

Abstract
Under the conditions of ruthenium catalyzed transfer hydrogenation, 1,1-disubstituted allenes 1a–1c and alcohols 2a–2g engage in redox-triggered generation of allylruthenium-aldehyde pairs to form products of hydrohydroxyalkylation 3a–3g, 4a–4g and 5a–5g with complete branched regioselectivity. By exploiting Curtin-Hammett effects, good to excellent levels of anti-diastereoselectivity (4:1 – >20:1) are obtained. Thus, all carbon quaternary centers are formed in a diastereoselective fashion upon carbonyl addition from the alcohol oxidation level in the absence of pre-metallated nucleophiles or stoichiometric byproducts. Exposure of allene 1b to equimolar quantities of alcohol 2a and aldehyde 6b under standard reaction conditions delivers adducts 4a and 4b in a 1:1 ratio. Similarly, exposure of allene 1b to equimolar quantities of aldehyde 6a and alcohol 2b provides adducts 4a and 4b in an identical equimolar ratio. Exposure of allene 1b to d2-para-nitrobenzyl alcohol, deuterio-2a, under standard reaction conditions delivers the product of hydrohydroxymethylation deuterio-4a, which incorporates deuterium at the carbinol position (>95% 2H) and the interior vinylic position (34% 2H). Competition experiments involving exposure of allene 1b to equimolar quantities of benzylic alcohols 2a and deuterio-2a reveal no significant kinetic effect. The collective data corroborate rapid, reversible alcohol dehydrogenation, allene hydrometallation and (E)-, (Z)-isomerization of the transient allylruthenium in advance of turn-over limiting carbonyl addition. Notably, analogous allene-aldehyde reductive C-C couplings employing isopropanol as the terminal reductant display poor levels of anti-diastereoselectivity, suggesting carbonyl addition is not turn-over limiting in reactions conducted from the aldehyde oxidation level.
Introduction
In the course of exploring C-C bond forming hydrogenations beyond hydroformylation,1 it was found that ruthenium2 and iridium3 based catalysts promote direct hydrohydroxyalkylation of π-unsaturated reactants.1,2,3 In such processes, primary alcohols engage π-unsaturated reactants as redox partners, where hydrogen exchange triggers generation of electrophile-nucleophile pairs en route to products of C-C coupling. In this way, carbonyl addition is achieved directly from the alcohol oxidation level in the absence of pre-metallated nucleophiles or stoichiometric byproducts. In nearly all systems studied, identical products of carbonyl addition may be formed from the aldehyde oxidation level under transfer hydrogenation conditions employing isopropanol or formic acid as terminal reductant.1,2,3
While control of relative and absolute stereochemistry has been achieved in iridium catalyzed hydrohydroxyalkylations,3 stereoselective ruthenium catalyzed processes have proven elusive. Indeed, under the conditions of ruthenium catalysis, high diastereoselectivities only have been observed in couplings of unsubstituted allenamides, where complete partitioning of (Z)- and (E)-σ-allylruthenium intermediates is achieved readily through steric differentiation of hydrogen and the sterically demanding NR1R2 moiety.2f,4 Exclusive carbonyl addition from the (E)-σ-allylruthenium through a chair-like transition structure accounts for the observed antidiastereoselectivity.
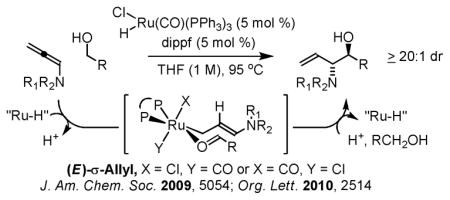
The energetic bias required for partitioning trisubstituted (Z)- and (E)-σ-allylruthenium intermediates derived upon hydrometallation of 1,1-disubstituted allenes is far more difficult to achieve. Studies on stoichiometric hydrometallation of 1,1-disubstituted allenes or dienes employing HXRu(CO)(PR3)3 (X = Cl, Br) reveal the resulting π-allylruthenium complexes Ru(η3-allyl)(X) (CO)(PR3)2 are highly fluxional.5,6 Rapid interconversion of π-allyl- and σ-allylruthenium complexes precludes kinetically controlled partitioning of (Z)- and (E)-σ-allylruthenium isomers via stereoselective hydrometallation. Therefore, it was postulated that crowding at the ruthenium center might result in energetic differentiation of the transient (Z)- and (E)-σ-allylruthenium isomers, or perhaps manifest in a Curtin-Hammett scenario, wherein a given σ-allyl isomer preferentially participates in carbonyl addition. However, while the feasibility of enacting Curtin-Hammett effects by virtue of configurationally dynamic allylmetal intermediates is suggested by stereoconvergence observed in the addition of γ-monosubstituted allylchromium reagents,7a,b γ,γ-disubstituted allylchromium reagents were found to act stereospecifically.7c Here, we report that exceptional levels of anti-diastereoselectivity may be obtained in the hydrohydroxyalkylation of 1,1-disubstituted allenes through exploitation of Curtin-Hammett effects. This method, which combines oxidation-construction events, enables diastereoselective formation of all-carbon quaternary centers under catalytic conditions in the absence of pre-metallated nucleophiles or stoichiometric byproducts.8,9
Results and Discussion
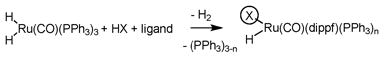
In an initial experiment, 1,1-disubstituted allene 1a was exposed to p-nitrobenzyl alcohol 2a in the presence of a ruthenium catalyst derived upon the combination of HClRu(CO)(PPh3)3 and dippf [dippf = bis(diisopropylphosphino)ferrocene] in THF solvent at 50 °C. Although the desired hydrohydroxyalkylation product 3a was isolated in excellent yield with complete branched regioselectivity, a modest 2:1 diastereoselectivity was observed. Based on the aforementioned line of reasoning, an assay of counterion was undertaken. Complexes HXRu(CO)(dippf)(PPh3)n are conveniently prepared in situ through the acid base reaction of H2Ru(CO)(PPh3)3 and HX (eqn. 1).10
 |
(1) |
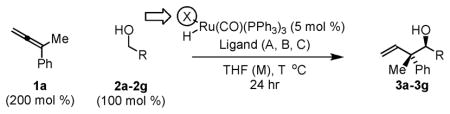
It was found for the specific combination of allene 1a and p-nitrobenzyl alcohol 2a, the ruthenium mesitylenesulfonate complex enforced complete levels of anti-diastereoselectivity. Encouraged by these results, the hydrohydroxyalkylation of 1,1-disubstituted allenes 1a–1c employing alcohols 2a–2g were explored. In many cases, the chloride counterion provided the highest levels of anti-diastereoselectivity. However, in other cases, for example allene 1b, the BINOL-modified phosphate counterion was most effective in promoting anti-diastereoselectivity.11 Finally, whereas dippf was the ligand of choice for allenes 1a and 1b, alternate ligands were required to achieved optimal levels of anti-diastereoselectivity for the phthalimido-substituted allene 1c. The assignment of relative stereochemistry for adducts 3a–3g, 4a–4g and 5a–5g is made in analogy to that determined for compound 5a, which was established via single crystal X-ray diffraction analysis, and compound 3c, which is reported in the literature (Table 1).8b
Table 1.
Oxidation level dependent anti-diastereoselectivity in ruthenium catalyzed couplings of allene 1c.
 | |||||
|---|---|---|---|---|---|
| Entry | X | Alcohol | R | Ligand, T (M) | Yield (dr) |
| 1 | OMes | 2a | p-NO2Ph | A, 50 °C (1 M) | 3a, 84% (>20:1)d |
| 2 | Cl | 2b | p-CF3Ph | A, 40 °C (0.5 M) | 3b, 85% (4:1) |
| 3 | Cl | 2c | Ph | A, 60 °C (0.5 M) | 3c, 99% (6:1) |
| 4 | Cl | 2d | 2-Furyl | A, 60 °C (0.5 M) | 3d, 99% (10:1) |
| 5 | Cl | 2e | HC=CHPh | A, 60 °C (0.2 M) | 3e, 99% (5:1) |
| 6 | Cl | 2f | n-Hexyl | A, 75 °C (1 M) | 3f, 77% (>20:1)c,d |
| 7 | Cl | 2g | (CH2)3OBn | A, 75 °C (1 M) | 3g, 67% (>20:1)c,d |
 | |||||
|---|---|---|---|---|---|
| Entry | X | Alcohol | R | Ligand, T (M) | Yield (dr) |
| 8 | O2PBinol | 2a | p-NO2Ph | A, 75 °C (1 M) | 4a, 97% (16:1) |
| 9 | O2PBinol | 2b | p-CF3Ph | A, 75 °C (1 M) | 4b, 93% (>20:1) |
| 10 | O2PBinol | 2c | Ph | A, 95 °C (1 M) | 4c, 85% (17:1) |
| 11 | O2PBinol | 2d | 2-Furyl | A, 95 °C (1 M) | 4d, 71% (15:1) |
| 12 | O2PBinol | 2e | HC=CHPh | A, 95 °C (1 M) | 4e, 94% (14:1) |
| 13 | Cl | 2f | n-Hexyl | A, 95 °C (1 M) | 4f, 86% (4:1)c,d |
| 14 | Cl | 2g | (CH2)3OBn | A, 95 °C (1 M) | 4g, 69% (4:1)c,d |
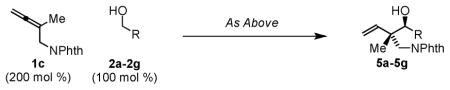 | |||||
|---|---|---|---|---|---|
| Entry | X | Alcohol | R | Ligand, T (M) | Yield (dr) |
| 15 | Cl | 2a | p-NO2Ph | C, 95 °C (0.5 M) | 5a, 83% (10:1) |
| 16 | Cl | 2b | p-CF3Ph | C, 95 °C (0.5 M) | 5b, 65% (14:1) |
| 17 | Cl | 2c | Ph | B, 75 °C (0.5 M) | 5c, 99% (8:1)d |
| 18 | O3SCam. | 2d | 2-Furyl | A, 85°C (1 M) | 5d, 84% (7:1) |
| 19 | Cl | 2e | HC=CHPh | B, 85°C (0.1 M) | 5e, 99% (5:1)d |
| 20 | Cl | 2f | n-Hexyl | A, 95°C (1 M) | 5f, 93% (>20:1)c,d |
| 21 | Cl | 2g | (CH2)3OBn | A, 85°C (1 M) | 5g, 72% (>20:1)d |
Yields are of isolated material. Diastereoselectivity was determined via 1H NMR analysis of crude reaction mixtures. See supporting information for further experimental details.
Ligands: A = dippf (5 mol %); B = dCypf, (5 mol%); C = PPh2Cy (15 mol %).
Three equivalents of allene.
48 hours.
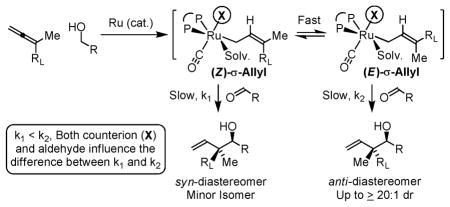
Under identical reaction conditions, a given allene can display substantially different levels of diastereoselectivity in response to the structure of its transient aldehyde partner. This observation suggests the relative thermodynamic stabilities of the (Z)- and (E)-σ-allylruthenium intermediates do not alone determine diastereocontrol. Rather, it appears that a Curtin-Hammett scenario is operative, wherein the transient aldehyde influences energetic partitioning of the diastereomeric transition structures. Kinetically controlled diastereoselection is suggested by the fact that the diastereomeric ratio does not change over the course of the reaction. This observation is significant, as reversible carbonyl addition or product oxidation, could potentially occur. However, although secondary alcohol oxidation is in general thermodynamically more favorable than primary alcohol oxidation, further oxidation of the coupling product is not observed. Our collective studies2 suggest that coordination of the homoallylic olefin to the catalyst provides a hexa-coordinate, 18-electron complex, suppressing β-hydride elimination through occupation of all available coordination sites. Indeed, in ruthenium catalyzed diene-carbonyl C-C couplings, coordinative saturation of the catalyst could be varied to partition formation of secondary alcohol and ketone products.2b In the present case, resubjecting adduct 3a to standard coupling conditions in the presence of allene 1a, which may serve as a hydrogen acceptor, does not result in any oxidation or erosion diastereomeric purity of recovered 3a, corroborating kinetically controlled carbonyl addition (eqn. 2).
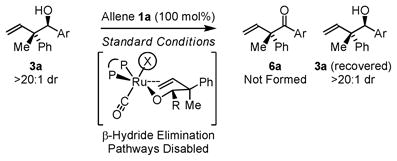 |
(2) |
Notably, when the allene coupling is conducted from the aldehyde oxidation level, diastereoselectivity is essentially absent. For example, whereas 10:1 and 8:1 anti-diastereoselectivities are observed in the coupling of allene 1c to alcohols 2a and 2c, respectively, corresponding couplings of aldehydes 6a and 6c employing isopropanol as terminal reductant under otherwise identical conditions are not diastereoselective (Table 2). These data suggest that carbonyl addition is no longer turnover-limiting in couplings conducted from the aldehyde oxidation level. There are two probable explanations for this. In reactions conducted from the aldehyde oxidation level, aldehyde concentration is higher throughout the course of the reaction, which should accelerate carbonyl addition. Alternatively, for such sterically congested ruthenium complexes, dehydrogenation of isopropanol, a secondary alcohol, may be slower than primary alcohol dehydrogenation. In either case, Curtin-Hammett effects can no longer be exploited to amplify diastereoselectivity. Consistent with this interpretation, in reactions conducted from the alcohol oxidation level, diastereoselectivity is found to be highly concentration dependent. Presumably, at lower concentration, carbonyl addition is sufficiently slow that the (E)-σ-allylruthenium consumed upon addition may be replenished via isomerization of the remaining (Z)-σ-allylruthenium isomer (Table 3).
Table 2.
Oxidation level dependent anti-diastereoselectivity in ruthenium catalyzed couplings of allene 1c.a
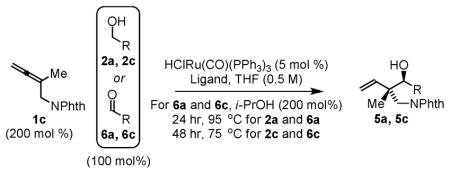 | ||||
|---|---|---|---|---|
| Entry | Oxidation Level | R | Ligand | Yield (dr) |
| 1 | Alcohol, 2a | p-NO2Ph | CyPPh2 (15 mol%) | 5a, 83% (10:1) |
| 2 | Aldehyde, 6a | p-NO2Ph | CyPPh2(15 mol%) | 5a, 99% (1:1) |
| 3 | Alcohol, 2c | Ph | dCypf (5 mol%) | 5c, 99% (8:1) |
| 4 | Aldehyde, 6c | Ph | dCypf (5 mol%) | 5c, 72% (1:1) |
As described in Table 1.
Table 3.
Concentration dependent anti-diastereoselectivity in ruthenium catalyzed couplings of allene 1c.
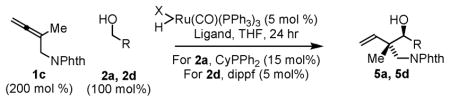 | |||||
|---|---|---|---|---|---|
| Entry | Alcohol | X | T°C | THF (conc.) | Yield (dr) |
| 1 | 2a | Cl | 95 °C | 1.0 M | 5a, 87% (6:1) |
| 2 | 2a | Cl | 95 °C | 0.5 M | 5a, 83% (10:1) |
| 3 | 2a | Cl | 95 °C | 0.2 M | 5a, 55% (> 20:1) |
| 4 | 2d | O3S-Camphor | 85 °C | 0.5 M | 5d, 72% (2:1) |
| 5 | 2d | O3S-Camphor | 85 °C | 0.2 M | 5d, 79% (4:1) |
| 6 | 2d | O3S-Camphor | 85 °C | 0.1 M | 5d, 82% (5:1) |
As described in Table 1.
To assess whether primary alcohol dehydrogenation is indeed more rapid than carbonyl addition the following competition experiment was performed. Allene 1b was exposed to equimolar quantities of alcohol 2a and aldehyde 6b under standard conditions employing the ruthenium catalyst generated in situ from H2Ru(CO)(PPh3)3, dippf and rac-BINOL-PO2H at 75 °C in THF solvent (1.0 M). The C-C coupling products 4a and 4b were produced in a 1:1 ratio. Under identical conditions employing equimolar quantities of aldehyde 6a and alcohol 2b, a nearly identical product ratio of coupling products 4a and 4b is observed. These data are consistent with rapid, reversible primary alcohol dehydrogenation in advance of turnover-limiting carbonyl addition (Scheme 1).12
Scheme 1.
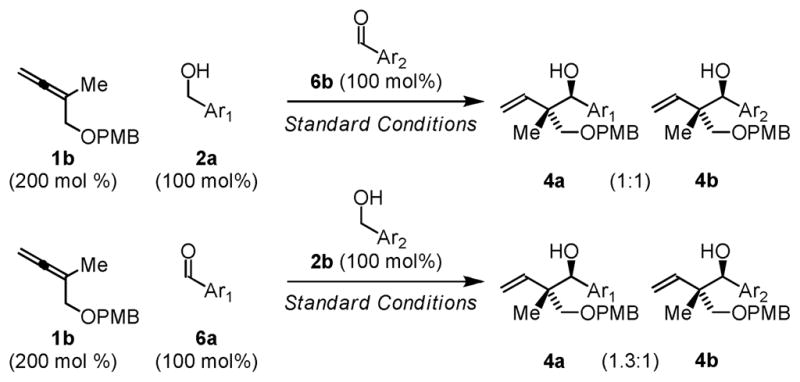
Competition experiments corroborating rapid, reversible dehydrogenation in advance of C-C coupling.a
aAs described in Table 1. Ar1 = p-NO2Ph, Ar2 = p-CF3Ph. Products 4a and 4b are obtained as 1:1.5 (syn:anti) diastereomeric mixtures.
Further insight into the catalytic mechanism is provided by deuterium labeling and competition kinetics experiments (Scheme 2).13 Exposure of allene 1b to d2-p-nitrobenzyl alcohol under standard coupling conditions delivers deuterio-4a, which incorporates deuterium at the carbinol methine (>99% 2H) and at the interior vinylic position (34% 2H), as determined by 1H and 2H NMR analyses. Complete retention of deuterium at the carbinol methine, along with kinetically controlled anti-diastereoselectivity (vida supra), corroborates resistance of the coupling products toward reversible dehydrogenation. Incomplete deuterium incorporation at the interior vinylic position suggests β-hydride elimination of the π-allylruthenium intermediate occurs to furnish diene byproducts. Such byproducts appear to be present in the crude reaction mixture and may account for the requirement of super-stoichiometric loadings of allene. Competition kinetic experiments involving exposure of allene 1b to equimolar quantities of p-nitrobenzyl alcohol and d2-p-nitrobenzyl alcohol reveal no significant kinetic effect (kH/kD = 1.06), within the error limits of the experiment, suggesting alcohol oxidation is not the turn-over limiting event. If carbonyl addition was the turn-over limiting event, an inverse secondary isotope effect would be anticipated. Given the error limits of the experiment, the absence of such an effect is inconclusive.
Scheme 2.
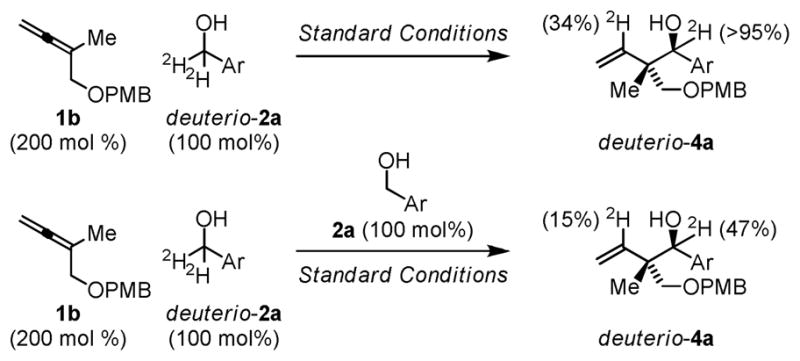
Deuterium labeling and competition kinetics experiments.a
aAs described in Table 1. Ar = p-NO2Ph,
Conclusion
In summary, by taking advantage of Curtin-Hammett effects in ruthenium catalyzed alcohol-allene C-C coupling, one bypasses the need to partition trisubstituted σ-allylmetal species in the ground state. Rather, from an equilibrating mixture of transient (Z)- and (E)-σ-allylruthenium isomers, preferential selection of the (E)-σ-allylruthenium species occurs upon energetic partitioning in the transition state for carbonyl addition. Such Curtin-Hammett effects provide a basis for diastereoselective carbonyl allylation to furnish secondary neopentyl homoallylic alcohols, which possess all carbon quaternary centers, thus setting the stage for development of related diastereo- and enantioselective processes.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445).
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem Int Ed. 2009;48:34. doi: 10.1002/anie.200802938.Han SB, Kim IS, Krische MJ. Chem Commun. 2009:7278. doi: 10.1039/b917243m.
- 2.For ruthenium catalyzed alcohol-unsaturate C-C coupling, see: Dienes: Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338. doi: 10.1021/ja801213x.Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:14120. doi: 10.1021/ja805356j.Han H, Krische MJ. Org Lett. 2010;12:2844. doi: 10.1021/ol101077v.Alkynes: Patman RL, Chaulagain MR, Williams VM, Krische MJ. J Am Chem Soc. 2009;131:2066. doi: 10.1021/ja809456u.Williams VM, Leung JC, Patman RL, Krische MJ. Tetrahedron. 2009;65:5024. doi: 10.1016/j.tet.2009.03.068.Allenes: Zbieg JR, McInturff EL. Org Lett. 2010;12:2514. doi: 10.1021/ol1007235.
- 3.For iridium catalyzed alcohol-allene C-C coupling, see: Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916. doi: 10.1021/ja902437k.For related couplings of dienes and allylic carboxylates see reference 1.
- 4.Skucas E, Zbieg JR, Krische MJ. J Am Chem Soc. 2009;131:5054. doi: 10.1021/ja900827p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For leading references on the stoichiometric reaction of HXRu(CO)(PR3)3 (X = Cl, Br) with allenes or dienes to furnish π-allylruthenium complexes, see: Hiraki K, Ochi N, Sasada Y, Hayashida H, Fuchita Y, Yamanaka S. J Chem Soc, Dalton Trans. 1985:873.Hill AF, Ho CT, Wilton-Ely DET. Chem Commun. 1997:2207.Xue P, Bi S, Sung HHY, Williams ID, Lin Z, Jia G. Organometallics. 2004;23:4735.
- 6.For studies involving π-allylruthenium complexes of the type Ru(η3-allyl)(X)(CO)(PR3)2, see: Barnard CFJ, Daniels BJA, Holland PR, Mawby RJ. J Chem Soc, Dalton Trans. 1980:2418.Hiraki K, Matsunaga T, Kawano H. Organometallics. 1994;13:1878.Sasabe H, Nakanishi S, Takata T. Inorg Chem Commun. 2002;5:177.Cadierno V, Crochet P, Diez J, Garcia-Garrido SE, Gimeno J. Organometallics. 2003;22:5226.
- 7.(a) Hiyama T, Okude Y, Kimura K, Nozaki H. Bull Chem Soc Jpn. 1982;55:561. [Google Scholar]; (b) Roush WR. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Heathcock CH, editors. Vol. 2. Pergamon; Oxford: 1991. pp. 1–53. [Google Scholar]; (c) Jubert C, Nowotny S, Kornemann D, Antes I, Tucker CE, Knochel P. J Org Chem. 1992;57:6384. [Google Scholar]
- 8.For selected examples of stereoselective carbonyl allylmetallation to furnish secondary neopentyl homoallylic alcohols, see reference 7c and the following: Sato M, Yamamoto Y, Hara S, Suzuki A. Tetrahedron Lett. 1993;34:7071.Habaue S, Yasue K, Yanagisawa A, Yamamoto H. Synlett. 1993:788.Nishigaichi Y, Takuwa A. Tetrahedron Lett. 1999;40:109.Denmark SE, Fu J. J Am Chem Soc. 2001;123:9488. doi: 10.1021/ja016552e.Denmark SE, Fu J. Org Lett. 2002;4:1951. doi: 10.1021/ol025971t.Ely RJ, Morken JP. J Am Chem Soc. 2010;132:2534. doi: 10.1021/ja910750b.
- 9.As described in reference 2c, ruthenium catalyzed C-C coupling of ethanol and 2-substituted dienes delivers anti-configured neopentyl homoallylic alcohols. However, incomplete regio- and diastereoselectivities are observed.
- 10.Dobson A, Robinson SR, Uttley MF. J Chem Soc, Dalton Trans. 1974:370. [Google Scholar]
- 11.Chiral counterions may prove effective in enantioselective C-C couplings of this type. For example, using the ruthenium catalyst generated in situ from H2Ru(CO)(PPh3)3, dippf and enantiomerically pure [(S)-BINOL]PO2H, allene 1b couples to alcohol 2a to deliver the secondary neopentyl alcohol 4a in 16% enantiomeric excess.”
- 12.For a related competition experiment, see: Bower JF, Skucas E, Patman RL, Krische MJ. J Am Chem Soc. 2007;129:15134. doi: 10.1021/ja077389b.
- 13.For selected examples of kinetic isotope effects in ruthenium catalyzed transfer hydrogenation, see: Casey CP, Johnson JB. J Org Chem. 2003;68:1998. doi: 10.1021/jo0205457.Sandoval CA, Ohkuma T, Muniz K, Noyori R. J Am Chem Soc. 2003;125:13490. doi: 10.1021/ja030272c.Pannetier N, Sortais J-B, Dieng PS, Barloy L, Sirlin C, Pfeffer M. Organometallics. 2008;27:5852.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.