

Abstract
Classification of mitochondrial DNA (mtDNA) into their respective haplogroups allows the addressing of various anthropologic and forensic issues. Unique to mtDNA is its abundance and non-recombining uni-parental mode of inheritance; consequently, mutations are the only changes observed in the genetic material. These individual mutations are classified into their cladistic haplogroups allowing the tracing of different genetic branch points in human (and other organisms) evolution. Due to the large number of samples, it becomes necessary to automate the classification process. Using 5-fold cross-validation, we investigated two classification techniques on the consented database of 21 141 samples published by the Genographic project. The support vector machines (SVM) algorithm achieved a macro-accuracy of 88.06% and micro-accuracy of 96.59%, while the random forest (RF) algorithm achieved a macro-accuracy of 87.35% and micro-accuracy of 96.19%. In addition to being faster and more memory-economic in making predictions, SVM and RF are better than or comparable to the nearest-neighbor method employed by the Genographic project in terms of prediction accuracy.
Keywords: mitochondrial DNA, ensemble learning, classification algorithms, support vector machines, random forest, genographic project
INTRODUCTION
Mitochondrial DNA (mtDNA) is the DNA located inside cell organelles called mitochondria. Whereas, regular nuclear DNA is present as a single copy per cell residing in the cell nucleus, mtDNA exists in multiple (2–10) copies within every mitochondrion present in the cell [10]. This means anywhere from hundreds of copies of mtDNA in regular cells to over 10 000 in liver cells, giving scientists easy access to vast numbers of samples as well as the higher likelihood of mtDNA surviving over time versus nuclear DNA. Further, differentiating mtDNA from nuclear DNA is its evolutionary origin. Due to its circular nature reminiscent of bacterial DNA, it is believed that the mtDNA is a component of ancestral bacterial DNA that was consumed by early eukaryotic cells: ancestors to modern cells. It is also believed that the vast majority of nuclear DNA originated from this bacterial origin before eventually transferring to the nucleus through evolution.
What makes the study of mtDNA intriguing is its uni-parental and non-recombining mode of inheritance. Normal nuclear DNA is passed from one generation to subsequent generation through meiosis, where genetic material in the form of chromosomes are halved from each parent, followed by fertilization where the two gametes are fused to the original number of chromosomes. During meiosis, genetic recombination occurs as chromosomes of each pair usually cross over. In this form of reproduction only half of the parental nuclear DNA makes it into the genetic code of the offspring.
In contrast, mtDNA is inherited almost exclusively from the mother. Mechanisms for this are attributed to simple dilution as each egg contains anywhere from 100 000 to 1 000 000 copies of mtDNA whereas sperm cells carry only 100 to 1000 (the majority of which resides wrapped around the tail that is oftentimes discarded as the sperm mates with the egg). Additionally, it has been shown that mammalian sperm cells are marked with ubiquitin during fertilization for destruction later on inside the embryo.
This mode of maternally exclusive inheritance allows the tracing of human lineage far back in time as the mtDNA remains constant from one generation to the next. This is further compounded by mtDNAs’ susceptibility to reactive oxidative species leading to a large number of mutations that allows detailed cladistic (a form of biological systematics that classifies living organisms based on shared ancestry) ancestral studies [1].
Launched in 2005, the Genographic project [2] under the direction of the National Geographic Society began assembly of a large database of mtDNA samples to address anthropological issues on a global level. A total of 78 590 typed mtDNA samples are collected with 21 141 samples released to the public at the participants’ consent.
To make use of mtDNA in anthropologic studies, the DNA is sequenced and classified into designated haplogroups (Hgs) which contain similar haplotypes that share a common ancestor based on single nucleotide polymorphism (SNP) mutations. Most recent sequencing technology sequence the first hypervariable region (HVR-I) of the circular DNA. Although many different definitions exist for the location of HVR-I, the nucleotides used in this project consists of those in the range from 16 024 to 16 569.
Due to mtDNAs’ susceptibility to mutation, that allows the possibility of numerous back mutations (a mutation that reverts to its original phenotype) as well as the occurrence of homoplasy (acquisition of identical traits in unrelated lineage), the Genographic project also conducted the typing of 22 coding region biallelic sites in addition to the standard extended sequencing of HVR-I; the Hgs utilized in the database are defined by the combined use of the 22-SNP panel results and the HVR-I haplotypes. A subset of 16 609 samples, known as the reference database, are used to train a function to automate the labeling and categorization process of the genetic information found in HVR-I.
The Genographic Project currently utilizes two 1-nearest neighbor (1-NN) based classification algorithms [3] for the task of categorizing HVR-I mtDNA sequences into their 23 basal Hgs. The 1-NN approach is a classification method in pattern recognition and instance-based learning (the algorithm constructs hypotheses from training instances that allows increasingly complex hypotheses in larger training sets) often considered one of the simplest machine learning algorithms.
The leave-one-out cross-validation accuracy on the reference database is determined to be 96.72–96.73%. This was compared to a rule based approach algorithm which only attained an accuracy of 85.3%. This can be mostly attributed to the sensitivity of rule based algorithms to the homoplasy and back mutations often associated with mtDNA HVR-I [2]. Since such parallel evolution is rampant in mtDNA, the ability of rule-based algorithms to classify HVR-I Hgs is expected to be unreliable [1].
In this study, we investigate two state-of-the-art classification algorithms. Namely, the random forest (RF) and support vector machines (SVM) algorithms. These two algorithms are promising because they often yield comparable accuracy to the 1-NN algorithm. Moreover, these two algorithms are more efficient in terms of time spent on predicting new samples. Experiments conducted on the consented database show that SVM is the most accurate one, correctly classifying 70 more samples than 1-NN. RF is slightly less accurate but still comparable to 1-NN. We further analyze the results on Hgs with low accuracy rates by examining the confusion matrices and discuss the possible causes.
This article is organized as follows. We introduce the classification algorithms in the next section. The results are presented and discussed in section ‘Results and Discussion’. We then give the concluding remarks in the last section.
MATERIALS AND METHODS
We first introduce the 1-NN algorithm employed by Behar et al. [2]. We then briefly describe two additional classification algorithms: RF and SVM. These classification algorithms are evaluated using the consented database, Database S1 [4], compiled by the Genographic project. This database consists of 21 141 mtDNA samples, each of which is genotyped, and whose HVR-I haplotype is provided. Each sample is transformed into a vector of 545 binary variables, each of which indicates the presence or absence of a SNP. The samples have been Hg-labeled into coarse Hgs and further sub-Hgs, with classification achieved through the use of a panel of 22 coding-region SNPs and hypervariable region I (HVR-I) motifs. These Hg-labels are recognized as the ‘gold-standard’, to which all other classifications will be compared. To facilitate comparison, the dataset is split into five subsets and 5-fold cross-validation of the algorithms is conducted using the same partition.
K-Nearest neighbor
Being an instance-based classification algorithm, the k-NN algorithm relies on a reference dataset, each sample in which is tagged with an Hg label. A new sample is classified by a majority vote of its k-NN. The distance between two samples is gauged by the Hamming distance, the number of letters that differ between the two. The best choice of k generally depends on the data; a larger k value will eliminate noise and errors found in the dataset but also blur the boundaries between distinct classes. An optimal k value can be obtained using a heuristic approach such as cross-validation. Due to the large amount of samples available to the project, the Genographic project utilizes the 1-NN method (where k = 1) [2].
The NN algorithm has the advantages of being easy to implement and interpret. It is in theory, the optimal classifier minimizing the expected squared prediction error [5] when there are a substantial number of reference samples uniformly distributed in space. The algorithm, however, becomes computationally intensive, especially as the sample size grows (as in the case of the Genographic project with over 70 000 samples).
Random forest
Leo Breiman and Adele Cutler’s RF algorithm [6] is a class of supervised, ensemble learning algorithms. RF grows ntree single decision trees, each tree submitting a ‘vote’ of classification. A given sample, in this case, a mtDNA HVR-1 haplotype, is input through all ntree trees. The Hg that receives majority vote is attached to the sample. Given a training dataset consisting of n samples and m features, a decision tree in RF is grown and propagated by (i) creating a bootstrap sample of equivalent size n by random sampling with replacement from the pool of n samples, (ii) selecting a designated mtry (<<m) features, sampling without replacement from the available pool of m for each tree, with one variable deciding the split at each node of a decision tree and (iii) growing the tree to full potential, without any pruning. Across a forest, bootstrap samples and composition of mtry nodal variables or features vary.
The random sampling inherent in RF accommodates several advantages. Each decision tree in the forest is built from a different bootstrap sample. Bootstrap samples typically represent approximately two-thirds of the available and full sample pool [7]. The training set for any particular classification tree leaves out a significant portion of the samples, thus called ‘out-of-bag’ (OOB) data. These excluded samples, because they were not used to construct a given tree, serve to provide an unbiased estimate of classification error. All OOB data is input through their respective trees and a classification for each OOB case is voted on. The collective OOB data serve as a formative test set, and each OOB case or sample assigned a series of test set classifications, the number equivalent to how many times that particular case was left out of the training set for a tree. OOB error is calculated by taking the proportion of classifications for a OOB case that do not agree with the true, ‘gold-standard’ classification over the total number of cases.
Though OOB error is stand-alone and sufficient indication of accuracy rates for a RF model, not all samples become OOB data when constructing the forest. Furthermore, predictions for cases are difficult to extract. In order to accommodate fine-tuned analysis of predicted versus observed Hg labels for every sample, using cross-validation ensures that all samples are eventually input into the model, and a classification provided for each sample.
The mtry and ntree parameters for our RF models have been carefully selected so as to minimize the OOB error. The tuneRF function of the RF package locates the optimal mtry value for each ntree value considered. The best pair of ntree and mtry is then selected to be the one with the lowest OOB error. In this study, the ntree values 300, 400 and 500 are considered.
Principal component analysis and RF
Especially with such a large dataset at hand, with 545 variables (SNPs), RF demands lots of memory and running time. With so many variables to consider, creating a straightforward and reliable model often becomes excruciatingly difficult. Training a model in RF becomes unwieldy and highly time consuming, also compromising accuracy and model efficacy due to the presence of so many variables, some of which may not even hold any relevance to the classification scheme. Principal component analysis (PCA) [5, 8] is a factor analysis technique that identifies the most meaningful basis in which to express a given dataset. PCA preserves the dynamics of the original dataset, but expresses it in another basis that may or may not be of reduced dimension. The new basis vectors represent the principal components (PCs) of the new subspace.
We achieve PCA through eigendecomposition [8]: (i) Let X be an n by m matrix, where n is the number of mtDNA samples and m is the number of variables in consideration. (ii) Find the sample covariance matrix CX of the original m variables in X. (iii) Perform eigendecomposition on CX such that PTCXP = CY, where P is an orthogonal matrix, whose columns are the PCs, and CY, a diagonal matrix containing the eigenvalues, is the sample covariance matrix of the new m variables defined by the PCs. (iv) Let Y = XP be the transformed dataset, X projected onto the new subspace defined by the PCs. Construction of RF models can then be based on the transformed dataset Y.
A further dimensional reduction step of PCA is to select k PCs out of the total m new variables or PCs. Oftentimes, k is varied by ordering PCs (eigenvectors) by their respective variances (eigenvalues). In this study, k = 64 when 90% of total variance is accounted for in the 64 PCs with the highest eigenvalues. Likewise, k is varied such that k PCs with the highest eigenvalues are selected.
Feature selection is a by-product of RF. A built-in function of the RF package, importance, ranks variables according to their respective Gini index, a numeric indicator of decrease in node impurity when a particular variable is purposely considered in the construction of an RF classification model [7]. After a RF model is trained on the transformed dataset, we obtain the importance value for each PC. The agreement between the eigenvalues and the importance values can then be accessed by computing the correlation between the two scores.
Support vector machines
Support vector machine classification is a binary classification algorithm, where only two classes are present in a training dataset [5, 9]. That is, a training dataset takes the following form:
| (1) |
where xi is a m-dimensional real vector and ci is the class to which the point xi belongs. Given a training dataset, SVM maps the samples to a high-dimensional space and seeks a maximal-margin separating hyperplane between the two classes of samples while mis-classification is allowed with penalty. Mapping the original space to a high-dimensional space, often called the feature space, is achieved by the use of a kernel function which implicitly maps two samples to the feature space and computes the inner product between them. Mapping samples to the feature space has the benefit of making those samples inseparable in the original space separable. The radial basis function (RBF) 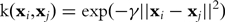 is the kernel of choice, where γ is a parameter.
is the kernel of choice, where γ is a parameter.
Despite being a binary classification algorithm, SVM is capable of handling multi-class data. The implementation we use, LIBSVM [11], takes the one-against-one approach, where a classifier is built for each pair of classes. Therefore, for a k-class classification problem, k(k-1)/2 classifiers are trained. This approach is more computation-intensive than others such the one-against-the-rest method, but it was shown empirically to be more effective in terms of prediction accuracy. In order to optimize the performance of SVM, 5-fold cross-validation is conducted on the consented database to tune the parameter C, the penalty constant, and γ on a grid [12]. Following the determination of the optimal parameters, the algorithm is trained again using the optimal parameters and the 5-fold cross-validation accuracy on the consented database is obtained.
RESULTS AND DISCUSSION
The RF package for R was used in this study [13]. RF was run through a 4 GB RAM Windows server and loaded through the RGUI programming environment. We performed seven runs of RF, with and without sub-selection of variables and with k = 64, 100, 200, 300, 400 and 545 variables considered for each model, as shown in Table 1 . The performance is compared in terms of micro-accuracy and macro-accuracy. The former is the weighted average of all Hg-wise accuracy rates, whereas the latter represents the non-weighted, raw average.
Table 1:
Micro-accuracy rates for the original and transformed datasets run through RF classifiers
| Classifier | Feature selection | Number of features | ntree | mtry | Micro- accuracy rate (%) |
|---|---|---|---|---|---|
| RF | Raw | 545 | 500 | 160 | 96.19 |
| RF | PCA | 64 | 400 | 20 | 95.98 |
| RF | PCA | 100 | 500 | 40 | 96.14 |
| RF | PCA | 200 | 300 | 160 | 96.00 |
| RF | PCA | 300 | 400 | 40 | 96.24 |
| RF | PCA | 400 | 500 | 80 | 96.18 |
| RF | PCA | 545 | 400 | 40 | 96.01 |
We ran importance on RF with the first 100 of 545 eigenvectors or PCs in descending order of the corresponding eigenvalues. A correlation coefficient of 0.64 was calculated between importance values and the eigenvalues, indicating strong correlation between the two scores.
We expected PCA to increase RF accuracy rates. Because multiple trials were not performed for each parameter set listed in Table 1, a difference in the accuracy rates cannot be statistically declared nor inferred. Since RF functions on random sampling of training samples and variables, given the same parameter set, a different RF model will be trained and accuracy rates will differ. Variability in RF accuracy rates might as well be due to inherent variability present in RF, not due to the application of PCA techniques or variations in number of variables considered in the model. Future work must involve multiple trials for each parameter set in order to generate a range of error and thus deduce statistically significant results.
The SVM trials were run with the LIBSVM-2.89 build on a Windows machine. By searching the set {(γ, C ) | γ, C ∈ {2−2, 2−1.5, … , 22}}, the optimal values of γ and C were determined to be 0.25 and 4, respectively. 5-fold cross-validation using the optimal parameters indicated the prediction accuracy of SVM to be 88.06% (macro-accuracy) and 96.57% (micro-accuracy).
Figure 1 summarizes the comparison of the Hg-wise accuracy rates by SVM, RF, RF-PCA and 1-NN algorithms, where RF-PCA denotes RF in conjunction with PCA with the best parameter set. The macro- and micro-accuracy rates of the classifiers are reported in Table 2. The individual Hg prediction accuracy rates by SVM, RF, RF-PCA and 1-NN are collected in Table 3.
Figure 1:

Comparison of SVM, RF, RF-PCA and 1-NN in Terms of Hg-wise Accuracy Rates. SVM, RF, RF-PCA and 1-NN denote SVM, RF, RF in conjunction with principal component analysis with the best parameter set and 1-nearest neighbor, respectively. The numbers of samples in individual Hgs are presented with bars.
Table 2:
Macro- and micro-accuracy rate of classifiers
| Classifier | Macro-accuracy rate (%) | Micro-accuracy rate (%) |
|---|---|---|
| 1-NN (LOO CV) | - | 96.73 |
| 1-NN (5F-CV) | 87.36 | 96.26 |
| RF (5F-CV) | 87.35 | 96.19 |
| RF-PCA (5F-CV) | 86.21 | 96.24 |
| SVM (5F-CV) | 88.06 | 96.59 |
NN = Nearest Neighbor, RF = Random Forest, RF-PCA = Random Forest with Principal Component Analysis, SVM = Support Vector Machines, LOO = Leave-one-out cross-validation, 5F-CV = 5-fold cross-validation
Table 3:
Unsorted individual Hg accuracy rate for 1-NN, RF and SVM
| Hg | Sample size | 1-NN accuracy rate (%) | RF accuracy rate (%) | RF-PCA accuracy rate (%) | SVM accuracy rate (%) |
|---|---|---|---|---|---|
| HV* | 465 | 64.73 | 64.73 | 59.78 | 62.58 |
| L0/1 | 279 | 98.57 | 98.92 | 99.64 | 99.64 |
| N* | 63 | 52.38 | 49.21 | 46.03 | 52.38 |
| M* | 336 | 69.64 | 75.60 | 77.98 | 82.44 |
| U* | 2809 | 96.05 | 95.83 | 96.33 | 96.55 |
| A | 373 | 99.46 | 98.66 | 99.73 | 100.00 |
| C | 230 | 99.57 | 98.70 | 99.57 | 99.13 |
| B | 297 | 96.97 | 95.62 | 97.64 | 97.64 |
| D | 193 | 85.49 | 89.64 | 84.97 | 88.60 |
| I | 475 | 97.47 | 98.11 | 97.68 | 97.89 |
| H | 7786 | 98.25 | 98.09 | 98.43 | 98.41 |
| K | 1759 | 99.55 | 99.37 | 99.37 | 99.37 |
| J | 1793 | 99.83 | 100.00 | 99.72 | 100.00 |
| R9 | 68 | 60.29 | 64.71 | 52.94 | 67.65 |
| L2 | 321 | 99.38 | 99.07 | 99.69 | 99.69 |
| T | 1880 | 99.84 | 99.84 | 99.89 | 99.84 |
| W | 407 | 98.03 | 97.54 | 95.58 | 96.31 |
| V | 610 | 89.67 | 88.85 | 89.34 | 89.18 |
| X | 338 | 99.41 | 97.34 | 97.63 | 98.22 |
| R* | 60 | 13.33 | 10.00 | 8.33 | 11.67 |
| L3* | 370 | 95.14 | 95.14 | 95.68 | 95.68 |
| N1* | 197 | 97.97 | 95.94 | 95.94 | 97.97 |
| R0* | 55 | 98.18 | 98.18 | 90.91 | 94.55 |
Training an RF model involves bootstrap sampling, which excludes certain samples or cases in construction of the forest. Whereas in training an SVM model, all cases in the training set are considered in building the model. This may account for some differences in predictive accuracy. However, because different bootstrap samples serve as the training set for each tree in a forest, across the forest, it is highly unlikely to find an OOB sample.
From Table 3, we can see that all four algorithms performed poorly on Hgs N*, M*, R9, R* and HV*. According to the confusion matrices in Figure 2, some N* samples are mis-classified as I or W, which is a relatively larger Hg than N*. Similarly, some R9 samples are mis-classified as H, and some R* samples are mis-classified as H or U*. Therefore, the low performance on Hgs N*, R9 and R* is likely due to sampling bias of the consented database.
Figure 2:

The Confusion Matrices of (A) 1-NN, (B) RF, (C) RF-PCA and (D) SVM. Each row in a matrix explains how samples in a particular Hg are classified by an algorithm. For example, out of the 279 samples in Hg L0/1, 1-NN labels 275 of them as L0/1, one of them as L2, and three of them as X. The darkness of a cell indicates the percentage of samples assigned to the cell. Therefore, the darker the diagonal of a matrix, the more accurate the corresponding algorithm.
Sampling bias, however, may not be the sole reason why Hgs with smaller sample sizes have low accuracy rates. Hg R0* has an accuracy rate of at least 94.55% despite having only 55 samples. Therefore, we suspected that some Hgs are intrinsically difficult to be distinguished from other Hgs. To better understand Hg M*, we plotted the samples in Hgs M*, D, W and L3* in the space spanned by the first three PCs, since an M* sample is sometimes mistaken for a D, W or L3*. The scatter plot in Figure 3 confirms our hypothesis.
Figure 3:

Scatter plot of samples in Hgs M*, D, W and L3*. PC1, PC2 and PC3 are the first three principal components extracted from the consented database. The plot shows that samples in these four Hgs cannot be easily separated in the space spanned by the first three principal components.
It is not surprising that some samples in Hg HV* are mistaken for samples in Hg H or V as seen in Figure 2. The three Hgs HV*, H and V are in the same sub-tree in the phylogeny of mtDNA Hgs presented in [2], where the three Hgs are distinguished by looking at three coding region SNPs. Their similarity is also observed by a scatter plot (not shown) with the first 3 PCs. Therefore, HV*, the smallest one among the three Hgs, is sacrificed by all three classification algorithms so as to maximize the micro-accuracy.
CONCLUSION
Underrepresented Hgs are likely under-sampled or even excluded all together, and overrepresented Hg tend to be randomly selected at higher frequencies, leading to an unbalanced dataset. Forming coarser-Hg divisions, reducing or increasing weights on relevant Hgs to compensate for the imbalance are possibilities to pursue.
In addition to the issue of sampling bias, we have demonstrated that some Hgs are intrinsically inseparable from one another provided only the HVR-I region is available. Ambiguous samples can be identified by examining the posterior probabilities, which are estimated by the SVM and RF implementations utilized in this study. Typing the coding region SNPs may be necessary to assist the categorization of these samples because of their important role in defining the Hgs.
We conclude that SVM outperforms 1-NN in terms of predictive performance. RF is likely to be the worst of the three. However, more experiments should be conducted with each parameter set for the conclusion to be statistically sound. We argue that SVM and RF are faster and more memory-economic than 1-NN in making inferences on new samples because they don’t rely on the entire training dataset. These are desirable features when Hg inference from mtDNA is implemented as a web service.
The algorithms and work presented in this paper involves categorization of mtDNA in the HVR-I region into their basal (coarse) Hgs. However, each Hg also contains much finer sub-Hgs which can further contribute to the addressing of anthropological questions. Future work may include using algorithms to categorize mtDNA samples into finer sub-Hgs provided enough samples are available for each sub-Hgs.
Key Point.
This article demonstrates that the prediction results by SVM and RF are better than or comparable to those achieved by the NN method employed by the Genographic project. SVM and RF afford a couple of desirable features when Hg inference from mtDNA is implemented as a web service. That is, they are faster and more memory-economic than 1-NN in labeling new samples.
FUNDING
National Science Foundation [CCF0755373 to C.W., Y.L., C.L. and C.-H.H.]; and the National Institutes of Health [R13LM008619 to C.-H.H.].
Acknowledgements
This research was carried out in conjunction with the Bio-Grid Initiatives and the REU program at the University of Connecticut.
Biographies
Carol Wong is currently an undergraduate student at the Department of Bioengineering, University of Pennsylvania. She was a recipient of the 2009 NSF Bio-Grid REU Fellowship.
Yuran Li is currently an undergraduate student at the Department of Chemistry and Biochemistry, University of Delaware. He was a recipient of the 2009 NSF Bio-Grid REU Fellowship.
Chih Lee received the B.S. and M.S. degree in computer science and information engineering in 2003 and 2005, respectively, from the National Taiwan University, Taipei, Taiwan. He is currently a doctoral student at the Department of Computer Science and Engineering, University of Connecticut. His research interest includes bioinformatics, computational biology, data mining, machine learning and natural language processing.
Chun-Hsi Huang received his PhD from the State University of New York at Buffalo in Computer Science in 2001. He is currently an Associate Professor at the Department of Computer Science and Engineering of the University of Connecticut. He is affiliated with the International Society of Computational Biology (ISCB) and the American Medical Informatics Association (AMIA).
References
- 1.Alexeyev MF, LeDoux SP, Wilson GL. Mitochondrial DNA and aging. Clin Sci. 2004;107:355–64. doi: 10.1042/CS20040148. [DOI] [PubMed] [Google Scholar]
- 2.Behar DM, Rosset S, Blue-Smith J, et al. The Genographic project public participation mitochondrial DNA database. PLoS Genet. 2007;3:e104. doi: 10.1371/journal.pgen.0030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dasarathy BV, editor. Nearest Neighbor (NN) Norms: NN Pattern Classification Techniques. Los Alamitos: IEEE Computer Society Press; 1991. [Google Scholar]
- 4.Behar DM, Rosset S, Blue-Smith J, et al. Correction: The Genographic project public participation mitochondrial DNA database. PLoS Genet. 2007;3:e169. doi: 10.1371/journal.pgen.0030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hastie T, Tibshirani R, Friedman JH. The Elements of Statistical Learning. 2nd edn. New York: Springer-Verlag; 2009. p. 746. [Google Scholar]
- 6.Breiman L. Random forests. Machine Learning. 1991;45:5–32. [Google Scholar]
- 7.Brieman L, Cutler A. [(15 November 2009, date last accessed)]. Random Forests [Internet] Berkeley, CA [cited 2010 Jan 24]. Available from: http://www.stat.berkeley.edu/∼breiman/RandomForests/cc_home.htm. [Google Scholar]
- 8.Shiens JA. [(15 November 2009, date last accessed)]. Tutorial on Principal Component Analysis [Internet] San Diego, CA [cited 2010 Jan 24]. Available from: http://www.snl.salk.edu/∼shlens/pub/notes/pca.pdf. [Google Scholar]
- 9.Steinwart I, Christmann A. Support Vector Machines. New York: Springer-Verlag; 2008. p. 602. [Google Scholar]
- 10.Wiesner RJ, Ruegg JC, Morano I. Counting target molecules by exponential polymerase chain reaction, copy number of mitochondrial DNA in rat tissues. Biochim Biophys Acta. 1992;183:553–9. doi: 10.1016/0006-291x(92)90517-o. [DOI] [PubMed] [Google Scholar]
- 11.Chang CC, Lin CJ. [(15 November 2009, date last accessed)]. LIBSVM: a library for support vector machines [Internet] Taipei, Taiwan [updated 2009 Nov 1; cited 2010 Jan 24]. Available from: http://www.csie.ntu.edu.tw/∼cjlin/libsvm. [Google Scholar]
- 12.Hsu CW, Chung CC, Lin CJ. [(15 November 2009, date last accessed)]. A practical guide to Support Vector Classification [Internet] Taipei, Taiwan [cited 2010 Jan 24]. Available from: http://www.csie.ntu.edu.tw/∼cjlin/papers/guide/guide.pdf. [Google Scholar]
- 13.Liaw A, Wiener M. Classification and Regression by random Forest. R News. 2002;2:18–22. [Google Scholar]