

Abstract
Patterns of codon usage have been extensively studied among Bacteria and Eukaryotes, but there has been little investigation of species from the third domain of life, the Archaea. Here, we examine the nature of codon usage bias in a methanogenic archaeon, Methanococcus maripaludis. Genome-wide patterns of codon usage are dominated by a strong A + T bias, presumably largely reflecting mutation patterns. Nevertheless, there is variation among genes in the use of a subset of putatively translationally optimal codons, which is strongly correlated with gene expression level. In comparison with Bacteria such as Escherichia coli, the strength of selected codon usage bias in highly expressed genes in M. maripaludis seems surprisingly high given its moderate growth rate. However, the pattern of selected codon usage differs between M. maripaludis and E. coli: in the archaeon, strongly selected codon usage bias is largely restricted to twofold degenerate amino acids (AAs). Weaker bias among the codons for fourfold degenerate AAs is consistent with the small number of tRNA genes in the M. maripaludis genome.
Keywords: codon usage, translation, selection, Archaea
1. Introduction
The frequencies of alternative synonymous codons vary among species and among genes within a genome, reflecting the combined effects of mutation bias, natural selection and random genetic drift [1,2]. Selection primarily favours translationally optimal codons—those best recognized by the most abundant tRNA species [3]. Selection has the greatest impact on highly expressed (HE) genes, because their translation has the largest effect on cellular efficiency during competitive growth [4]. The strength of selected codon usage bias varies among species, and in some it is weak or absent. Across Bacteria, the strength of selected bias is positively correlated with the copy numbers of rRNA and tRNA genes, and negatively correlated with generation time [5–8], implying a co-adapted suite of genome characteristics necessary for achieving fast growth rates.
While codon usage has been extensively studied in Bacteria, as well as in some groups of Eukaryotes, there has been little work on patterns of codon bias in the third domain of life, the Archaea. Karlin et al. [9] used codon-usage analyses to predict HE genes in 19 archaeal genomes. However, their approach (i) assumed translational selection without first testing for its presence and (ii) is expected to give anomalous results, even in species where selection has shaped codon usage [10]. Some Archaea have been included in larger analyses mainly focused on genomes from Bacteria. For example, Dos Reis et al. [11] claimed that six of 16 species of Archaea showed evidence of translational selection, but (at least among Bacteria) their measure of selection does not correlate well with a population genetics-based approach [6].
Here, we investigate variation in patterns of synonymous codon usage across the genes of Methanococcus maripaludis, one of the most extensively studied archaeal species. Methanococcus maripaludis is a mesophilic methanogen isolated from salt marsh sediment [12]. A complete genome sequence [13] and genome-wide expression data [14] have been determined for this species. Methanococcus maripaludis has a fastest doubling time of 2.3 h at 37°C [12], a near-minimal complement of tRNA genes, and only three rRNA operons. These features seem typical of many Archaea and, by comparison with Bacteria, it might be predicted that such species should have little or no selected codon usage bias. Nevertheless, we find clear evidence of translational selection in HE genes in M. maripaludis, although the pattern of codon bias is rather different from those seen in most Bacteria.
2. Material and methods
Protein-coding sequences from the genome of M. maripaludis strain S2 [13] were obtained from the GenBank database (accession no. BX950229) using the ACNUC retrieval system [15]. Six genes with fewer than 50 codons were excluded from subsequent analyses. The numbers of tRNA genes, with their predicted anti-codon sequences, were obtained from the tRNA scan SE database [16]. Gene expression level was estimated from protein abundance data [14], as the signal intensity (n1 values) normalized by protein molecular weight.
For each gene the deviation from random codon usage was measured using the effective number of codons, Nc [17]; Nc values potentially range from 20, when only one synonym is used for each amino acid (AA), to 61 when all codons are used randomly. A plot of Nc values against GC3s, the G + C content at synonymously variable third positions of codons, is useful to explore variation in patterns of codon bias. Within-group correspondence analysis was also used to identify any major trends among genes (see electronic supplementary material).
A dataset of 51 expected HE genes encoding ribosomal proteins was identified on the basis of genome annotation, expression level data [14] and conservation across Archaea (see electronic supplementary material). Optimal codons were identified as those occurring significantly more frequently in the HE genes than across all genes, using χ2-tests with sequential Bonferroni correction [10]. Codon adaptation index (CAI; [18]) values were computed using codon fitness values from the HE gene set. Finally, the strength of selected codon usage bias (S) in the HE genes was estimated with the method used for Bacteria by Sharp et al. [6]; the method was adapted to obtain analogous values for individual AAs in both M. maripaludis and Escherichia coli.
3. Results
(a). Variation in codon bias among genes in Methanococcus maripaludis
The A + T richness of the M. mariplaudis genome dominates its overall codon usage (table 1), with an average G + C content at synonymously variable third positions (GC3s) of 0.23. The standard deviation of GC3s values across the 1716 genes analysed (0.040) is only a little higher than the value expected from random binomial variation (0.030). In contrast, Nc values range quite widely (figure 1), indicating variability owing to some additional source(s) of bias. Nearly all of the expected HE genes have very low Nc values given their GC3s, indicating more strongly biased codon usage. Correspondence analysis indicates that there is a single major trend among genes, with the HE genes lying towards one extreme of that trend (see electronic supplementary material).
Table 1.
Codon usage in Methanococcus maripaludis, summed across across all genes (all) and for 51 HE genes (high). The second value in each case is the relative synonymous codon usage (the observed number divided by the average for that AA).
| high |
all |
high |
all |
high |
all |
high |
all |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 72 | 0.75 | 16 089 | 1.56 | Ser | UCU | 28 | 0.45 | 6178 | 1.21 | Tyr | UAU | 20 | 0.21 | 9940 | 1.06 | Cys | UGU | 33 | 0.85 | 4047 | 1.21 |
| Phe | UUCa | 121 | 1.25 | 4522 | 0.44 | Ser | UCC | 42 | 0.67 | 2430 | 0.48 | Tyr | UACa | 171 | 1.79 | 8795 | 0.94 | Cys | UGCa | 45 | 1.15 | 2633 | 0.79 |
| Leu | UUAa | 304 | 3.64 | 18 877 | 2.53 | Ser | UCAa | 182 | 2.90 | 11 554 | 2.26 | Ter | UAA | 50 | 2.94 | 1460 | 2.54 | Ter | UGA | 0 | 0.00 | 134 | 0.23 |
| Leu | UUG | 66 | 0.79 | 5553 | 0.74 | Ser | UCG | 4 | 0.06 | 2102 | 0.41 | Ter | UAG | 1 | 0.06 | 130 | 0.23 | Trp | UGG | 44 | 1.00 | 3186 | 1.00 |
| Leu | CUU | 58 | 0.69 | 12 587 | 1.69 | Pro | CCUa | 143 | 1.89 | 5691 | 1.37 | His | CAU | 23 | 0.34 | 3105 | 0.84 | Arg | CGU | 1 | 0.01 | 799 | 0.32 |
| Leu | CUC | 53 | 0.63 | 3383 | 0.45 | Pro | CCC | 4 | 0.05 | 1465 | 0.35 | His | CACa | 114 | 1.66 | 4255 | 1.16 | Arg | CGC | 0 | 0.00 | 251 | 0.10 |
| Leu | CUA | 13 | 0.16 | 2426 | 0.33 | Pro | CCA | 150 | 1.98 | 7671 | 1.84 | Gln | CAAa | 133 | 1.42 | 4631 | 1.01 | Arg | CGA | 5 | 0.06 | 1338 | 0.53 |
| Leu | CUG | 7 | 0.08 | 1939 | 0.26 | Pro | CCG | 6 | 0.08 | 1840 | 0.44 | Gln | CAG | 54 | 0.58 | 4546 | 0.99 | Arg | CGG | 1 | 0.01 | 641 | 0.25 |
| Ile | AUU | 225 | 1.40 | 23 812 | 1.55 | Thr | ACU | 87 | 1.00 | 8311 | 1.36 | Asn | AAU | 51 | 0.36 | 17 956 | 1.30 | Ser | AGU | 54 | 0.86 | 5047 | 0.99 |
| Ile | AUCa | 195 | 1.21 | 8136 | 0.53 | Thr | ACC | 63 | 0.72 | 3585 | 0.59 | Asn | AACa | 229 | 1.64 | 9706 | 0.70 | Ser | AGCa | 67 | 1.07 | 3383 | 0.66 |
| Ile | AUA | 62 | 0.39 | 14 168 | 0.92 | Thr | ACAa | 189 | 2.17 | 9532 | 1.57 | Lys | AAAa | 803 | 1.90 | 38 675 | 1.78 | Arg | AGAa | 440 | 5.45 | 8771 | 3.47 |
| Met | AUG | 207 | 1.00 | 12 597 | 1.00 | Thr | ACG | 9 | 0.10 | 2934 | 0.48 | Lys | AAG | 43 | 0.10 | 4709 | 0.22 | Arg | AGG | 37 | 0.46 | 3376 | 1.33 |
| Val | GUUa | 361 | 2.33 | 17 585 | 2.05 | Ala | GCUa | 293 | 1.82 | 6944 | 0.98 | Asp | GAU | 164 | 1.15 | 19 381 | 1.41 | Gly | GGUa | 227 | 1.50 | 8138 | 0.98 |
| Val | GUC | 25 | 0.16 | 2199 | 0.26 | Ala | GCC | 12 | 0.07 | 1714 | 0.24 | Asp | GACa | 122 | 0.85 | 8095 | 0.59 | Gly | GGC | 76 | 0.50 | 4103 | 0.50 |
| Val | GUA | 208 | 1.34 | 12 102 | 1.41 | Ala | GCA | 322 | 2.00 | 17 607 | 2.47 | Glu | GAA | 514 | 1.87 | 36 441 | 1.83 | Gly | GGA | 285 | 1.89 | 17 312 | 2.09 |
| Val | GUG | 26 | 0.17 | 2439 | 0.28 | Ala | GCG | 16 | 0.10 | 2221 | 0.31 | Glu | GAG | 36 | 0.13 | 3438 | 0.17 | Gly | GGG | 16 | 0.11 | 3571 | 0.43 |
aThe codons occurring at significantly higher frequencies in the HE dataset.
Figure 1.

The effective number of codons, Nc, and G + C content at synonymously variable third codon positions, GC3s, for Methanoccus maripaludis genes. The line indicates the expected Nc value with random codon usage. The subset of 51 HE genes is shown in red.
Eighteen codons occur at significantly higher frequencies in the HE genes than in coding sequences as a whole (table 1). The frequency of these putative optimal codons (Fop) in each gene is highly correlated with experimental estimates of protein abundance (r = 0.59; figure 2). CAI values are similarly highly correlated with protein abundance (r = 0.59).
Figure 2.
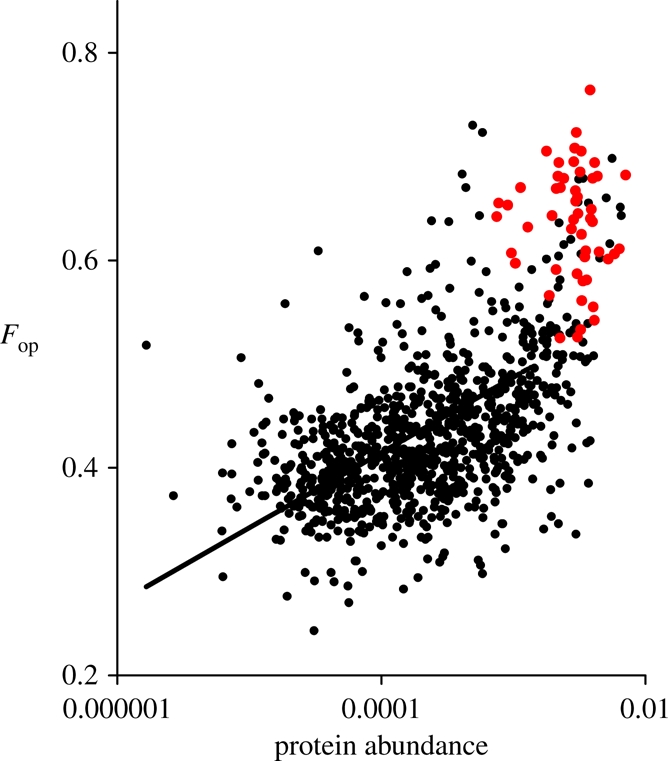
The frequency of optimal codons in a gene (Fop) as a function of gene expression level, as estimated from protein abundance data (from [14]). The subset of 51 HE genes is shown in red.
(b). The strength of selected codon bias in Methanococcus maripaludis
To estimate the strength of selected codon bias in the HE genes, we calculated S (the product of the selective difference between codons and the long-term effective population size) as defined previously for Bacteria [6]. The S value of 1.63 for M. maripaludis is surprisingly high; it is similar to that for the bacterium E. coli (S = 1.49), which has a much faster growth rate and more than twice as many rRNA and tRNA genes [6].
The previous S value estimates the strength of selected bias across four pairs of codons for twofold degenerate AAs. Calculating S values for these AAs individually and contrasting M. maripaludis and E. coli (table 2), we find that S is not significantly different between species (paired t-test, p = 0.42). However, analogous S values for fourfold degenerate AAs (see electronic supplementary material) are significantly lower in M. maripaludis than in E. coli (p < 0.01). Comparing between AAs within species, S values are significantly lower (p = 0.03) for fourfold than for twofold degenerate AAs in M. maripaludis, but not significantly different (p = 0.36) in E. coli.
Table 2.
The strength of selected codon usage bias (S) in HE genes, for each AA in M. maripaludis and E. coli. Only AAs with twofold or fourfold degeneracy, and only those for which both species exhibit preference for an optimal codon (shown), are included.
| twofold degenerate AAs |
fourfold degenerate AAs |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
M. maripaludis |
E. coli |
M. maripaludis |
E. coli |
||||||
| AA | codon | S | codon | S | AA | codon | S | codon | S |
| Phe | UUC | 1.79 | UUC | 1.79 | Pro | CCU | 0.55 | CCG | 0.79 |
| Tyr | UAC | 2.27 | UAC | 1.39 | Thr | ACA | 0.61 | ACU | 1.52 |
| His | CAC | 1.28 | CAC | 1.16 | Val | GUU | 0.28 | GUU | 1.14 |
| Asn | AAC | 2.12 | AAC | 1.74 | Ala | GCU | 0.95 | GCU | 1.49 |
| Asp | GAC | 0.58 | GAC | 1.17 | Gly | GGU | 0.61 | GGU | 1.21 |
4. Discussion
Codon usage varies across the M. maripaludis genome, with a single major trend among genes. Various observations are consistent with selection for translationally optimal codons being the cause of this variation: (i) HE genes lie towards one end of this major trend, (ii) putative translationally optimal codons can be identified for all AAs except Glu (table 1), including codons (UUC, UAC, AUC, AAC) that are optimal in all species owing to their complementarity to the only tRNA anti-codons available [6], and (iii) the frequency of these codons in a gene (Fop) is highly correlated with abundance of the encoded protein (figure 2). Strangely, Xia et al. [14] reported that CAI values did not correlate well with their protein abundance data; we find the opposite (see electronic supplementary material).
Two additional points arise from the gene expression data. First, data are available for only 967 genes (56% of the total). This may indicate that some predicted open reading frames do not in fact encode proteins, or that many genes were not expressed to a measurable extent under the growth conditions used to estimate protein abundance. Consistent with this, the genes lacking expression level data have lower Fop values (median 0.39) than others (median 0.44). Second, codon usage bias is expected to be most strongly selected during periods of exponential growth, and so Fop values may correlate less well with expression data (such as those used here) collected under other growth conditions. Interestingly, the HE genes have stronger bias than would be predicted given the overall observed relationship of Fop with expression level (figure 2), as expected if these HE genes are even more highly expressed, relative to other genes, during periods at maximum growth rate.
In a recent analysis of selected codon usage bias (S) in the genomes of 214 prokaryotes, the 26 Archaea seemed to conform to the same trends as the 188 Bacteria [8], but here the S value for M. maripaludis is high in comparison with bacterial species with a similarly long minimal doubling time (more than 2 h) or with similarly small numbers of rRNA operons and tRNA genes [6,7]. Interestingly, the pattern of bias in M. maripaludis differs from that in E. coli, the archetypal example of selected codon usage bias in Bacteria (table 2). While the strength of selected bias is similar in the two species for twofold degenerate AAs, the bias is reduced for fourfold degenerate AAs in M. maripaludis. For twofold degenerate AAs there is typically only one form of tRNA and one codon is favoured over another because it better matches the anti-codon. For fourfold degenerate AAs there are usually multiple species of tRNA, with abundances largely determined by gene copy number [19]. In E. coli (and other Bacteria), strongly selected codon usage bias for fourfold degenerate AAs is associated with increased copy numbers of cognate tRNA genes; E. coli K-12 has 26 tRNA genes for these AAs. In contrast, M. maripaludis has only 10 genes—one copy for each of two different tRNAs for each of the five AAs; if this explains the weak selected codon usage bias, it remains unclear why tRNA gene duplication has not been favoured.
In conclusion, it is surprising that translational selection in M. maripaludis has had a strong impact on codon usage for some (twofold degenerate) but not other (fourfold degenerate) AAs. It will be interesting to extend this study to other species to see whether this is a general difference between Bacteria and Archaea.
Acknowledgements
We thank Kai Zeng for discussion of the extension of S values to four codon families. L.R.E. was funded by a studentship from the BBSRC.
References
- 1.Bulmer M. 1991. The selection–mutation–drift theory of synonymous codon usage. Genetics 129, 897–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharp P. M., Li H. 1986. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 24, 28–38 10.1007/BF02099948 (doi:10.1007/BF02099948) [DOI] [PubMed] [Google Scholar]
- 3.Ikemura T. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 2, 13–34 [DOI] [PubMed] [Google Scholar]
- 4.Ehrenberg M., Kurland C. G. 1984. Costs of accuracy determined by a maximal growth rate constraint. Quart. Rev. Biophys. 17, 45–82 10.1017/S0033583500005254 (doi:10.1017/S0033583500005254) [DOI] [PubMed] [Google Scholar]
- 5.Rocha E. P. C. 2004. Codon usage bias from tRNA's point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 14, 2279–2286 10.1101/gr.2896904 (doi:10.1101/gr.2896904) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharp P. M., Bailes E., Grocock R. J., Peden J. F., Sockett R. E. 2005. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 33, 1141–1153 10.1093/nar/gki242 (doi:10.1093/nar/gki242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharp P. M., Emery L. R., Zeng K. 2010. Forces that influence the evolution of codon bias. Phil. Trans. R. Soc. B 365, 1203–1212 10.1098/rstb.2009.0305 (doi:10.1098/rstb.2009.0305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vieira-Silva S., Rocha E. P. C. 2010. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 6, e1000808. 10.1371/journal.pgen.1000808 (doi:10.1371/journal.pgen.1000808) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlin S., Mrazek J., Ma J., Brocchieri L. 2005. Predicted highly expressed genes in archaeal genomes. Proc. Natl Acad. Sci. USA 102, 7303–7308 10.1073/pnas.0502313102 (doi:10.1073/pnas.0502313102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henry I., Sharp P. M. 2007. Predicting gene expression level from codon usage bias. Mol. Biol. Evol. 24, 10–12 10.1093/molbev/msl148 (doi:10.1093/molbev/msl148) [DOI] [PubMed] [Google Scholar]
- 11.Dos Reis M., Savva R., Wernisch L. 2004. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 32, 5036–5044 10.1093/nar/gkh834 (doi:10.1093/nar/gkh834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones W. J., Paynter M. J. B., Gupta R. 1983. Characterization of Methanococcus maripaludis sp. nov., a new methanogen isolated from salt marsh sediment. Arch. Microbiol. 135, 91–97 10.1007/BF00408015 (doi:10.1007/BF00408015) [DOI] [Google Scholar]
- 13.Hendrickson E. L., et al. 2004. Complete genome sequence of the genetically tractable hydrogenotrophic methanogen Methanococcus maripaludis. J. Bacteriol. 186, 6956–6969 10.1128/JB.186.20.6956-6969.2004 (doi:10.1128/JB.186.20.6956-6969.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia Q., et al. 2006. Quantitative proteomics of the archaeon Methanococcus maripaludis validated by microarray analysis and real time PCR. Mol. Cell. Proteomics 5, 868–881 10.1074/mcp.M500369-MCP200 (doi:10.1074/mcp.M500369-MCP200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gouy M., Gautier C., Attimonelli M., Lanave C., Di Paola G. 1985. ACNUC—a portable retrieval system for nucleic acid sequence databases: logical and physical design and usage. Comp. Appl. Biosci. 1, 167–172 [DOI] [PubMed] [Google Scholar]
- 16.Chan P. P., Lowe T. M. 2009. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97 10.1093/nar/gkn787 (doi:10.1093/nar/gkn787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright F. 1990. The ‘effective number of codons’ used in a gene. Gene 87, 23–29 10.1016/0378-1119(90)90491-9 (doi:10.1016/0378-1119(90)90491-9) [DOI] [PubMed] [Google Scholar]
- 18.Sharp P. M., Li H. 1987. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295 10.1093/nar/15.3.1281 (doi:10.1093/nar/15.3.1281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanaya S., Yamada Y., Kudo Y., Ikemura T. 1999. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 238, 143–155 10.1016/S0378-1119(99)00225-5 (doi:10.1016/S0378-1119(99)00225-5) [DOI] [PubMed] [Google Scholar]