

Abstract
Metformin is the most commonly used pharmacological therapy for type 2 diabetes. We carried out a GWA study on glycaemic response to metformin in 1024 Scottish patients with type 2 diabetes. Replication was in two cohorts consisting of 1783 Scottish patients and 1113 patients from the UK Prospective Diabetes Study. In a meta-analysis (n=3920) we observed an association (P=2.9 *10−9) for a SNP rs11212617 at a locus containing the ataxia telangiectasia mutated (ATM) gene with an odds ratio of 1.35 (95% CI 1.22 to 1.49) for treatment success. In a rat hepatoma cell line, inhibition of ATM with KU-55933 attenuated the phosphorylation and activation of AMPK in response to metformin. We conclude that ATM, a gene known to be involved in DNA repair and cell cycle control, plays a role in the effect of metformin upstream of AMPK, and variation in this gene alters glycaemic response to metformin.
In treating Type 2 diabetes metformin is recommended as first line therapy in most national and international guidelines1,2. Despite the clinical use of metformin for over 50 years, its mechanism of action has not been fully elucidated. Whilst it is established that metformin activates AMP-activated protein kinase (AMPK)3 by inhibition of the mitochondrial respiratory chain4, causing an increase in cellular AMP5, it remains uncertain whether AMPK is its sole therapeutic target.
There is considerable variability in glycaemic response to metformin. No clinical phenotype usefully predicts response6 yet there has been little pharmacogenetic investigation of metformin with no consistently replicated genetic variant established. We hypothesised that a GWA approach could be applied to the glycaemic response to metformin to gain insight into the mechanism of metformin’s action in humans, and to identify variants that may be useful clinically to predict efficacy or adverse outcome.
As part of the Wellcome Trust Case Control Consortium 2 study (WTCCC2), a GWA study of 15 complex traits and disorders, we carried out the first GWA study on metformin response in patients with type 2 diabetes, using a large Scottish observational genetic cohort (GoDARTS) of European ancestry. As our principal outcome phenotype, we used the ability to reduce HbA1c (the most widely used measure of medium term glycaemic control) in the first 18 months of therapy to below 7%, this being a key measure of success in many treatment algorithms. Covariates shown to alter metformin response, such as baseline HbA1c and creatinine clearance were included in a logistic regression model (supplementary methods). Full details of the cohorts and models used are available in the online methods and baseline characteristics of the cohorts are shown in supplementary figure 1 and supplementary table 1. Samples were genotyped using the Affymetrix 6.0 microarray. After strict quality control we analysed 705,125 SNPs in 1024 metformin treated patients (supplementary methods). The quantile-quantile plot is shown in supplementary figure 2; the genomic inflation factor was 1.003. The Manhattan plot is shown in supplementary figure 3. We found that 14 SNPs with a p-value <1*10−6 mapped to a 340 kb strong LD block on chromosome 11q22 (figure 1). No stronger association was observed around this locus after imputing the data to the 2.2 million HapMap II CEU panel. SNPs at other loci, that are potentially associated with metformin response, did not achieve a p-value lower than 1*10−6 and have not been followed up in the current study (supplementary table 2). The minor allele (C) of the most strongly associated SNP, rs11212617, had a frequency of 44% and was associated with treatment success (achieving an HbA1c below 7%) with an allelic odds ratio of 1.64 (95% Confidence interval 1.37 to 1.99, p=1.9*10−7) (table 1). The full model is shown in supplementary table 3a.
Figure 1.

Regional association plots around the ATM locus for the logistic regression analysis. The solid and open triangles are from directly typed and imputed SNPs respectively
Table 1.
Association analysis results between rs11212617 and glycaemic response to metformin in the discovery and internal replication cohorts, and the combined sample. The reference allele for rs11212617 is A. For the UKPDS samples, results are for rs609261, which was genotyped in this cohort due to technical difficulties, but was a proxy for rs11212617 (r2=0.997 in WTCCC2 controls). The logistic regression analysis shows allelic odds ratio (OR) for the ability to achieve a treatment HbA1c <=7% in the 18 months after starting metformin. The linear regression analysis shows the per-allele increase in treatment HbA1c in the treatment period after starting metformin. Covariates included in the model were baseline HbA1c, gap between treatment starting and baseline HbA1c, dose, adherence, creatinine clearance, and treatment group. Full models are shown in supplementary table 3. 95% confidence intervals of the beta or OR are shown in square brackets.
| Study | Sample size |
Logistic | Linear | ||
|---|---|---|---|---|---|
| OR | p | beta | p | ||
| Discovery | 1024 | 1.64 [1.37,1.99] | 1.9E-07 | −0.18 [−0.26,−0.10] | 1.8E-05 |
| Replication 1 | 1783 | 1.21 [1.05,1.38] | 0.007 | −0.07 [−0.13,−0.01] | 0.022 |
| Replication 2 (UKPDS) |
1113 | 1.37 [1.10,1.72] | 0.006 | −0.12 [−0.23,−0.02] | 0.021 |
| Combined | 3920 | 1.35 [1.22,1.49] | 2.9E-09 | −0.11 [−0.16,−0.07] | 6.6E-07 |
Our primary analysis used a binary treatment target as its endpoint. To check the robustness of this, we also analysed the treatment HbA1c as a quantitative trait in a linear regression. In parallel with the primary analysis we found the C allele of rs11212617 was associated with lower treatment HbA1c (per allele Beta −0.18% [95% confidence intervals −0.26 to −0.1], p=1.8*10−5) (table 1).
Two replication cohorts were used. SNP rs11212617 was genotyped in an independent GoDARTS cohort of 1783 metformin-treated patients with type 2 diabetes (replication 1). The minor allele (C) of rs11212617 was associated with treatment success (allelic OR 1.21 95%CI 1.05 to 1.38; p=0.007) (table 1). The second replication cohort was 1113 UK patients prospectively treated with metformin in the UKPDS (UK Prospective Diabetes) cohort (replication 2). The UKPDS was a prospective randomised clinical trial of intensive vs conventional treatment in type 2 diabetes. In the UKPDS, where, for technical reasons, we typed the proxy SNP rs609261 (r2=0.997 with rs11212617 in 5197 WTCCC2 controls) the minor allele was associated with treatment success (allelic OR 1.37 95%CI 1.1 to 1.72; P=0.006) (table 1). The combined p-value achieved significance of p=2.9*10−9 for 3920 metformin-treated patients (table 1). The full models for each cohort are shown in supplementary table 3a-c. In the combined linear regression, each copy of the rs11212617 minor allele C is associated with 0.11% (p=6.6*10−7) lower absolute treatment HbA1c (table 1).
Metformin can be used as monotherapy alone, or added in to other therapies. However, current prescribing practice is for metformin to be used first line; we therefore analysed the monotherapy subgroup separately. Most of the association signal at rs11212617 with metformin response for the full group arises from the monotherapy subgroup (supplementary table 4). In a meta-analysis of this monotherapy group (n=2264) the combined odds-ratio for treatment success was 1.42 (95% CI 1.26 to 1.62), p=4*10−8. To assess the clinical impact of rs11212617, we studied the UKPDS cohort that was randomly assigned to metformin monotherapy (n=284) and followed up prospectively, and therefore not prone to treatment selection bias. In this subgroup, the 19% of patients with two copies of the C allele at rs11212617 have a 3.3-fold greater likelihood of achieving an HbA1c <=7%. This equates to a model adjusted difference in treatment HbA1c of 0.61% between those who are CC vs AA at this SNP. Adding genotype to the full linear regression model in this UKPDS group randomised to metformin increased the variance in the treatment HbA1c explained by the model from 27.5% to 30% (p=0.007).
To ensure that the genotypic effect on metformin response was not related to an effect on HbA1c per se, and to assess the association with fasting insulin and HOMA derived insulin resistance, we analysed summary statistics for rs11212617 from the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC)7. We found no association of rs11212617 with HbA1c (p=0.82), HOMA-IR (p=0.99) and fasting insulin (p=0.73) in at least 35,914 non-diabetic individuals (supplementary methods). We found no association between baseline HbA1c and rs11212617 in the GoDARTs discovery or replication dataset. In addition there was no association of rs11212617 with lipid parameters, blood pressure, height, weight, BMI, adiponectin and leptin in up to 6148 Scottish controls (supplementary table 5); nor with type 2 diabetes risk in a case-control study of 5788 Scottish GoDARTS patients with type 2 diabetes and 6357 non-diabetic Scottish GoDARTS controls (p=0.64).
The SNP rs11212617 falls within a large block of linkage disequilibrium that includes the genes CUL5, ACAT1, NPAT, ATM, C11orf65, KDELC2, EXPH5. Of these, ATM was considered a possible candidate gene. Firstly, homozygous loss of function mutations in ATM cause Ataxia Telangiectasia (A-T; OMIM #208900) which is a neurodegenerative disorder characterized by loss of muscle coordination and progressive ataxia, radiosensitivity, immunodeficiency and a predisposition to cancer8; additionally, patients with A-T have been reported to have marked insulin resistance and increased risk of diabetes9,10. Secondly, previous laboratory reports suggest that activation or inhibition of ATM alters AMPK activation11-13. None of the other genes at the locus have been reported to be associated with diabetes or insulin action.
ATM encodes a 370 kDa protein that is a Ser/Thr protein kinase of the atypical phosphoinositide 3-kinase-related protein kinase (PIKK) family. ATM is activated by double-stranded DNA breaks, and acts to induce cell-cycle arrest and facilitate DNA repair14. To investigate if ATM was the causal gene affecting the glycaemic response to metformin we studied the effects of a selective ATM inhibitor, KU-55933, on the activation of AMPK by metformin in rat hepatoma (H4IIE) cells. Figure 2 shows that KU-55933 markedly reduced AMPK activation by metformin. Similarly, figure 3 shows that phosphorylation of AMPK and its downstream target, ACC, by metformin was inhibited by KU-55933. These results are supported by previous reports of ATM involvement in the activation of AMPK by stimuli other than metformin11-13. We conclude that ATM acts upstream of AMPK, and is required for a full response to metformin. ATM is also reported to be involved in insulin signalling and pancreatic β-cell dysfunction, both of which may influence metformin action: apoE −/− mice heterozygous for loss of atm function were insulin-resistant compared to apoE−/− mice with normal atm15; however, mice lacking atm develop diabetes due to β cell dysfunction16.
Figure 2.
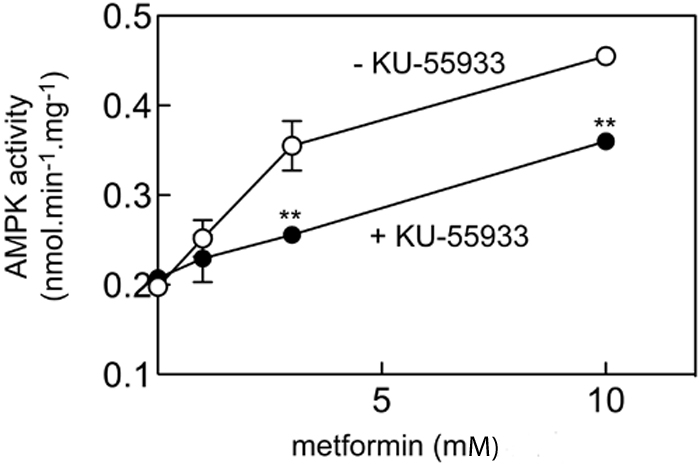
Effect of KU-55933 on AMPK activation by metformin
H4IIE cells were pre-treated with or without 10 μM KU-55933 for 30 min and then with various concentrations of metformin for 1 hr, and AMPK activity measured; Results are mean ± S.D. (n = 2); **significantly different from incubation without KU-55933 by 2-way ANOVA (p<0.01).
Figure 3.
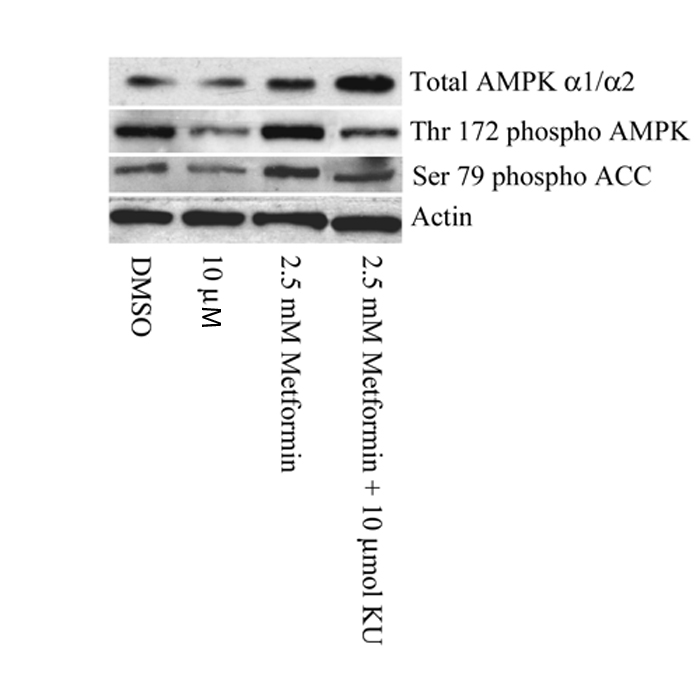
A Western Blot comparing the phosphorylation status of Thr-172 of AMPK and Ser-79 of ACC (a well characterized marker of AMPK activation). H4IIE cells were pre-treated with or without 10 μM KU-55933 (KU) for 1 hour and then for 3 hours with or without 2.5mmol/L metformin. Metformin induced phosphorylation of AMPK and subsequent phosphorylation of ACC was partially reduced by KU-55933.
Potential functionality of all the 98 SNPs with strong linkage disequilibrium (r2 >0.8 according to the HapMap CEU panel) to rs11212617 was assessed (supplementary table 6). SNP rs228589, which is in intron 1 of the NPAT gene, is in a predicted promoter of ATM17. Two SNPs, rs227092 and rs4585, are located in the ATM 3′UTR region. Variant rs4585 lies 24bp downstream of a polyadenylation site, and is predicted to alter efficiency of polyadenylation (supplementary table 6). The 3′UTR region of ATM is among the longest known mammalian 3′UTRs and has been suggested to influence ATM mRNA translation allowing rapid response to stimuli at the post-transcriptional level18,19.
Type 2 diabetes is associated with increased cancer risk, and an overlap between genes predisposing to prostate cancer and type 2 diabetes has been described20. Metformin has been shown in epidemiological studies to be associated with decreased cancer risk21, and to decrease tumour burden in pten deficient mice22. Activation of AMPK by metformin requires the known tumour suppressor LKB1. In this study the implication of ATM, a gene known to be involved in DNA repair and cancer, in the glycaemic response to metformin establishes a further link between cancer pathways, type 2 diabetes and metformin activation of AMPK.
In this study, we have established the utility of a genome wide approach to study the pharmacogenomics of metformin response, and the utility of large genetics resources linked to routinely collected clinical data for pharmacogenetic studies. We have identified the first robustly replicated variant to be associated with metformin response. Whilst this observation may not be of immediate clinical utility, explaining only 2.5% of variance in metformin response, this study is an example of how genome-wide association studies can be applied to pharmacogenomic models to identify novel pathways and mechanisms, and has established an unexpected link between glucose homeostasis and the DNA damage response.
Supplementary Material
Acknowledgements
We are grateful to all the participants who took part in this study, to the general practitioners, to the Scottish School of Primary Care for their help in recruiting the participants, and to the whole team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. The Wellcome Trust provides support for Wellcome Trust United Kingdom Type 2 Diabetes Case Control Collection (GoDARTS) and informatics support is provided by the Chief Scientist Office. The Wellcome Trust funds the Scottish Health Informatics Programme, provides core support for the Wellcome Trust Centre for Human Genetics in Oxford and funds the Wellcome Trust Case Control Consortium 2. This research was specifically funded by Diabetes UK (07/0003525), MRC (G0601261) and the Wellcome Trust (084726/Z/08/Z, 085475/Z/08/Z, 085475/B/08/Z). We also acknowledge support from the NIHR award to Moorfields Eye Hospital NHS Foundation Trust and University College London Institute of Ophthalmology for a Specialist Biomedical Research Centre for Ophthalmology (ACV). P. Donnelly was supported in part by a Wolfson–Royal Society Merit Award. KZ holds a Henry Wellcome Post-Doctoral Fellowship. SAH and DGH were supported by the EXGENESIS consortium (LSHM-CT-2004-005272) funded by the European Commission.
Appendix
On Line Methods
Samples
The discovery cohort and first replication cohort were both ascertained from the Diabetes Audit and Research Tayside Study (DARTS)23. Validated prescribing data, biochemistry data as well as clinical phenotypes back to 1992 can be retrieved from central databases for all the DARTS patients. Prospective longitudinal data were also collected on these patients. Since October 1997, all patients with diabetes have been invited to give written informed consent to DNA as part of the Wellcome Trust United Kingdom Type 2 Diabetes case control collection. As of June 2009, 8000 cases and 7000 controls of European ancestry have participated in this Genetics of DARTS (GoDARTS) study.
As part of the WTCCC2, 4134 GoDARTS cases were selected primarily for a genome wide association study of statin response, but also for a study of response to oral hypoglycaemic agents (OHA). Following the WTCCC2 genotyping quality control (see below), 1024 patients were identified who were initiated on metformin and had a definable metformin response (discovery cohort). Of the GoDARTS cases not in the WTCCC2 discovery cohort metformin response could be defined in 1783 patients and this was used for the first round replication.
The second replication cohort was patients with type 2 diabetes either randomised to metformin, or treated with metformin as per protocol, in the UK Prospective Diabetes Study (UKPDS)24. A total number of 1113 white European patients, who were exposed to metformin and passed the genotyping quality control, were identified in the UKPDS cohort. These were either primarily randomized to metformin (n=284), were randomized second line as add-in to sulphonylureas (n=231), or had metformin added to sulphonylurea per protocol for symptomatic hyperglycaemia or when fasting glucose was greater than 15mmol/L (n=598).
Phenotypes
Because over 92% of the OHA prescriptions issued in GoDARTS cohort are either metformin (51.4%) or sulphonylurea (41.2%), we focused on two treatment schemes of metformin monotherapy (metformin added in following failure of dietary control) or dual therapy (metformin added to stable sulphonylurea treatment).
Following initiation of oral hypoglycaemic agents in type 2 diabetes, there is an initial reduction in HbA1c, followed by a gradual deterioration. This can be seen in both the UKPDS study and other diabetes trials such as ADOPT25. The gradual deterioration in HbA1c will reflect both drug efficacy (or inefficacy) to control HbA1c, and the underlying diabetes progression. To target the drug response alone we focused on the first 18 months of metformin therapy to minimize the response window but ensure minimal exclusion due to lack of HbA1c data.
In this observational study, the patient’s physician will be treating to achieve an HbA1c target, which over the majority of the study period would have been 7%. We therefore defined our primary drug response phenotype as a dichotomous trait of treatment success which was the ability to achieve an HbA1c below 7% in the first 18 months of treatment, with censoring if diabetes therapy was changed prior to this. Our secondary analysis took the quantitive phenotype as treatment HbA1c which was the lowest HbA1c observed between 1 and 18 months after metformin treatment or prior to a change in therapy (cessation of metformin or addition of further oral hypoglycaemic therapy).
The two phenotypes were modelled with multiple linear or logistic regression using the same set of covariates which includes baseline HbA1C, adherence, daily dose, creatinine clearance, baseline gap and treatment group. More details of the models and covariates were provided in supplementary methods.
In the UKPDS the HbA1c was measured yearly, and the HbA1c measure closest to 1 year after commencing metformin was taken as the treatment HbA1c. In the cases randomized to metformin this would have been approximately one year after starting metformin; in those starting metformin as per protocol due to hyperglycaemia the time between starting metformin and the treatment HbA1c was variable so an additional covariate (treatment gap) was included in the model. Treatment success was a treatment HbA1c <=7%. Other covariates included were as for the GoDARTS models, except adherence and dose were not included due to missing data.
GWAS genotyping and Quality Control (QC)
Samples were genotyped at Affymetrix’s service laboratory on the Genome-Wide Human SNP Array 6.0. Genotype data quality control was via the standard protocol that was established for the WTCCC2 studies26 (supplementary methods). Specifically, concordance check was performed on 116 SNPs by 1779 individuals overlapped between this GWA data and the WTCCC1 T2D case control study27. Based on the concordance rate of 99.73%, individuals with more than 10% discordance were removed from the current study. After such stringent QC, the clean data set included 705125 autosomal SNPs on 3736 samples, of whom 1024 have definable metformin response.
Replication Genotyping
In the first round replication, following assay optimization of the top two SNPs from the GWA (rs11212617 and rs624366, r2=0.997 in 3736 GoDARTS samples), rs11212617 had better genotyping performance and was genotyped by the standard Taqman-based allelic discrimination method (Applied Biosystems) in the whole GoDARTS cohort, which included 7000 non-diabetic controls. The overall call rate was 98.3% with a concordance rate of 99.9% to the GWA genotypes. There was no deviation from Hardy Weinberg Equilibrium (p=0.44). Minor allele C had a frequency of 43.9% in the 1783 GoDARTS replication sample.
Taqman was also used for genotyping the UKPDS replication sample. Following optimization of 4 highly correlated SNPs (rs11212617 and rs624366 as well as two proxies rs609261 and rs2345801) in a subset of UKPDS samples, SNP rs609261 was selected and genotyped based on better genotyping performance than the other SNPs. rs609261 is almost a perfect proxy for rs11212617 (r2=0.997 in 5197 WTCCC2 controls) and as such, results are presented as for rs11212617 in the UKPDS cohort. Genotyping of all available UKPDS DNAs (n=3400) was carried out in duplicate using standard conditions. Discrepancy between the duplicate genotyping runs was 0.4%, and only samples for which the duplicate genotypes were concordant were analysed. The final UKPDS replication cohort included 1113 Caucasian patients and the minor allele frequency was 42.1% with no deviation from HWE (p=0.98).
Statistical Analysis
Logistic and linear regression modelling was performed with PLINK and SNPTEST28,29 assuming an additive genetic model. All the results presented were unadjusted for population stratification as a genomic inflation factor of 1.003 and 0.998 was observed for logistic and linear regression analyzes respectively.
Data from the discovery cohort were also imputed to the HapMap II CEU panel of 2.2 million SNPs with program IMPUTE and tested for association with SNPTest taking into account the imputed genotype probabilities30.
As the two SNPs rs11212617 and rs609261 that were genotyped in the Go-DARTS sample and the UKPDS sample respectively were in near complete LD (r2=0.997), we directly combined the single marker association test results from the discovery cohort and the two replication cohorts with the inverse variance fixed effect method as implemented in the R package of GenABEL31.
Functional studies
KU-55933 is a cell-permeable ATP-competitive inhibitor of ATM (IC50 = 13 nM; Ki = 2.2 nM) with selectivity over other PIKK family kinases (IC50 = 2.5, 9.3, 16.6 μM for DNA-PK, mTOR, PI 3-K, respectively; IC50 > 100 μM for PI 4-K and ATR)32. It is reported to have little activity towards a panel of 70 conventional serine/threonine kinases. It has been shown to inhibit ATM-dependent cellular protein phosphorylation following ionizing radiation (IR) and sensitizes cells that express wild-type ATM (but not mutant ATM) to the cytotoxic effects of IR and DNA-damaging agents33,34.
H4IIE cells were cultured in Dulbecco’s modified Eagle’s medium containing 5% (v/v) fetal bovine serum to 80% confluence. For AMPK assays, cells were pretreated with 10 μM KU55933 (30 min) prior to treatment with various concentrations of metformin for 1 hr. Lysates were prepared (rapid lysis method35), centrifuged (4°C, 10 min 21,000 x g), and the supernatants frozen for later analysis. AMPK activity was assayed in immunoprecipitates as described35 using the AMARA peptide36.
To establish that the change in AMPK activity was due to regulation of phosphorylation of AMPK and that this translated into changes in substrate phosphorylation, we used Western Blotting to compare the phosphorylation status of Thr-172 of AMPK and Ser-79 of ACC (a well characterized marker of AMPK activation). In both cases metformin induced phosphorylation was partially reduced (Figure 3).
For these experiments, H4IIE cells were cultured to 80% confluence as above, then serum starved for 16 hr, pretreated with KU55933 or vehicle (DMSO) for 1 hr then treated for 3 hr with or without metformin. They were harvested as above. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose. The membrane was blocked in TBST containing 5% milk for 1 hour, then incubated overnight at 4°C with anti-phospho-Thr172-AMPK antibody (Cell Signaling, Beverly, MA), anti-phospho-Ser79 ACC antibody (Cell Signaling) or anti-β-actin antibody (Sigma, Dorset, UK). Membranes were washed in TBST, incubated with goat anti-rabbit IgG conjugated to horseradish peroxidase (Pierce, Chester, UK) then washed again in TBST. The membrane was then developed using an ECL Western Blotting Detection Kit (Amersham Biosciences) before exposure to X-Ray film (Thermo Scientific, Waltham, MA).
Footnotes
A full list of WTCCC2 members and MAGIC investigators is listed in the supplementary notes.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Nathan DM, et al. Medical management of hyperglycaemia in type 2 diabetes mellitus: a consensus algorithm for the initiation and adjustment of therapy : A consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia. 2009;52:17–30. doi: 10.1007/s00125-008-1157-y. [DOI] [PubMed] [Google Scholar]
- 2.NICE clinical guideline 87 . Type 2 diabetes: the management of type 2 diabetes. National Institute for Health and Clinical Excellence; 2009. [Google Scholar]
- 3.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 5.Hawley SA, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donnelly LA, Doney AS, Hattersley AT, Morris AD, Pearson ER. The effect of obesity on glycaemic response to metformin or sulphonylureas in Type 2 diabetes. Diabet Med. 2006;23:128–133. doi: 10.1111/j.1464-5491.2005.01755.x. [DOI] [PubMed] [Google Scholar]
- 7.Dupuis J, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boder E. Ataxia-telangiectasia: an overview. Kroc Found Ser. 1985;19:1–63. [PubMed] [Google Scholar]
- 9.Schalch DS, McFarlin DE, Barlow MH. An unusual form of diabetes mellitus in ataxia telangiectasia. N Engl J Med. 1970;282:1396–1402. doi: 10.1056/NEJM197006182822503. [DOI] [PubMed] [Google Scholar]
- 10.Bar RS, et al. Extreme insulin resistance in ataxia telangiectasia: defect in affinity of insulin receptors. N Engl J Med. 1978;298:1164–1171. doi: 10.1056/NEJM197805252982103. [DOI] [PubMed] [Google Scholar]
- 11.Sun Y, Connors KE, Yang DQ. AICAR induces phosphorylation of AMPK in an ATM-dependent, LKB1-independent manner. Mol Cell Biochem. 2007;306:239–245. doi: 10.1007/s11010-007-9575-6. [DOI] [PubMed] [Google Scholar]
- 12.Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS One. 2008;3:e2009. doi: 10.1371/journal.pone.0002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanli T, et al. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010;78:221–229. doi: 10.1016/j.ijrobp.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 15.Schneider JG, et al. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4:377–389. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Miles PD, Treuner K, Latronica M, Olefsky JM, Barlow C. Impaired insulin secretion in a mouse model of ataxia telangiectasia. Am J Physiol Endocrinol Metab. 2007;293:E70–74. doi: 10.1152/ajpendo.00259.2006. [DOI] [PubMed] [Google Scholar]
- 17.Trinklein ND, Aldred SJ, Saldanha AJ, Myers RM. Identification and functional analysis of human transcriptional promoters. Genome Res. 2003;13:308–312. doi: 10.1101/gr.794803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukao T, et al. ATM is upregulated during the mitogenic response in peripheral blood mononuclear cells. Blood. 1999;94:1998–2006. [PubMed] [Google Scholar]
- 19.Savitsky K, et al. Ataxia-telangiectasia: structural diversity of untranslated sequences suggests complex post-transcriptional regulation of ATM gene expression. Nucleic Acids Res. 1997;25:1678–1684. doi: 10.1093/nar/25.9.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gudmundsson J, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 21.Libby G, et al. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang X, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 23.Morris AD, et al. The diabetes audit and research in Tayside Scotland (DARTS) study: electronic record linkage to create a diabetes register. DARTS/MEMO Collaboration. Bmj. 1997;315:524–528. doi: 10.1136/bmj.315.7107.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 25.Kahn SE, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- 26.Barrett JC, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeggini E, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 29.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 32.Hickson I, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 33.Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Invest. 2007;117:2723–2734. doi: 10.1172/JCI31604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crescenzi E, Palumbo G, de Boer J, Brady HJ. Ataxia telangiectasia mutated and p21CIP1 modulate cell survival of drug-induced senescent tumor cells: implications for chemotherapy. Clin. Cancer Res. 2008;14:1877–1887. doi: 10.1158/1078-0432.CCR-07-4298. [DOI] [PubMed] [Google Scholar]
- 35.Hardie DG, Salt IP, Davies SP. Analysis of the role of the AMP-activated protein kinase in the response to cellular stress. Methods Mol. Biol. 2000;99:63–75. doi: 10.1385/1-59259-054-3:63. [DOI] [PubMed] [Google Scholar]
- 36.Dale S, Wilson WA, Edelman AM, Hardie DG. Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett. 1995;361:191–195. doi: 10.1016/0014-5793(95)00172-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.