

Abstract
Connexins (Cx) and gap junction (GJ) mediated communication has been linked with the regulation of cell cycle traverse. However, it is not clear whether Cx expression or GJ channel function are the key mediators in this process or at what stage this regulation may occur. We therefore tested the hypothesis that enhanced Cx expression could alter the rate of cell cycle traverse independently of GJ channel function. Sodium butyrate (NaBu) or anti-arrhythmic peptide (AAP10) were used to enhance Cx expression in HeLa cells stably expressing Cx43 (HeLa-43) and primary cultures of human fibroblasts (HFF) that predominantly express Cx43. To reduce GJ mediated communication, 18α-glycyrrhetinic acid (GA) was used. In HeLa-43 and HFF cells, NaBu and AAP10 enhanced Cx43 expression and increased channel function, while GA reduced GJ mediated communication but did not significantly alter Cx43 expression levels. Timelapse microscopy and flow cytometry of HeLa-WT (wild type, Cx deficient) and HeLa-43 cells dissected cell cycle traverse and enabled measurements of intra-mitotic time and determined levels of G1 arrest. Enhanced Cx43 expression increased mitotic durations corresponding with a G1 delay in cell cycle, which was linked to an increase in expression of the cell cycle inhibitor p21waf1/cip1 in both HeLa-43 and HFF cells. Reductions in Cx43 channel function did not abrogate these responses, indicating that GJ channel function was not a critical factor in reducing cell proliferation in either cell type. We conclude that enhanced Cx43 expression and not GJ mediated communication, is involved in regulating cell cycle traverse.
Keywords: Connexin 43, gap junction, cell cycle traverse, cell migration, mitotic duration
Introduction
Many classical tumour cell lines, including HeLa and C6 glioma cells, are deficient in connexin (Cx) expression and are generally referred to as ‘communication deficient’. Introduction of Cx genes can ‘normalise’ cell growth leading to proposals that these proteins have tumour suppressor like characteristics and can exert growth regulatory control [Hellmann et al., 1999; Mesnil et al., 1995; Zhu et al., 1992]. Such systems have provided extensive information on the role of Cxs in cell growth responses and numerous studies now suggest that Cx43 may play a major role in proliferative disorders including diverse carcinomas [Kanczuga-Koda et al., 2006], vascular diseases such as atherogenesis [Johnstone et al., 2009; Kwak et al., 2003] and in non-healing diabetic wounds [Wang et al., 2007]. Indeed a recent review documented over 70 alterations in Cx expression and signalling properties attributed to cellular proliferative disorders [Mesnil et al., 2005].
Connexins are the constitutive proteins of gap junction (GJ) channels that permit the exchange of small metabolites and signalling molecules between neighbouring cells. The most widely expressed connexin is Cx43, which in addition to being highly permissive to solutes, has an expression pattern that appears to follow the cell cycle. For example, during normal cell cycle progression, Cx43 expression is decreased, the protein internalised and differentially phosphorylated at sites on its carboxyl terminus [Solan et al., 2003; Solan and Lampe, 2009]. Furthermore Cx43 GJ mediated communication decreases during the G1 stage of the cell cycle [Lampe et al., 1998; Lampe and Lau, 2004]. However, based on current literature there is no consensus as to the importance of expression over GJ channel function in cell cycle mediated events with varying reports in different systems [Avanzo et al., 2007; Sanchez-Alvarez et al., 2006]. For example, in mouse lung carcinoma models the maintenance of GJ mediated communication is important in tumour suppression [Avanzo et al., 2004] and a decrease in GJ channel function, linked to improper assembly and targeting of Cx43, promotes cancer progression in mouse skin carcinogenesis models [Holden et al., 1997]. Conversely, decreases in Cx43 expression have been associated with increased cell proliferation in cervical and metastatic breast carcinoma cell lines [Aasen et al., 2005] and Cx43 mediated rescue of the malignant phenotype of metastatic breast cancer cells have been shown to be independent of GJ function [McLachlan et al., 2006]. Further studies suggest that protein interactions independent of Cx43 GJ mediated communication may be important in the suppression of cell growth [Olbina and Eckhart, 2003], with cytoplasmic localization of Cx43 inhibiting cellular proliferation [Moorby and Patel, 2001]. Control over the cell cycle is exerted by proteins classed as tumour suppressor proteins including p21waf1/cip1 and retinoblastoma proteins [Schafer, 1998] and Cx43 is considered by many as a tumour suppressor protein [Avanzo et al., 2004; Mesnil et al., 2005; Zhang et al., 2003]. However, the precise mechanism and signalling pathways through which Cx43 mediates cell cycle traverse is currently unknown.
In the present work we adopted a timelapse microscopy approach of multiple parameters to dissect the impact of enhanced Cx43 expression over GJ mediated communication on cell cycle events in both HeLa cells transfected to express Cx43 (HeLa-43) [Mesnil et al., 1995] and primary human foreskin fibroblasts (HFF) that predominantly express Cx43 [Wright et al., 2009]. The data reveal that enhanced Cx43 expression delays mitotic durations and this correlates with an accumulation of cells in the G1 stage that is associated with increased levels of the cell cycle inhibitor p21waf1/cip1. By contrast, inhibiting channel function had no effect on mitotic fate regardless of enhanced Cx43 expression. These provide strong evidence for the hypothesis that Cx43 can mediate cell cycle traverse in a non-GJ dependent manner.
Materials and Methods
Cell culture
Non-transfected HeLa-WT cells which do not express connexins, were maintained in Dulbecco’s Modified Eagle’s Media (DMEM) supplemented with 10% v/v FBS, penicillin/streptomycin 100 μg/ml and L-glutamine 2 mM (cDMEM). HeLa cells stably expressing Cx43 (HeLa-43) under the control of the SV40 early promoter were maintained in cDMEM supplemented with puromycin (0.5 μM, Sigma) [Mesnil et al., 1995]. Dermal fibroblasts (HFF) which predominantly express Cx43, were derived from human infant foreskin tissue sections using standard techniques for explant culture, cells were cultured and maintained in cDMEM [Wright et al., 2009]. Tissue sections used in this study were obtained with the appropriate ethical approval (Yorkhill research ethics committee, dermatological research version 6 03/07/01) and with patient/parental consent. All cells were maintained and used at 60–80% confluency.
Drug treatments
All agents were diluted in pre-warmed media at their final concentrations; sodium butyrate 0.5 mM (NaBu, Sigma) which enhances transcription through the SV40 early promoter in the HeLa-43 cells, 18-α-glycerrhetinic acid 25 μM (GA, Sigma), anti-arrhythmic peptide 50 nM (AAP10, Zealand Pharma [Easton et al., 2009]) and Colcemid 0.5μg/mL (Gibco [Marquez et al., 2003]). The levels of NaBu, GA and AAP10 used were non-toxic to the cells and did not alter cell morphology following 24 h exposure where <1% of cells took up propidium iodide (data not shown). All treatment time courses were as indicated, with the exception of G1 emptying experiments where cells were pre-treated for 12 h with the relevant agent prior to addition of Colcemid. In all experiments indicated as control, cells were grown in their respective media (as described) without drug treatment, with the exception of G1 emptying experiments where Colcemid was applied as described.
Antibodies
Immunocytochemistry and Western blot analysis of samples was performed using primary polyclonal antibodies directed against Cx43 designated as rCx43 (Kind gift Dr E. Rivedal [Rivedal and Leithe, 2005]) or polyclonal Cx43 (Sigma [Johnstone et al., 2009]). Monoclonal antibodies against p21waf1/cip1 (Calbiochem [Kurash et al., 2008]) and β-tubulin (Sigma) were also used. Western blot primary antibodies were visualized using anti mouse or rabbit secondary antibodies as required: for chemiluminescent detection horse radish peroxidise (HRP) (BioRad) was used and for detection using the LiCor Odyssey imaging system IRDye-680/800 (LiCor) secondary antibodies were used. For immunofluorescence primary antibodies were visualised using goat anti mouse-Alexa 488 or goat anti rabbit-Alexa 594 secondary antibodies as required (Invitrogen).
Western blot
All HeLa and HFF cells were grown to 60–70% confluency prior to treatment in 6 well plates. Cells were lysed in PBS containing SDS (1% w/v), Na3VO4 (20 mM), DTT (1 μM), PMSF (100 μM) and 1× complete protease inhibitor cocktails (Sigma). Total protein lysates (100μg) were separated on 12.5% polyacrylamide gels and transferred to nitrocellulose before detection with the relevant primary antibodies. Band densities were quantified on film (Chemiluminesence, BioRad image analyser FX, BioRad Quantity One) or by fluorescence on nitrocellulose membranes using a Li-Cor Odyssey infrared imaging system and software (LiCor [Johnstone et al., 2009]). Band intensity was subtracted from background levels and normalized against β-tubulin levels. Statistical analysis was performed, using one-way ANOVA with Bonferroni post-hoc tests comparison of density versus controls.
Immunofluorescence
Cells grown on 16mm2 coverslips were fixed with ice cold methanol following treatment, washed in PBS, permeabilized using 0.1% v/v Triton X-100 (Sigma) and blocked using 5% w/v milk-PBS. Proteins were detected using primary and secondary antibodies (as described). All images were captured on a Zeiss Axiovert 200 inverted confocal microscope linked to a Zeiss LSM 510 Meta laser scanning system as previously described [Easton et al., 2009]. Image capture and processing was performed using Carl Zeiss AIM software and semi-quantitative analysis of fluorescence of plaque sizes was performed using MetaMorph software (Universal Imaging Corp [Singh et al., 2005]).
To determine significance for each treatment compared to controls, values from 3 fields of view per slide per treatment (n=3) were used. Channel thresholds remained constant for analysis and regions around all plaques and immunofluorescent staining were created automatically by the software. The area (pixels) and fluorescent intensity (0–255nm) were recorded and logged for each image and pixel sizes converted to μm for analysis.
Microinjection
Microinjection was performed as previously described [Easton et al., 2009]. Briefly, HeLa-43 cells were grown to 80% confluency on 60mm2 culture dishes and exposed to either NaBu and or GA or in combination for 24 h. HFF cells were exposed to AAP10 for 5 h. Individual cells were microinjected with Alexa 594 dye (charge -2, Mw 759Da (Invitrogen, UK)), using an Eppendorf 5120 femtojet system linked to a Cairns monochromator with ~50 cells injected per plate for each experimental group. After 5 min cells were washed in PBS and fixed in 3.75% formaldehyde. Data are presented as the percentage of injected cells per plate transferring dye to <4, 5–9 or >10 neighbouring cells ± SE. In each grouping, comparisons to non-treated control levels were made using one way ANOVA with Dunnets multiple comparison analysis. Experiments were repeated in triplicate in one sitting and then on two further occasions with ~50 cells injected per experiment, giving n = 9, in each case *= p<0.05 and ** = p<0.01.
Timelapse analysis
Experimental set up for timelapse was performed as previously described [Errington et al., 2005]. Briefly, media containing treatments was replaced in wells immediately before the plate was placed on a timelapse microscope, (Zeiss Axiovert 100, with heated motorized stage, a mounted incubator unit and humidified CO2). Static phase contrast images were recorded every 5 min for 24 h at 3 regions per well on each six-well plate using a microscope mounted CCD camera (Nikon Eclipse TS120). Image capture was controlled by AQM 2000 software (Kinetic Imaging Ltd; Nottingham, UK). Images from timelapse capture were analysed using MetaMorph image analysis software (Universal Imaging Corp.). Measured parameters included 1) total cell numbers, 2) the number of mitotic events, 3) intra-mitotic duration (IrMD), 4) the occurrence of polyploid nuclei and 5) apoptotic/ necrotic occurrences [Marquez et al., 2003].
Statistical analysis was based on approximately 3000–4000 mitotic events for each treatment (n=3) and cell type, with all data expressed as the mean ± SE. The IrMD (defined by cells rounding up, splitting and flattening back down) was determined for all cells in view (Fig. 2).
Figure 2. Extraction of intra-mitotic duration from timelapse analysis of HeLa cells.
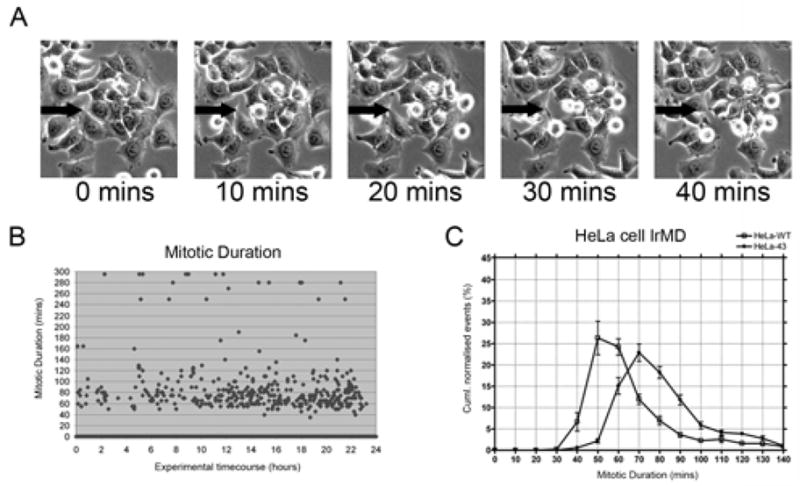
Cells plated in 6 well dishes were visualised on a Zeiss Axiovert microscope with heated stage and mounted incubator to maintain 5% CO2 and 37°C. In A, a small group of HeLa-43 cells from a field of view are shown, black arrow points to a dividing cell recorded as going through mitosis. Collated images were analysed using MetaMorph and the intra-mitotic duration (IrMD) was calculated as the time taken for a cell to enter mitosis, split into two daughter cells and flatten back down for every cell in view (A). Mitotic event end frames were recorded against the start frame of each corresponding event, the difference converted into a time format and plotted against the timecourse of the experiment (B). The IrMDs were grouped according to their duration (i.e. 1–10 mins, 11–20 mins etc), taken as a percentage of the overall mitotic events and then plotted in a line graph in Prism to allow comparison of the IrMD between treatment groups (C). In C plots of HeLa-WT and HeLa-43 cell IRMDs are shown.
Cell Cycle analysis
Cells grown to 60–70% confluency were pre-treated for 12 h with the appropriate drug. Following incubation, media was changed with fresh media containing Colcemid plus required experimental treatments. At the specified time points cells were harvested by trypsinisation and cell pellets recovered by centrifugation at 0.5 g for 5 min followed by re-suspension in PBS and fixed overnight at 4°C in 70% ethanol. Fixed cells were washed in PBS and approximately 1× 106 cells resuspended in a solution of PBS plus Trion X-100 (0.1% v/v), RNase (200 μg/mL) and propidium iodide (20 μg/mL). Cells (n=5000) were collected by FACS (BD FACScalibre) and sequential gating was applied to FSC/SSC then FL2-A/W plots for analysis of cell cycle (as described [Saragovi et al., 1999]). The rate of release from G1 was determined on FL2-H plots using FlowJo software [Jaksch et al., 2008].
Cell migration and proliferation
Fibroblasts were seeded at 1×106 cells in 6well plates and grown to confluence. Prior to experimentation the media on cells was replace with serum free media for 60 min, followed by application of the appropriate drug treatments 90 mins prior to wounding. Scrape wounds (~600μm) were introduced using 100μl pipette tips, throughout the centre of the monolayer and images recorded by timelapse microscopy (as above, 15 min capture for 48 h). Media with fresh treatments was replaced every 12 hours (h) throughout experimentation. Determination of wound closure was performed using MetaMorph analysis software at 12 h timepoints images. At each timepoint regions were created around the edge of the wound and grown to confluent monolayers. Region measurements (in pixels) of the total area inside the wound were normalised to starting wound areas and comparisons were made between control and treated cells at 12 h timepoints. Statistical analysis of differences was measured using one way Anova test with Dunnets post-test, ** = p<0.01.
Results
Sodium butyrate enhances Cx43 expression and function in HeLa-43 cells
To investigate whether enhanced Cx43 expression mediates cell cycle traverse we first determined the effects of NaBu and GA on Cx43 expression and function. Following 24 h exposure to GA, Cx43 expression in HeLa-43 cells was comparable to un-treated cells (Fig. 1A). By contrast, treatment of HeLa-43 cells for 24 h with NaBu significantly increased Cx43 protein expression levels (256 ± 51%) which was not altered in cells co-treated with NaBu and GA (185 ± 27%, Fig. 1A). Under control conditions immunofluorescent analysis identified Cx43 at points of cell to cell contact that measured on average 0.46±0.01 μm in plaque size. The size and localization of these plaques were not altered as a result of exposure to GA, with plaques measuring on average 0.46±0.04 μm (Fig. 1B, Table 1). The relative size of GJ plaques at areas of cell to cell contact were significantly enhanced following exposure to NaBu alone and in combination with GA as compared to the amount of Cx43 detected at membrane locations in control cells (0.86±0.03 μm and 0.74±0.05 μm respectively, Fig. 1B–C). Expression of Cx43 was not detected in HeLa-WT cells (data not shown).
Figure 1. Cx43 expression and function in HeLa-43 cells following exposure to NaBu and/or GA.

Representative Western blots from HeLa-43 cells demonstrate alterations in Cx43 protein expression following 24 h treatments (as labelled) with β-tubulin used for loading controls (A). In B representative immunofluorescent images show Cx43 expression and sub-cellular localization in HeLa-43 cells following treatments (as labelled). Arrows indicate GJ plaques, bar = 20 μm and are representative for all panels. Differences between the total size of GJ plaques following treatments were assessed using Metamorph software (C). In D dye transfer in HeLa-43 cells was measured following treatments (as indicated). HeLa-43 cells were microinjected with Alexa 594, data are expressed as the percentage (of total cells) transferring dye to: 0–4; 5–9; >10 neighbouring cells (n=3, with ~50 cells injected per sitting). Data of the mean ±SE were analysed for significance compared to controls *=p<0.05, **=p<0.01.
To assess Cx43 channel function following exposure to NaBu and/or GA, cells were microinjected with the fluorescent tracer Alexa 594. Under control conditions 15% of injected HeLa-43 cells transferred the dye to >10 neighbouring cells (Fig. 1D). Exposure to NaBu significantly increased dye transfer with ~75% of injected cells transferring dye to >10 cells (Fig. 1D). By contrast, treatment of the cells with GA or NaBu in combination with GA attenuated coupling with ~90% of cells exhibiting restricted transfer to <4 neighbouring cells (Fig. 1D). HeLa-WT cells did not transfer the Alexa 594 under any of the conditions (data not shown). Thus, although GA had no impact on the level of Cx43 expression it effectively attenuated dye transfer under control and NaBu conditions. Cell viability following 24 h exposure to NaBu and GA was confirmed by PI uptake and FACS analysis where no significant alterations were observed between control and treated conditions (data not shown).
Cx43 expression increases intra-mitotic durations independently of GJ channel function
To determine the impact of Cx43 expression on cell cycle traverse we initially adopted a timelapse microscopy approach that enabled tracking of cell cycle progression at the single cell level. Cells were imaged every 5 min for up to 24 h under control or treated conditions. The intra-mitotic duration (IrMD) for each cell in the field of view was calculated as the time taken for a cell to enter mitosis, split into two daughter cells and flatten back down, providing a graphical analysis of the mitotic duration throughout the course of the experiment (Fig. 2A, B). To enable direct comparisons to be made between cell types e.g. HeLa-WT and HeLa-43 and treatment groups the IrMDs were grouped according to their duration and expressed as a percentage of the overall mitotic events (Fig. 2C), thus permitting the mean intra-mitotic duration to be extracted and direct comparisons between cell types and treatment groups to be made (Fig 2; see materials and methods for further details). This initial analysis revealed that HeLa-WT cells had a shorter IrMD duration than HeLa-43 cells (Fig. 2C). The mean IrMD of HeLa-43 cells under control conditions was approximately 70 min which was unaffected by blocking GJ mediated communication with GA (Fig. 3A). Following exposure to NaBu, and upregulation of Cx43 expression, the IrMD increased to 80 min in the presence or absence of GA (Fig. 3A). To control for effects of both these agents in the absence of Cx43 expression equivalent experiments were performed in parental HeLa-WT cells. IrMD was significantly shorter in these cells at approximately 60 mins and was not affected by exposure to NaBu or GA (Fig. 3B). Together these results indicate that Cx43 expression can delay cell cycle progression by extending the time taken to progress through mitosis by a mechanism that depends on the level of Cx43 expression and not channel function. This data supports the emerging concept that enhanced Cx43 expression can slow down cell growth rates independently of channel function [Ionta et al., 2009] and dissects this for the first time at a single cell level.
Figure 3. Mean IrMD of HeLa-43 and HeLa-WT cells following treatment with NaBu and or GA.
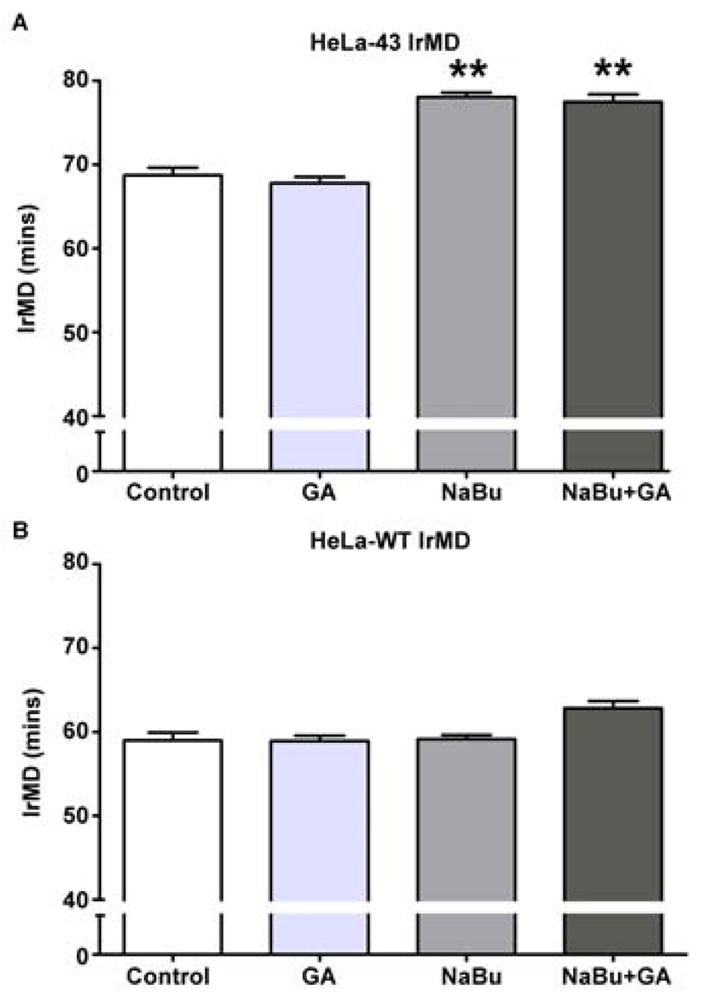
HeLa-43 (A) and HeLa-WT (B) cells treated with NaBu and/ or GA were analysed by 24 h timelapse microscopy. Data of the mean IrMD ±SE for each treatment were analysed for significance as compared to controls (A, B). In each experiment **=p<0.01, n=3 for all treatments.
Enhanced Cx43 expression induces G1 arrest
The impact of altered Cx43 expression on cell cycle dynamics were further dissected by analysis of G1 traverse by capturing cells in G2/M following colcemid treatment and flow cytometry analysis. Following colcemid treatment of HeLa-WT cells the population of cells in G1 was reduced to 5.6±0.4% of the starting population after 24 h (Fig. 4A). Exposure to GA and or NaBu did not alter the rate of emptying of the G1 pool (Fig. 4A), with 50% of cells exiting G1 by 7 h. Under control conditions in HeLa-43 cells, colcemid treatment reduced the G1 population to 6.9±0.2% after 24 h (Fig. 4B), with 50% of cells exiting G1 by ~7 h and this was not altered by exposure to GA (Fig. 4B). By contrast, treatment of HeLa-43 cells with NaBu significantly reduced the rate of G1 traverse with 34.7±2.6% of cells remaining in G1 following colcemid exposure and required up to 13 h for 50% of cells to be released from G1. Inhibiting GJ mediated communication with GA did not alter the rate of emptying of the G1 pool (Fig. 4B). This G1 delay was associated with a significant increase in the expression of Cx43 and the cell cycle inhibitor protein p21waf1/cip1 (Fig. 5A–B). By contrast, HeLa-WT cells under control conditions did not express p21waf1/cip1 and treatments with NaBu induced only low levels of p21waf1/cip1 expression (Fig. 5A–B). Thus enhanced Cx43 expression produced a G1 delay in the HeLa-43 cells with a temporal increase in p21waf1/cip1 expression and suggests that the G1/S checkpoint is induced by enhanced Cx43 expression independently of channel function.
Figure 4. Cell cycle analysis in HeLa-43 and HeLa-WT cells.
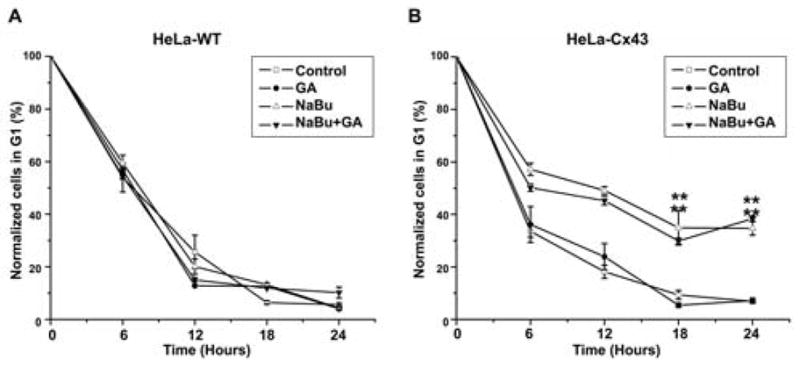
Cell cycle was assessed using flow cytometry of cells treated with NaBu and/ or GA and co-incubated with colcemid. Data of percentage cells (±SE) in G1 at specified timepoints were plotted for each treatment in HeLa-WT cells (A) and HeLa-43 (B).
Figure 5. The effect of NaBu on Cx43 and p21 expression in HeLa cells.
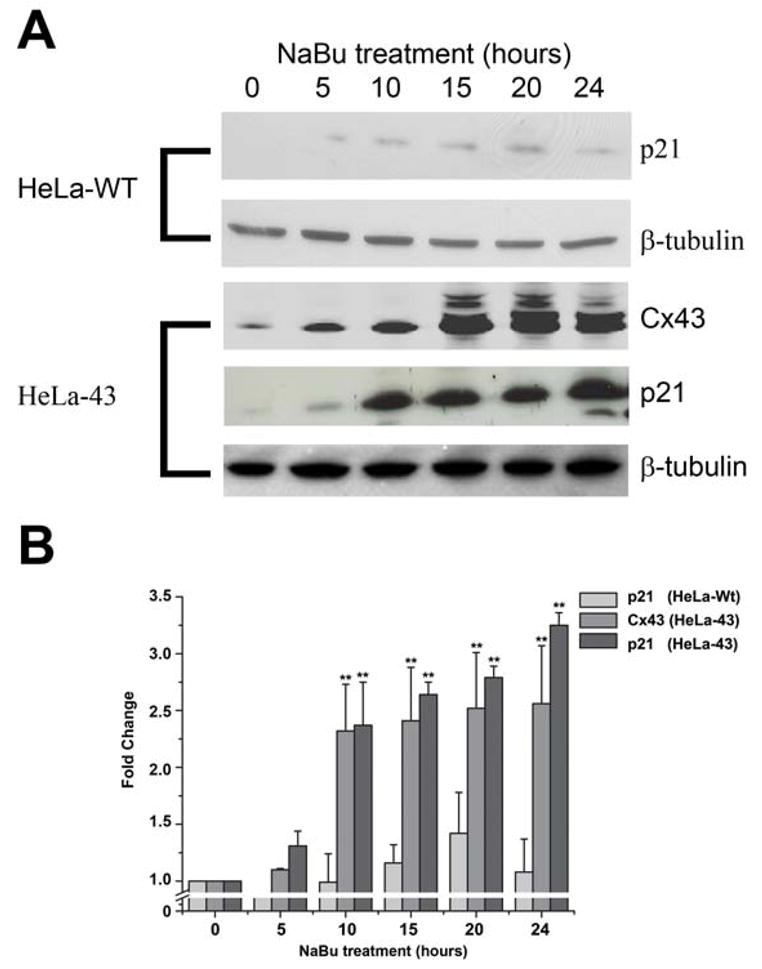
Representative Western blots show Cx43 and p21 expression in HeLa-WT and HeLa-43 cells treated with NaBu over a 24 h timecourse (A). Levels of Cx43 and p21 protein expression as compared to controls were determined for each treatment, with β-tubulin was used as an internal standard control, (n=3, B). In each experiment **=p<0.01, n=3 for all treatments.
Enhanced Cx43 expression reduces rates of cell cycle traverse
To further examine the effects of enhanced Cx43 expression on the rate of cell cycle traverse HeLa cells were treated with AAP10, a peptide that post-translationally enhances Cx43 expression [Easton et al., 2009], as opposed to NaBu which exerts transcriptional control. Timelapse microscopy analysis revealed that exposure of the HeLa-43 cells to AAP10 over 24 h significantly reduced the rate of cell cycle traverse with an elongation in IrMD, producing similar results to that observed following exposure to NaBu (Fig. 6A). Treatment of HeLa-43 cells with AAP10 resulted in a transient upregulation of Cx43 protein expression (Fig. 6B–C) and enhanced targeting of Cx43 to regions of cell to cell contact (data not shown, e.g. [Easton et al., 2009]). A significant but transient increase in the expression of p21waf1/cip1 was also observed, that reduced in line with Cx43 expression (Fig. 6B–C). AAP10 treatments produced no alteration on p21waf1/cip1 expression levels in HeLa-WT cells, (Fig. 6B–C).
Figure 6. The effect of AAP10 on IrMD, Cx43 and p21 expression profiles in HeLa cells and primary fibroblasts.

HeLa-43 cells treated with AAP10 were analysed by timelapse microscopy for changes in mean IrMD as compared to non-treated cells (A). Representative Western blots show induction of transient increases in Cx43 expression following AAP10 treatments in both HeLa-43 cells (B–C) and to a lesser extent in HFF cells (D–E). In both cases this was associated with transient increases in p21 expression, which was not found in HeLa-WT cells (B–C). In B, HeLa-43 cells treated with NaBu and HeLa-Wt cells under control conditions were used as p21+ and p21− controls (respectively) as previously described (B). In each experiment *=p>0.05, **=p<0.01, n=3 for all treatments.
To demonstrate these effects were not restricted to the behaviour of exogenously expressed Cx43 we characterized the effects of AAP10 in primary human dermal fibroblasts (HFF) that primarily express Cx43 [Wright et al., 2009]. Microinjection analysis determined that exposure of HFF cells to AAP10 enhanced GJ mediated communication with ~40% of cells transferring Alexa594 to >10 cells compared to only 5% of non treated cells (Fig. 7A). We have previously reported that GA can block AAP10 induced GJ mediated communication [Clarke et al., 2006; Easton et al., 2009]. Following introduction of a ‘scrape wound’ [Wright et al., 2009] to a confluent monolayer of HFF cells, cell migration and proliferation into the ‘gap’ was monitored over 48 h. Cells treated with AAP10 took up to 6 h longer than control cells to achieve 50% ‘scrape wound’ closure, illustrating delays in cell migration and or proliferation potential (Fig. 7B). Similar effects were seen following treatment of HFF cells with NaBu. These events correlated with a transient increase in Cx43 and p21Waf1/Cip1 expression levels, peaking at 15 h (~2.5 fold, and ~4 fold, respectively) (Fig. 6D–E), again reinforcing the concept that enhanced Cx43 expression delays cell proliferation events in diverse cell types.
Figure 7. The effect of AAP10 on Cx43 expression and function and cell growth dynamics in HFF.
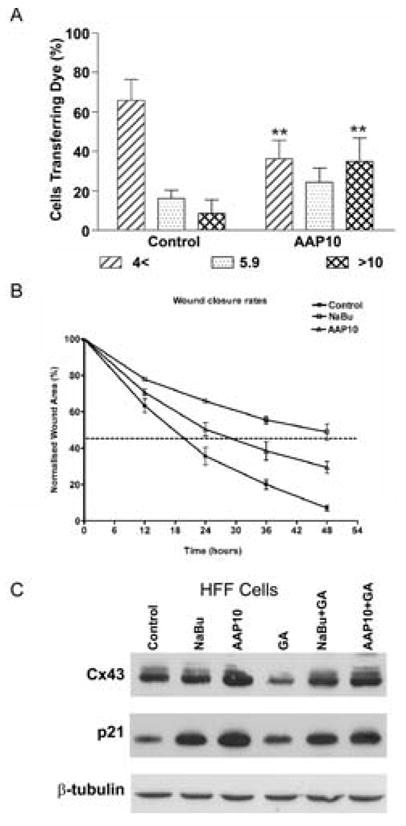
Dye transfer in HFF cells following treatment with AAP10 was assessed by the extent of transfer of Alexa 594 following microinjection. Data of the mean percentage (of total cells ± SE) transferring dye to: 0–4; 5–9; >10 neighbouring cells (n=3, with ~50 cells injected per sitting) were analysed for significance compared to controls (A). A ~600 μm scrape wound was introduced to confluent monolayers of HFF cells in the presence or absence of AAP10 or NaBu and wound closure monitored over 48 h by timelapse microscopy with wound closure was assessed at 5 h intervals (B). Both AAP10 and NaBu significantly impeded fibroblast wound closure rates with the time to reach 50% closure (dotted lines) increased by 6 h and 30 h (respectively) as compared to control cells (B, n=3 and **=p<0.01). HFF cells exposed to NaBu or AAP10 in the presence or absence of GA for 15 h were analysed by Western blot for the expression of Cx43, p21 and β-tubulin as a loading control determined (C).
Finally, to correlate the observed NaBu and GA effects in HeLa-43 cells, HFF cells were treated with NaBu or AAP10 in the presence or absence of GA and Cx43 and p21Waf1/Cip1 expression levels assessed. Exposure of the cells to NaBu or AAP10 for 15 h enhanced Cx43 expression with corresponding increases in p21Waf1/Cip1 expression (Fig. 7C). Treatment of these cells with GA corresponded to a slight reduction in Cx43 expression. Exposure of the cells to NaBu or AAP10 in the presence of GA maintained elevated levels of Cx43 and p21Waf1/Cip1 (Fig. 7C). This data further suggests a correlation between Cx43 expression and p21Waf1/Cip1 as seen in the HeLa-43 cells.
Discussion
In pathophysiological states such as carcinogenesis, atherosclerotic disease and chronic wound healing events, Cx43 expression is altered suggesting that it may be a factor in pathological cell proliferation [Johnstone et al., 2009; Wang et al., 2007; Wright et al., 2009]. Alterations in both Cx43 protein expression and its ability to pass low molecular weight fluorescent dyes (i.e., GJ mediated communication) have been implicated in control of cell growth responses yet it remains unclear what the key factors are in controlling cell cycle events. In the present work we increased Cx43 protein expression and simultaneously down regulated channel function, thus dissecting individual effects on cell cycle progression. Using timelapse microscopy we demonstrated that while Cx43 protein expression could be enhanced by transcriptional activation using NaBu or post translationally using AAP10, both correlated with reduced mitotic delivery rates independently of Cx43 channel function; these studies identified that this delay in cell cycle traverse corresponded with 1. increased p21waf1/cip1 expression, 2. accumulation of cells in G1-phase and 3. an increase in cellular mitotic duration.
Throughout our studies we used NaBu to increase Cx43 expression, in keeping with previous studies that the HDAC inhibitor can exert transcriptional activation of Cx43 in immortalized and primary cells [Khan et al., 2007; Martin et al., 2001]. Using our model systems we confirmed that NaBu can enhance Cx43 expression via the SV40 early promoter in HeLa-43 and from the endogenous Cx43 gene in HFF cells, and that this correlated with enhanced channel function, in keeping with previous studies [Martin et al., 2001]. Further, our data also show that enhanced Cx43 expression correlated with a delay and eventually an arrest in the G1-phase of the cell cycle. Previous investigations have shown that cell cycle specific changes in Cx43 expression are typified by internalisation and specific phosphorylation of sites on the Cx43 carboxyl tail resulting in significantly decreased GJ function in G0 [Solan et al., 2003; Xie et al., 1997]. The carboxyl tail of Cx43 may play an important role in its ability to inhibit cell proliferation, with emphasis on the MAPK phosphorylated sites including Serines 262/279/282 [Dang et al., 2006; Johnstone et al., 2009]. In keeping with our findings, previous studies by Alexander et al. also demonstrated that re-introduction of Cx43 protein rescued growth control independently of GJ function, further emphasizing that Cx43 expression is more important than channel function in control over cell proliferation events [Alexander et al., 2004]. Our studies show that cell cycle inhibition and upregulation of p21Waf1/Cip1 protein expression associated with increased Cx43 expression was not affected by attenuating channel function. Indeed, treatment of cells with GA, reducing GJ mediated communication, did not alter p21waf1/cip1 in either NaBu or AAP10 treated cells in keeping with previous reports [Park et al., 2008]. This further implicates enhanced Cx43 expression but not GJ mediated communication as an important factor in reducing cell proliferation rates. Our data therefore strongly suggests a link between Cx43 upregulation as an inhibitor of cell cycle through enhanced p21Waf1/Cip1 and confirm previous findings that p21Waf1/Cip1 expression associated with reduced Ki67 expression in Cx43 over-expression models regulates cell proliferation [Herrero-Gonzalez et al., 2009]. While p21Waf1/Cip1 is generally considered a cell cycle inhibitor protein, low levels of p21Waf1/Cip1 may act to promote cyclin-cdk complex formation and aid progression through the cell cycle, potentially explaining why p21Waf1/Cip1 expression did not correlate with reduced cell proliferation in our HeLa-WT cells [LaBaer et al., 1997]. The level of p21Waf1/Cip1 present can significantly affect the binding and kinase activity of these complexes with high levels acting to inhibit cyclin-cdk interactions resulting in reduced proliferation in keeping with our data [LaBaer et al., 1997].
Connexin 43, p21waf1/cip1 and the SV40 early promoter, driving Cx43 expression in the HeLa-43 cells, are regulated through Sp1 binding sites in their promoter regions, therefore we must consider the possibility of transcriptional upregulation by the pharmacological agents as opposed to interaction pathways between Cx43 and p21Waf1/Cip1 [Albrecht et al., 1989; Gartel and Tyner, 1999; Teunissen et al., 2003]. Our NaBu data was inconclusive in this regard, but data from AAP10 treated cells clearly demonstrated that p21Waf1/Cip1 was only enhanced when Cx43 was present in HeLa cells. Furthermore, the transient nature of the Cx43 induction was mirrored by a transient increase in p21Waf1/Cip1 expression. We have recently demonstrated that Cx43 upregulation via AAP10 is post-translationally mediated [Easton et al., 2009] and real time PCR analysis confirms that there are no transcriptional changes (data not shown). AAP10 and its stable analogue Rotigaptide have been intensely studied in the cardiovascular system where their anti-arrhythmic properties and acute impact on Cx43 coupling were first observed [Kjolbye et al., 2007]. The acute enhancement of coupling can be blocked by channel blockers such as GA [Clarke et al., 2006] and is dependent on a protein kinase C (PKC) mechanism [Easton et al., 2009]. By contrast, the enhanced expression of Cx43 that occurs following prolonged exposure to AAP10 is independent of PKC activity [Easton et al., 2009]. The present data points to the peptide having further properties in controlling cell growth rates through maintaining enhanced Cx43 expression, although the mechanism underlying this event remains unresolved. This adds credence to suggestions that Cx43 expression may provide a ‘focal platform’ or hub for controlling and co-ordinating gene expression networks related to cell growth responses in cellular populations [Johnstone et al., 2009]. Indeed Cx43 does not exist in the plasma membrane in a niche of its own, but rather as an integral member of a complex ‘junctional nexus’ rich in other cell to cell adhesion proteins including cadherins and occludin, all of which interact with cytoskelatal linking proteins including Z0-1, Z0-2, alpha and beta catenins [Laird, 2006]. The rapid half life of Cx43 may be delayed by its increased presence thereby having subsequent consequences on signalling cascades alerting a transition to enter the mitotic state.
Both NaBu and AAP10 treated cells produced increases in p21Waf1/Cip1 that correlated not only with reduced proliferation rates but an increase in IrMD. While p21Waf1/Cip1 is generally considered an inhibitor of the G1 phase of the cell cycle, studies by others have shown that p21Waf1/Cip1 induced cell cycle checkpoints lead to a reduced ability of cells to enter mitosis [Chang et al., 2000]. Timelapse and necrosis assays demonstrated that NaBu and AAP10 induced p21Waf1/Cip1 expression in a cell death-independent manner in keeping with previous reports that p53 independent pathways also promote such increases [Miyazaki et al., 2008]. These observations remain consistent with those shown in Phase 2 clinical trials of the HDAC inhibitors Suberoylanidide hydroxamic acid (SAHA) and NaBu, which enhance p21Waf1/Cip1 expression in patients and are not associated with alterations in p53, Bax or BCl-2 apoptotic proteins [Duvic et al., 2007].
Previous lines of investigation suggest that decreased Cx43 expression results in increased neoplastic cell growth in mouse lung pneumocytes isolated from Cx43 heterozygous mice [Avanzo et al., 2007]. The consequences of a loss of these interactions can be clearly seen with decreased Cx43 expression resulting in uncontrolled cellular proliferation in Cx43 knockout mice [Avanzo et al., 2007], by Cx43-siRNA in cell culture [Herrero-Gonzalez et al., 2009] and in many forms of cancer [Mesnil et al., 2005]. Taken together with the data presented, we propose that upregulation of Cx43 expression delays the rate of cell cycle traverse by inhibiting the rate of delivery from G1 thereby extending the time between mitotic events and in so doing reducing cell proliferation rates.
In conclusion, our data supports the hypothesis that Cx43 is an important regulator of cell cycle progression. Furthermore, by dissecting these events at the single cell level we show for the first time that this is related to increased mitotic durations and not likely due to changes in gap junctional communication. Understanding how Cx43 regulates p21Waf1/Cip1 expression and other cell cycle inhibitors will be important in dissecting the mechanisms underlying aberrant cell proliferation in a variety of pathophysiological conditions including carcinogenesis, atherogenesis and a variety of epidermal hyperproliferative disorders.
Acknowledgments
We would like to thank Dr Susan Jamieson for supply of the HeLa cells, Dr Michael Edward for the supply of tissue for isolation of human skin samples, Dr Eve Kandyba for help with isolation of the primary HFF cells. We thank Zealand Pharma for the supply of AAP10 and we gratefully acknowledge the expertise and assistance provided by the School of Medicines Flow Cytometry Core at the University of Virginia. SJ was supported by a GCU studentship and research award from The Carnegie Trust for the Universities of Scotland (SJ). This work was also supported by the BBSRC (Grant no C23049, PEM and RJE), an American Heart Association Scientist Development Grant (BEI), NIH RO1088554 (BEI), and Chief Scientist Office (CZB/606/04, PEM).
Abbreviations
- Cx
Connexin
- GJ
Gap junction
- NaBu
Sodium butyrate
- GA
18-α-glycyrrhetinic acid
- AAP10
Anti-arrhythmic peptide
- IrMD
Intra-mitotic duration
- HFF
Human foreskin fibroblasts
Footnotes
The Authors have no conflict of interest to declare
References
- Aasen T, Graham SV, Edward M, Hodgins MB. Reduced expression of multiple gap junction proteins is a feature of cervical dysplasia. Mol Cancer. 2005;4:31. doi: 10.1186/1476-4598-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht GR, Cavallini B, Davidson I. Detection of specific protein binding to the SV40 early promoter in vivo. Nucleic Acids Res. 1989;17:7945–63. doi: 10.1093/nar/17.19.7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DB, Ichikawa H, Bechberger JF, Valiunas V, Ohki M, Naus CC, Kunimoto T, Tsuda H, Miller WT, Goldberg GS. Normal cells control the growth of neighboring transformed cells independent of gap junctional communication and SRC activity. Cancer Res. 2004;64:1347–1358. doi: 10.1158/0008-5472.can-03-2558. [DOI] [PubMed] [Google Scholar]
- Avanzo JL, Mennecier G, Mesnil M, Hernandez-Blazquez FJ, Fukumasu H, da Silva TC, Rao KV, Dagli ML. Deletion of a single allele of Cx43 is associated with a reduction in the gap junctional intercellular communication and increased cell proliferation of mouse lung pneumocytes type II. Cell Prolif. 2007;40:411–421. doi: 10.1111/j.1365-2184.2007.00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzo JL, Mesnil M, Hernandez-Blazquez FJ, Mackowiak II, Mori CM, da Silva TC, Oloris SC, Garate AP, Massironi SM, Yamasaki H, Dagli ML. Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis. 2004;25:1973–1982. doi: 10.1093/carcin/bgh193. [DOI] [PubMed] [Google Scholar]
- Chang BD, Broude EV, Fang J, Kalinichenko TV, Abdryashitov R, Poole JC, Roninson IB. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000;19:2165–2170. doi: 10.1038/sj.onc.1203573. [DOI] [PubMed] [Google Scholar]
- Clarke TC, Thomas D, Petersen JS, Evans WH, Martin PE. The antiarrhythmic peptide rotigaptide (ZP123) increases gap junction intercellular communication in cardiac myocytes and HeLa cells expressing connexin 43. Br J Pharmacol. 2006;147:486–495. doi: 10.1038/sj.bjp.0706631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang X, Jeyaraman M, Kardami E. Regulation of connexin-43-mediated growth inhibition by a phosphorylatable amino-acid is independent of gap junction-forming ability. Mol Cell Biochem. 2006;289:201–207. doi: 10.1007/s11010-006-9162-2. [DOI] [PubMed] [Google Scholar]
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton JE, Petersen JS, Martin PE. Naunyn Schmiedebergs Arch Pharmacol. 2009. The anti-arrythmic peptide AAP10 remodels Cx43 and Cx40 expression and functioneditor^editors. [DOI] [PubMed] [Google Scholar]
- Errington RJ, Marquez N, Chappell SC, Wiltshire M, Smith PJ. Curr Protoc Cytom. Unit. Chapter 12. 2005. Time-lapse microscopy approaches to track cell cycle progression at the single-cell level. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res. 1999;246:280–289. doi: 10.1006/excr.1998.4319. [DOI] [PubMed] [Google Scholar]
- Hellmann P, Grummer R, Schirrmacher K, Rook M, Traub O, Winterhager E. Transfection with different connexin genes alters growth and differentiation of human choriocarcinoma cells. Exp Cell Res. 1999;246:480–90. doi: 10.1006/excr.1998.4332. [DOI] [PubMed] [Google Scholar]
- Herrero-Gonzalez S, Valle-Casuso JC, Sanchez-Alvarez R, Giaume C, Medina JM, Tabernero A. Connexin43 is involved in the effect of endothelin-1 on astrocyte proliferation and glucose uptake. Glia. 2009;57:222–233. doi: 10.1002/glia.20748. [DOI] [PubMed] [Google Scholar]
- Holden PR, McGuire B, Stoler A, Balmain A, Pitts JD. Changes in gap junctional intercellular communication in mouse skin carcinogenesis. Carcinogenesis. 1997;18:15–21. doi: 10.1093/carcin/18.1.15. [DOI] [PubMed] [Google Scholar]
- Ionta M, Ferreira RA, Pfister SC, Machado-Santelli GM. Exogenous Cx43 expression decrease cell proliferation rate in rat hepatocarcinoma cells independently of functional gap junction. Cancer Cell Int. 2009;9:22. doi: 10.1186/1475-2867-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaksch M, Munera J, Bajpai R, Terskikh A, Oshima RG. Cell cycle-dependent variation of a CD133 epitope in human embryonic stem cell, colon cancer, and melanoma cell lines. Cancer Res. 2008;68:7882–7886. doi: 10.1158/0008-5472.CAN-08-0723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone SR, Ross J, Rizzo MJ, Straub AC, Lampe PD, Leitinger N, Isakson BE. Oxidized phospholipid species promote in vivo differential cx43 phosphorylation and vascular smooth muscle cell proliferation. Am J Pathol. 2009;175:916–24. doi: 10.2353/ajpath.2009.090160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanczuga-Koda L, Sulkowski S, Lenczewski A, Koda M, Wincewicz A, Baltaziak M, Sulkowska M. Increased expression of connexins 26 and 43 in lymph node metastases of breast cancer. J Clin Pathol. 2006;59:429–433. doi: 10.1136/jcp.2005.029272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan Z, Akhtar M, Asklund T, Juliusson B, Almqvist PM, Ekstrom TJ. HDAC inhibition amplifies gap junction communication in neural progenitors: potential for cell-mediated enzyme prodrug therapy. Exp Cell Res. 2007;313:2958–2967. doi: 10.1016/j.yexcr.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Kjolbye AL, Haugan K, Hennan JK, Petersen JS. Pharmacological modulation of gap junction function with the novel compound rotigaptide: a promising new principle for prevention of arrhythmias. Basic Clin Pharmacol Toxicol. 2007;101:215–230. doi: 10.1111/j.1742-7843.2007.00123.x. [DOI] [PubMed] [Google Scholar]
- Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, Wall D, Li E, Gaudet F. Methylation of p53 by Set7/9 Mediates p53 Acetylation and Activity In Vivo. Molecular Cell. 2008;29:392–400. doi: 10.1016/j.molcel.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Kwak BR, Veillard N, Pelli G, Mulhaupt F, James RW, Chanson M, Mach F. Reduced connexin43 expression inhibits atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice. Circulation. 2003;107:1033–1039. doi: 10.1161/01.cir.0000051364.70064.d1. [DOI] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe PD, Kurata WE, Warn-Cramer BJ, Lau AF. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J Cell Sci. 1998;111(Pt 6):833–841. doi: 10.1242/jcs.111.6.833. [DOI] [PubMed] [Google Scholar]
- Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–1186. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez N, Chappell SC, Sansom OJ, Clarke AR, Court J, Errington RJ, Smith PJ. Single cell tracking reveals that Msh2 is a key component of an early-acting DNA damage-activated G2 checkpoint. Oncogene. 2003;22:7642–7648. doi: 10.1038/sj.onc.1206876. [DOI] [PubMed] [Google Scholar]
- Martin PE, Blundell G, Ahmad S, Errington RJ, Evans WH. Multiple pathways in the trafficking and assembly of connexin 26, 32 and 43 into gap junction intercellular communication channels. J Cell Sci. 2001;114:3845–3855. doi: 10.1242/jcs.114.21.3845. [DOI] [PubMed] [Google Scholar]
- McLachlan E, Shao Q, Wang HL, Langlois S, Laird DW. Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res. 2006;66:9886–9894. doi: 10.1158/0008-5472.CAN-05-4302. [DOI] [PubMed] [Google Scholar]
- Mesnil M, Crespin S, Avanzo JL, Zaidan-Dagli ML. Defective gap junctional intercellular communication in the carcinogenic process. Biochim.Biophys. Acta. 2005;1719:125–145. doi: 10.1016/j.bbamem.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Mesnil M, Krutovskikh V, Piccoli C, Elfgang C, Traub O, Willecke K, Yamasaki H. Negative growth control of HeLa cells by connexin genes: connexin species specificity. Cancer Res. 1995;55:629–639. [PubMed] [Google Scholar]
- Miyazaki H, Shiozaki A, Niisato N, Ohsawa R, Itoi H, Ueda Y, Otsuji E, Yamagishi H, Iwasaki Y, Nakano T, Nakahari T, Marunaka Y. Chloride ions control the G1/S cell-cycle checkpoint by regulating the expression of p21 through a p53-independent pathway in human gastric cancer cells. Biochem Biophys Res Commun. 2008;366:506–512. doi: 10.1016/j.bbrc.2007.11.144. [DOI] [PubMed] [Google Scholar]
- Moorby C, Patel M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp Cell Res. 2001;271:238–248. doi: 10.1006/excr.2001.5357. [DOI] [PubMed] [Google Scholar]
- Olbina G, Eckhart W. Mutations in the second extracellular region of connexin 43 prevent localization to the plasma membrane, but do not affect its ability to suppress cell growth. Mol Cancer Res. 2003;1:690–700. [PubMed] [Google Scholar]
- Park JH, Lee MY, Heo JS, Han HJ. A potential role of connexin 43 in epidermal growth factor-induced proliferation of mouse embryonic stem cells: involvement of Ca2+/PKC, p44/42 and p38 MAPKs pathways. Cell Prolif. 2008;41:786–802. doi: 10.1111/j.1365-2184.2008.00552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivedal E, Leithe E. Connexin43 synthesis, phosphorylation, and degradation in regulation of transient inhibition of gap junction intercellular communication by the phorbol ester TPA in rat liver epithelial cells. Exp Cell Res. 2005;302:143–152. doi: 10.1016/j.yexcr.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Sanchez-Alvarez R, Paino T, Herrero-Gonzalez S, Medina JM, Tabernero A. Tolbutamide reduces glioma cell proliferation by increasing connexin43, which promotes the up-regulation of p21 and p27 and subsequent changes in retinoblastoma phosphorylation. Glia. 2006;54:125–134. doi: 10.1002/glia.20363. [DOI] [PubMed] [Google Scholar]
- Saragovi HU, Rebai N, Di Guglielmo GM, Macleod R, Sheng J, Rubin DH, Greene MI. A G1 cell cycle arrest induced by ligands of the reovirus type 3 receptor is secondary to inactivation of p21ras and mitogen-activated protein kinase. DNA Cell Biol. 1999;18:763–770. doi: 10.1089/104454999314908. [DOI] [PubMed] [Google Scholar]
- Schafer KA. The cell cycle: a review. Vet Pathol. 1998;35:461–478. doi: 10.1177/030098589803500601. [DOI] [PubMed] [Google Scholar]
- Singh D, Solan JL, Taffet SM, Javier R, Lampe PD. Connexin 43 interacts with zona occludens-1 and -2 proteins in a cell cycle stage-specific manner. J Biol Chem. 2005;280:30416–30421. doi: 10.1074/jbc.M506799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Fry MD, TenBroek EM, Lampe PD. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J Cell Sci. 2003;116:2203–2211. doi: 10.1242/jcs.00428. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–72. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunissen BE, Jansen AT, van Amersfoorth SC, O’Brien TX, Jongsma HJ, Bierhuizen MF. Analysis of the rat connexin 43 proximal promoter in neonatal cardiomyocytes. Gene. 2003;322:123–136. doi: 10.1016/j.gene.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Wang CM, Lincoln J, Cook JE, Becker DL. Abnormal connexin expression underlies delayed wound healing in diabetic skin. Diabetes. 2007;56:2809–2817. doi: 10.2337/db07-0613. [DOI] [PubMed] [Google Scholar]
- Wright CS, van Steensel MA, Hodgins MB, Martin PE. Connexin mimetic peptides improve cell migration rates of human epidermal keratinocytes and dermal fibroblasts in vitro. Wound Repair Regen. 2009;17:240–249. doi: 10.1111/j.1524-475X.2009.00471.x. [DOI] [PubMed] [Google Scholar]
- Xie H, Laird DW, Chang TH, Hu VW. A mitosis-specific phosphorylation of the gap junction protein connexin43 in human vascular cells: biochemical characterization and localization. J Cell Biol. 1997;137:203–210. doi: 10.1083/jcb.137.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Kaneda M, Morita I. The gap junction-independent tumor-suppressing effect of connexin 43. J Biol Chem. 2003;278:44852–44856. doi: 10.1074/jbc.M305072200. [DOI] [PubMed] [Google Scholar]
- Zhu D, Kidder GM, Caveney S, Naus CC. Growth retardation in glioma cells cocultured with cells overexpressing a gap junction protein. Proc Natl Acad Sci USA. 1992;89:10218–10221. doi: 10.1073/pnas.89.21.10218. [DOI] [PMC free article] [PubMed] [Google Scholar]