be productively evaluated under a priori epistatic hypotheses, which may also ease issues related to multiple testing. On this account, it is also striking that the epistatic network identified in Stylianou et al. (2006) as Chr4-Adip11 is centered on a region also identified here as contributing to the epistatic architecture of fatpad weight.
The results of composite interval mapping also suggest that adjacent slQTL and epiQTL impact the mapping process. For example, there is a dramatic and unexpected increase in significance (nearly three orders of magnitude) for the additive slQTL peak at 134.82 Mb on chromosome 1 when composite interval mapping was applied (Figure 1A). While this is the most dramatic example, such effects were repeatedly observed (Figure S1) and on chromosomes 7 and 9, this resulted in the identification of two novel loci. Interestingly, this suggests that adjacent functional variants with opposite effects were fixed in the original parental lines during their production. Indeed, inspection of the regression coefficients from the full linear model shows that the epistatic peak closest to the slQTL signal at 134.82 Mb on chromosome 1 (DD with 12:73.42–89.12 Mb) and the marginal signal itself share a positive sign. However, the two slightly centromeric interactions involving the additive value on chromosome 1 (AA with 13:0–24.24 Mb and AD with 6:133.92–142.67 Mb) are both negative. Conditioning on these adjacent markers is indeed expected to enhance the signal of the neighboring additive effect, consistent with our observations. Thus, comparing the results of a conventional single-locus mapping model and composite interval mapping may be an indirect means of identifying neighboring functional variants. Further mapping in later generations of this advanced intercross will provide a great deal of additional information on the sign, magnitude, and physiological basis for these observed effects as recombination is expected to further separate their statistical signatures.
Conclusions:
The application of multiple mapping approaches, including an epistatic model, is a vital strategy for characterizing complex genetic architectures. Contrary to suggestions based on human genome-wide association study findings, we found substantial numbers of pairwise epistatic interactions involving many more loci than show single-locus effects that account for an important portion of trait variation. This is likely due to the genetic structure of our experimental population where allele frequencies are intermediate; there are no rare alleles in our mapping system. This is critical since epistasis is known to produce predominantly additive and dominance variance when relatively rare alleles are involved (Cheverud and Routman 1995; Cheverud 2000).
Here, the use of a combination of techniques was further enhanced by the improved genetic resolution offered by AI lines. While single-locus scans remain the most tractable, pairwise epistatic relationships can now be dissected in great detail as well, and the identification of candidate loci for such interactions is possible. This is especially true for characters for which a large body of literature exists describing the mechanistic relationships among candidate genes and related pathologies. In such cases, incorporating a priori information regarding functional interactions can be used to help focus epistatic mapping studies and both ease the difficulties associated with multiple comparisons and facilitate the physiological interpretation of statistical results. It is an exciting prospect that even more fine-scale mapping of these loci will be possible in later generations of the LG,SM AI line. Undoubtedly future analyses, coupled with the incorporation of sequence information from the parental lines, will aid in further refining the physiological hypotheses presented here for fatpad variation and greatly contribute to our understanding of the statistical signatures of complex genetic architectures.
Acknowledgments
The authors acknowledge G. L. Fawcett and C. A. Lambert for offering insightful comments and suggestions on earlier drafts of this manuscript. This work was supported by a grant from the National Institutes of Health (DK-055736) and a doctoral dissertation improvement grant from the National Science Foundation (DEB-0608352).
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.123505/DC1 and the QTL archive at http://qtlarchive.org.
References
- Bartlett, M. S., and J. B. S. Haldane, 1935. The theory of inbreeding with forced heterozygosity. J. Genet. 31 327–340. [Google Scholar]
- Bera, T. K., X-F. Liu, M. Yamada, O. Gavrilova, E. Mezey et al., 2008. A model for obesity and gigantism due to disruption of the Ankrd26 gene. Proc. Natl. Acad. Sci. USA 105(1): 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachya, G., C. Yanay and M. Linial, 2006. Synaptic proteins as multi-sensor devises of neurotransmission. BMC Neurosci. 7(Suppl 1): S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann, G. A., C. S. Haley, U. Renne, S. A. Knott and M. Schwerin, 1998. Quantitative trait loci affecting body weight and fatness from a mouse line selected for extreme high growth. Genetics 150 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann, G. A., J. Kratzsch, C. S. Haley, U. Renne, M. Schwerin et al., 2000. Single QTL effects, epistasis, and pleiotropy account for two-thirds of the phenotypic F(2) variance of growth and obesity in DU6i x DBA/2 mice. Genome Res. 10(12): 1941–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, Y., 2007. Angiogenesis modulates adipogenesis and obesity. J. Clin. Invest. 117(9): 2362–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai, C., 1956. a Analysis of quantitative inheritance of body size in mice. I. Hybridization and maternal influence. Genetics 41 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai, C., 1956. b Analysis of quantitative inheritance of body size in mice. II. Gene action and segregation. Genetics 41 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chehab, F. F., 2008. Minireview: obesity and lipodystrophy—Where do the circles intersect? Endocrinology 149 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud, J. M., 2000. Detecting epistasis among quantitative trait loci, pp. 58–81 in Epistasis and the Evolutionary Process, edited by J. B. Wolf, E. D. Brodie and M. J. Wade. Oxford University Press, Oxford.
- Cheverud, J. M., and E. J. Routman, 1995. Epistasis and its contribution to genetic variance components. Genetics 139 1455–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud, J. M., E. J. Routman, F. A. M. Duarte, B. van Swinderen, K. Cothran et al., 1996. Quantitative trait loci for murine growth. Genetics 142 1305–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud, J. M., L. S. Pletscher, T. T. Vaughn and B. Marshall, 1999. Differential response to dietary fat in large (LG/J) and small (SM/J) inbred mouse strains. Physiol. Gen. 1 33–39. [DOI] [PubMed] [Google Scholar]
- Cheverud, J. M., T. T. Vaughn, L. S. Pletscher, A. C. Peripato, E. S. Adams et al., 2001. Genetic architecture of adiposity in the cross of LG/J and SM/J inbred mice. Mamm. Genome 12 3–12. [DOI] [PubMed] [Google Scholar]
- Cheverud, J. M., T. H. Ehrich, J. P. Kenney, L. S. Pletscher and C. F. Semenkovich, 2004. a Genetic evidence for discordance between obesity and diabetes-related traits in the LGXSM recombinant inbred mouse strains. Diabetes 53 2700–2708. [DOI] [PubMed] [Google Scholar]
- Cheverud, J. M., T. H. Ehrich, T. Hrbek, J. P. Kenney, L. S. Pletscher et al., 2004. b Quantitative trait loci for obesity and diabetes-related traits and their dietary responses to high fat feeding in the LGXSM recombinant inbred mouse strains. Diabetes 53 3328–3336. [DOI] [PubMed] [Google Scholar]
- Cheverud, J. M., G. L. Fawcett, J. P. Jarvis, E. A. Norgard, M. Pavlicev et al., 2010. Calpain-10 is a component of the obesity-related quantitative trait locus Adip1. J. Lipid Res. 51 907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christians, J. K., A. Hoeflich and P. D. Keightley, 2006. PAPPA2, an enzyme that cleaves an insulin-like growth-factor-binding protein, is a candidate gene for a quantitative trait locus affecting body size in mice. Genetics 173 1547–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corva, P. M., S. Horvat and J. F. Medrano, 2001. Quantitative trait loci affecting growth in high growth (hg) mice. Mamm. Genome 12(4): 284–290. [DOI] [PubMed] [Google Scholar]
- Darvasi, A., and M. Soller, 1995. Advanced intercross lines, an experimental population for fine genetic mapping. Genetics 141 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw van Weenen, J. E., L. Hu, K. Jansen-Van Zelm, M. G. de Vries, J. T. Tamsma et al., 2009. Four weeks high fat feeding induces insulin resistance without affecting dopamine release or gene expression patterns in the hypothalamus of C57Bl6 mice. Brain Res. 1250 141–148. [DOI] [PubMed] [Google Scholar]
- Doria, A., M. Patti and C. Kahn, 2008. The emerging genetic architecture of type 2 diabetes. Cell Metab. 8 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr, R. H., K. D. Taylor, S. R. Brant, J. D. Rioux, M. S. Silverberg et al., 2006. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314 1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbatarny, H. S., S. J. Netherton, J. D. Ovens, A. V. Ferguson, and D. H. Maurice et al., 2007. Adiponectin, ghrelin, and leptin differentially influence human platelet and human vascular endothelial cell functions: implication in obesity-associated cardiovascular diseases. Euro. J. Pharmacol. 558 7–13. [DOI] [PubMed] [Google Scholar]
- Fawcett, G. L., C. C. Roseman, J. P. Jarvis, B. Wang, J. B. Wolf et al., 2008. Genetic architecture of adiposity and organ weight using combined generation QTL analysis. Obesity 16 1861–1868. [DOI] [PubMed] [Google Scholar]
- Fawcett, G. L., J. P. Jarvis, C. C. Roseman, B. Wang, J. B. Wolf et al., 2010. Fine-mapping of obesity-related quantitative trait loci in an F9/10 advanced intercross line. Obesity 18(7): 1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenske, T. S., C. McMahon, D. Edwin, J. C. Jarvis, J. M. Cheverud et al., 2006. Identification of candidate alkylator-induced cancer susceptibility genes by whole genome scanning in mice. Cancer Res. 66 5029–5038. [DOI] [PubMed] [Google Scholar]
- Gabory, A., M-A. Ripoche, A. Le Digarcher, F. Watrin, A. Ziyyat et al., 2009. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 136 3413–3421. [DOI] [PubMed] [Google Scholar]
- Gardner, M., J. Bertranpetit and D. Comas, 2008. Worldwide genetic variation in dopamine and serotonin pathway genes: Implications for association studies. Am. J. Med. Genet. B 147B(7): 1070–1075. [DOI] [PubMed] [Google Scholar]
- Gat-Yablonski, G., and M. Phillip, 2008. Leptin and regulation of linear growth. Curr. Opin. Clin. Nutr. Metab. Care 11 303–308. [DOI] [PubMed] [Google Scholar]
- Haldane, J. B. S., and C. H. Waddington, 1931. Inbreeding and linkage. Genetics 16 357–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69 315–324. [DOI] [PubMed] [Google Scholar]
- Hanlon, P., W. A. Lorenz, Z. Shao, J. M. Harper, A. T. Galecki et al., 2006. Three-locus and four-locus QTL interactions influence mouse insulin-like growth factor-I. Physiol. Genomics 26 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, W. D., 1959. a The theoretical distribution of lengths of parental gene blocks in the gametes of an F1 individual. Genetics 44 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, W. D., 1959. b Theoretical distribution of the initial linkage block lengths intact in the gametes of a population intermated for n generations. Genetics 44 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, W. D., 1959. c Early generation analysis of lengths of heterozygous chromosome segments around a locus held heterozygous with backcrossing or selfing. Genetics 44 833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, W. D., 1959. d The breakup of initial linkage blocks under selected mating systems. Genetics 44 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, S., M. Yamanaka, H. Kawachi, T. Matsui and H. Yano, 2005. Acitin A inhibits differentiation of 3T3–L1 preadipocyte. Mol. Cell. Endocrinol. 232 21–26. [DOI] [PubMed] [Google Scholar]
- Hoffmann, A-C., R. Mori, D. Vallbohmer, J. Brabender, E. Klein et al., 2008. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia 10(7): 674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvat, S., L. Bunger, V. M. Falconer, P. Mackay, A. Law et al., 2000. Mapping of obesity QTLs in a cross between mouse lines divergently selected on fat content. Mamm. Genome 11(1): 2–7. [DOI] [PubMed] [Google Scholar]
- Ichihara, S., and Y. Yamada, 2008. Genetic factors for human obesity. Cell. Mol. Life Sci. 65 1086–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimori, N., R. Li, P. M. Kelmenson, R. Korstanje, K. A. Walsh et al., 2004. Quantitative trait loci that determine plasma lipids and obesity in C57BL/6J and 129S1/SvImJ inbred mice. J Lipid Res. 45(9): 1624–1632. [DOI] [PubMed] [Google Scholar]
- Jannink, J.-L., M. C. A. M. Bink and R. C. Jansen, 2001. Using complex plant pedigrees to map valuable genes. Trends Plant Sci. 6 337–342. [DOI] [PubMed] [Google Scholar]
- Jarvis, J. P., and J. M. Cheverud, 2009. Epistatis and the evolutionary dynamics of measured genotypic values during simulated serial bottlenecks. J. Evol. Biol. 22 1658–1668. [DOI] [PubMed] [Google Scholar]
- Kahn, Z., S. Vijayakumar, T. Villanueva de la Torre, S. Rotolo and A. Bafico, 2007. Analysis of endogenous LRP6 function reveals a novel feedback mechanism by which Wnt negatively regulates its receptor. Mol. Cell. Biol. 27 7291–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., T. Hardge, L. May and G. Bulfield, 1996. A genetic map of quantitative trait loci for body weight in the mouse. Genetics 142 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., K. H. Morris, A. Ishikawa, V. M. Falconer and F. Oliver, 1998. Test of candidate gene–quantitative trait locus association applied to fatness in mice. Heredity 81(Pt 6): 630–637. [DOI] [PubMed] [Google Scholar]
- Kenney-Hunt, J. P., B. Wang, E. A. Norgard, G. Fawcett, D. Falk et al., 2008. Pleiotropic patterns of quantitative trait loci for 70 murine skeletal traits. Genetics 178 2275–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H., S. Sen, C. S. Avery, E. Simpson, P. Chandler et al., 2001. Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Genomics 74(3): 273–286. [DOI] [PubMed] [Google Scholar]
- Klein, R. J., C. Zeiss, E. Y. Chew, J-Y. Tsai, R. S. Sackler et al., 2005. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science 308 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, M. G., T. T. Vaughn, L. S. Pletscher, K. King-Ellison, E. Adams et al., 1998. Genetic variation in body weight growth and composition in the intercross of Large (LG/J) and Small (SM/J) inbred strains of mice. Genet. Mol. Biol. 21 211–218. [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping mendelian factors underlying quantitative traits using RF.LP linkage maps. [published erratum appears in Genetics 136: 705] Genetics 121 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani, A., J. Radhakrishnan, H. Wang, A. Mani, M-A Mani et al., 2007. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 315(5816): 1278–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrabian, M., P. Z. Wen, J. Fisler, R. C. Davis and A. J. Lusis, 1998. Genetic loci controling body fat, lipoprotein metabolism, and insulin levels in a multifactorial mouse model. J. Clin. Invest. 101(11): 2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgard, E. A., J. P. Jarvis, C. C. Roseman, T. J. Maxwell, J. P. Kenney-Hunt et al., 2009. Replication of long bone length QTL in the F9-F10 LG,SM advanced intercross. Mamm. Genome 20: 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papst, C., M. Bohn, H. F. Utz, A. E. Melchinger, D. Klein et al., 2004. QTL mapping for European corn borer resistance (Ostrinia nubilalis Hb.), agronomic and forage quality traits of testcross progenies in early-maturing European maize (Zea mays L.) germplasm. Theor. Appl. Genet. 108 1545–1554. [DOI] [PubMed] [Google Scholar]
- Phillips, P. C., 2008. Epistasis-the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 9(11): 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team, 2009. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org.
- Rockman, M. V., and L. Kruglyak, 2008. Breeding designs for recombinant inbred advanced intercross lines. Genetics 179 1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen, C. J., C. Ackert-Bicknell, W. G. Beamer, T. Nelson, M. Adamo et al., 2005. Allelic differences in a quantitative trait locus affecting insulin-like growth factor-I impact skeletal acquisition and body composition. Pediatr. Nephrol. 20(3): 255–260. [DOI] [PubMed] [Google Scholar]
- Silver, L. M., 1995. Mouse Genetics: Concepts and Applications. Oxford University Press, New York.
- Sladek, R., G. Rocheleau, J. Rung, C. Dina, L. Shen et al., 2007. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445 881–885. [DOI] [PubMed] [Google Scholar]
- Smith Richards, B. K., B. N. Belton, A. C. Poole, J. J. Mancuso, G. A. Churchill et al., 2002. QTL analysis of self-selected macronutrient diet intake: fat, carbohydrate, and total kilocalories. Physiol. Genomics 11(3): 205–217. [DOI] [PubMed] [Google Scholar]
- Sokal, R. S., and F. J. Rohlf, 1995. Biometry. W. H. Freeman, New York.
- Stylianou, I. M., R. Korstanje, R. Li, S. Sheehan, B. Paigen et al., 2006. Quantitative trait locus analysis for obesity reveals multiple networks of interacting loci. Mamm. Genome 17 22–36. [DOI] [PubMed] [Google Scholar]
- Taylor, B. A., and S. J. Phillips, 1996. Detection of obesity QTLs on mouse chromosomes 1 and 7 by selective DNA pooling. Genomics 34(3): 389–398. [DOI] [PubMed] [Google Scholar]
- Taylor, B. A., C. Wnek, D. Schroeder and S. J. Phillips, 2001. Multiple obesity QTLs identified in an intercross between the NZO (New Zealand obese) and the SM (small) mouse strains. Mamm. Genome 12(2): 95–103. [DOI] [PubMed] [Google Scholar]
- Togawa, K., M. Moritani, H. Yaguchi and M. Itakura, 2006. Multidimensional genome scans identify the combinations of genetic loci linked to diabetes-related phenotypes in mice. Hum. Mol. Genet. 15(1): 113–128. [DOI] [PubMed] [Google Scholar]
- Tropea, A., F. Tiberi, F. Minici, M. Orlando, M. F. Gangale et al., 2007. Ghrelin affects the release of luteolytic and luteotropic factors in human luteal cells. J. Clin. Endocrinol. Metab. 92(8): 3239–3245. [DOI] [PubMed] [Google Scholar]
- Uchida, T., T. Nakamura, N. Hashimoto, T. Matsuda, K. Kotani et al., 2005. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat. Med. 11(2): 175–182. [DOI] [PubMed] [Google Scholar]
- Vaughn, T. T., L. S. Pletscher, A. Peripato, K. King-Ellison, E. Adams et al., 1999. Mapping quantitative trait loci for murine growth: a closer look at genetic architecture. Genet. Res. 74 313–322. [DOI] [PubMed] [Google Scholar]
- Warden, C. H., J. S. Fisler, S. M. Shoemaker, P. Z. Wen, K. L. Svenson et al., 1995. Identification of four chromosomal loci determining obesity in a multifactorial mouse model. J. Clin. Invest. 95(4): 1545–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzell, A. M., H. C. Rowe, B. G. Hansen, C. Ticconi, B. A. Halkier et al., 2007. Linking Metabolic QTLs with Network and cis-eQTLs Controlling Biosynthetic Pathways. PLoS Genet. 3 1687–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, S., and C. Alzheimer, 2006. Roles of activin in tissue repair, fibrosis and inflammatory disease. Cytokine Growth Factor Rev. 17(3): 157–171. [DOI] [PubMed] [Google Scholar]
- West, D. B., J. Goudey-Lefevre, B. York and G. E. Truett, 1994. Dietary obesity linked to genetic loci on chromosomes 9 and 15 in a polygenic mouse model. J. Clin. Invest. 94(4): 1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, R. H., T. J. Morgan and T. F. C. Mackay, 2006. High-resolution mapping of quantitative trait loci affecting increased life span in Drosophila melanogaster. Genetics 173 1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff, T. K., and J. P. Mather, 1995. Inhibin, Activin and the female reproductive axis. Annu. Rev. Physiol. 57 219–244. [DOI] [PubMed] [Google Scholar]
- Yamauchi, T., Y. Nio, T. Maki, M. Kobayashi, T. Takazawa et al., 2007. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 13 332–339. [DOI] [PubMed] [Google Scholar]
- Yi, N., A. Diament, S. Chiu, K. Kim, D. B. Allison et al., 2004. Characterization of epistasis influencing complex spontaneous obesity in the BSB model. Genetics 167(1): 399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., D. K. Zinniel, K. Kim, E. J. Eisen, A. Bartolucci et al., 2006. Bayesian analyses of multiple epistatic QTL models for body weight and body composition in mice. Genet. Res. 87(1): 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini, E., L. J. Scott, R. Saxena, B. F. Voight, J. L. Marchini et al., 2008. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 40(5): 638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z. B., 1994. Precision mapping of quantitative trait loci. Genetics 136 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemke, F., and C. S. Mantzoros, 2010. Adiponectin in insulin resistance: lessons from translational research. Am. J. Clin. Nutr. 91(Suppl): 258S–261S. [DOI] [PMC free article] [PubMed] [Google Scholar]