

Abstract
Primary hepatocellular carcinoma (HCC) is a significant human cancer globally, with poor prognosis. New and efficacious therapy strategies are needed as well as new biomarkers for early detection of at-risk patients. In this review, we discuss select microarray studies of human HCCs, and propose a gene signature that has promise for clinical/translational application. This gene signature combines the proliferation cluster of genes and the hepatic cancer initiating/stem cell gene cluster for identification of HCCs with poor prognosis. Evidence from cell-based assays identifies the existence of a mechanistic link between these two gene clusters, involving the proliferation cluster gene Polo-like kinase 1 (PLK1). We propose that PLK1 is a promising therapy target for HCC.
Keywords: Microarray gene signatures, proliferation genes, Polo-like kinase 1 (PLK1), Epithelial Cell Adhesion Molecule (EpCAM), Hepatic progenitors and cancer initiating/stem cells, Polycomb Repressive Complex 2 (PRC2)
1. Introduction
Primary liver cancer, hepatocellular carcinoma (HCC), is the fifth most common cancer world-wide [1]. In the Unites States, liver cancer, relative to other cancers, has the most rapidly growing mortality rate [2]. Major etiologic agents in HCC pathogenesis are chronic infection with hepatitis B virus or hepatitis C virus [3], [4], [5]. Other causal factors of lower incidence include alcohol abuse, metabolic disorders, and environmental agents, e.g., exposure to aflatoxin B1 [6]. Regarding HBV-mediated HCC, despite availability of the HBV vaccine, the World Health Organization estimates that globally 400 million people are chronically infected with HBV. Moreover, the HBV vaccine is not always protective and children born of infected mothers also become chronically infected. There is no vaccine for HCV. Current treatments for chronic HBV infection include antiviral nucleoside analogs that eventually result in viral resistance [7]. Treatment for HCV infection includes a combination of interferon and ribavirin [8]. When diagnosed at early stage, HCC remains eligible for potential curative options such as surgical resection, orthotopic liver transplantation or percutaneous destructions. However, most of HCCs have widespread dissemination within the liver at diagnosis (intermediary stage) or show extrahepatic dissemination within the portal tract, lymph nodes or distant visceral metastasis (advanced stage) [9]. Either transarterial hepatic chemoembolization for intermediary stage HCCs, or systemic targeted therapies such as sorafenib – i.e. the anti-angiogenic and anti-MAPK pathway agent – for advanced stage HCCs, are of modest, although significant benefit [10]. As recommended by Llovet and Bruix [11], new and efficacious therapies are needed, along with new diagnostic biomarkers for early detection of liver cancer.
Microarray studies of human tumor samples and bioinformatics meta-analyses continue to provide a wealth of information regarding genes differentially expressed in various types of cancer [12], [13], [14], [15], [16]. For liver cancer, more than 300 microarray studies have been published [17] identifying genes deregulated in HCC, although some of the published studies provide more transparent data than others. The ongoing challenge is to identify and characterize clinically relevant genes that can serve as early biomarkers for detection and classification of the disease or serve as targets for designing mechanism-based therapies. This review focuses on select microarray studies of human liver tumors. Herein we highlight specific HCC gene signatures we consider promising for translational application in diagnosis of at-risk patients. We base this assessment on the link of the proposed HCC gene signatures to established mechanisms of cancer pathogenesis as well as to liver physiology and development. We will present “cancer” and “liver-specific” gene signatures associated with HCC pathogenesis.
1.1. Cancer gene signature: the proliferation gene cluster
Chen and colleagues in 2002 [18] analyzed 102 primary HCC tumor samples and 74 non-tumor samples for differentially expressed genes, using a cDNA microarray representing 17,400 human genes. They demonstrated increased expression of the proliferation cluster of genes required for cell cycle progression. Up-regulated genes include those involved in DNA replication, e.g., the minichromosome maintenance3–7 (MCM3–7) proteins, thymydilate synthase (TYMS), proliferating cell nuclear antigen (PCNA), and those involved in G2/M progression, such as mitotic kinases CDC2, CDC20, Polo-like kinase 1 (PLK1), and mitotic regulators MAD2 and Bub1. Also, they observed decreased expression of liver-specific genes, indicative of hepatocyte de-differentiation and/or loss of liver function. This proliferation gene signature distinguished liver tumors with mutant p53 from those with wild type (WT) p53, and liver tumors with vascular invasion.
In 2004, Thorgeirsson’s group [19] analyzed the gene expression profile of 91 human primary HCC tumors by microarray analyses. They also identified enhanced expression of the proliferation cluster of genes as the best predictor for an unfavorable outcome. This proliferation gene signature convincingly distinguished two groups of HCC patients, Clusters A and B, having significant differences in survival of 30 months vs. 90 months, respectively. The proliferation cluster of genes included: PCNA, Bub3, MCM2, 6, and 7, and cell cycle regulators cyclinA2 (CCNA2), cyclinB1 (CCNB1), CDC2 associated protein2 (CKS2), and cyclin-dependent kinase 4 (CDK4). Additional features of the poor survival HCC Cluster A include reduced expression of liver specific genes, similar to the observations by Chen et al. [18], and enhanced expression of genes involved in proteasomal degradation and the ubiquitin pathway. The ubiquitin pathway is often deregulated in cancer [20]. Together, these genes were termed the survival gene expression signature.
In a subsequent study [21], Thorgeirsson’s group identified by global gene expression analyses of 139 human HCCs two subtypes of HCC. The one subtype exhibited features of hepatoblasts (HB) and the other of differentiated hepatocytes (HC). The strategy for this study involved comparison of gene expression profiles from three different species (human, rat and mouse). Specifically, the gene expression profiles of human HCCs were compared to those of fetal and adult rat hepatocytes, and to mouse HCCs that originated from adult hepatocytes. The mouse hepatocyte-originating HCCs were obtained from the Myc/E2F1 and Myc/TGFα mouse liver cancer models. In these animal models transgenes were expressed from the albumin promoter which is transcriptionally active in differentiated hepatocytes. Importantly, the gene expression profile of rat hepatoblasts in comparison to that of mouse hepatocytes (from the two mouse HCC models) was distinct and well-separated from each other. Interestingly, the gene expression profiles of several human HCC samples co-clustered with rat hepatoblasts suggesting a similar cell developmental stage. This subtype was referred to as the HB subtype and was shown to express markers of hepatic progenitors or “oval cells,” including keratin 7 (KRT7) and keratin 19 (KRT19). Both HB and HC subtypes expressed α-Fetoprotein (AFP) to similar levels. Further analysis of the gene expression profiles by hierarchical cluster analyses, demonstrated that the HB subtype co-clustered with the proliferation group of genes which characterized the poor survival Cluster A of human HCCs [19]. The HC subtype was further subdivided into Cluster A (proliferation signature-positive) and Cluster B (proliferation signature-negative). The poorest survival between HB and HC subtypes in Cluster A was exhibited by the HB subtype.
Interestingly, another microarray study by the Zucman-Rossi group [22] came to the same conclusion. Specifically, this microarray analysis also identified two major clusters of HCC tumors which were sub-grouped into six distinct subgroups (G1–G6). Significantly, these G1–G6 subgroups were also characterized by their association with distinct clinical and molecular/genetic alterations, including viral infection, activation of the PI3K/AKT pathway and p53 mutations, thereby providing additional descriptors for precise classification of HCCs. Subgroups G1–G3 exhibited high rate of chromosomal instability and were associated with poor prognosis. Importantly, HCC subgroups G1–G3 over-expressed proliferation, cell cycle and DNA metabolism genes, similar to the cluster A subtype of HCCs described by Lee et al [19]. Interestingly, subgroups G1 and G2 that originated from patients with chronic HBV infection, low HBV copy number vs. high, respectively, also exhibited over-expression of fetal genes including AFP and parentally imprinted genes, thus resembling the poor prognosis, HB cluster A subtype [21].
1.2. Proliferation gene signature and Polo-like kinase 1 (PLK1)
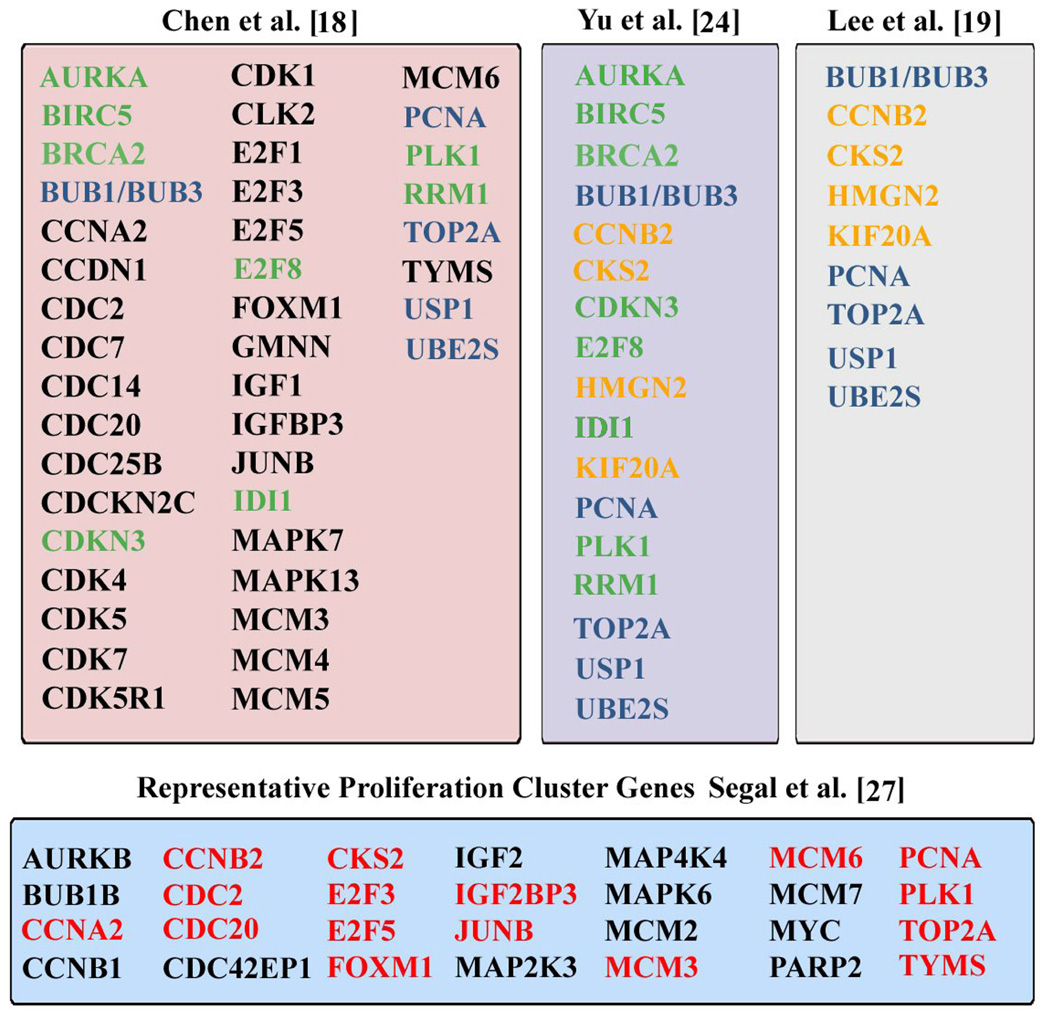
The proliferation gene signatures identified by Chen et al. [18] and Lee et al. [19, 21] overlap the c-myc up-regulated gene signature identified using the non-transgenic mouse model of B-cell lymphoma [23]. The significance of this animal model is that the oncogenic transformation of p53-null bone marrow cells occurred in vivo, following infection with a conditionally active c-myc encoding retrovirus [23], [24]. Accordingly, this experimental design permitted identification of a broader set of proliferation genes, those expressed at earlier times in the process of oncogenic transformation. The relevance of this animal model to liver cancer is that c-myc over-expression also characterizes human HCC [18], [19], [25], including hepatoblastoma, a rare liver cancer in children [26]. Furthermore, the proliferation cluster is shared across diverse human malignancies indicating a common mechanism in tumor progression [27]. Table I shows the proliferation cluster of genes identified in human HCCs by the studies of Chen et al. [18] and Lee et al. [19]. The central column (Table I) includes c-myc up-regulated proliferation genes identified by Yu et al. [24] in the non-transgenic mouse model of B-cell lymphoma. In addition, Table I shows the proliferation cluster of genes identified by Segal et al [27] via meta-analysis of 1,975 microarrays from multiple types of human tumors.
Table I.
Proliferation Cluster Gene Signatures identified using human HCCs [18], [19] and c-myc up-regulated proliferation genes [24]. Green designates the overlap of the proliferation cluster gene signatures in the studies by Chen et al. [18] and Yu et al. [24]. Orange designates overlap of the proliferation cluster gene signatures in Lee et al. [19] and Yu et al. [24]. Blue designates overlap of proliferation genes from the three studies [18], [19], [24]. Red designates overlap between the study by Segal et al. [27] and genes found in the other three columns.
 |
In HBV-mediated hepatocarcinogenesis, studies in animal models [28], [29] have demonstrated that the viral X protein acts as a weak oncogene or a co-factor in liver cancer pathogenesis. The HBV X protein activates cellular mitogenic pathways and consequently deregulates hepatocyte gene expression [30], [31], [32]. Employing a cell-based assay of HBV X protein-induced hepatocyte transformation, we also quantified enhanced expression of the proliferation genes MCM3–7, TYMS, PCNA, and PLK1 as pX-expressing hepatocytes undergo oncogenic transformation [33], [34]. Significantly, we have shown that inhibition of PLK1 suppressed pX-mediated hepatocyte transformation [34]. PLK1 has important roles in cell cycle progression, including regulation of many aspects of mitosis including mitotic entry, centrosome maturation, formation of bipolar spindle, chromosome segregation, activation of the anaphase promoting complex/cyclosome, exit from mitosis, and cytokinesis [35]. In normal cells, PLK1 mediates in the G2/M transition recovery from the DNA damage checkpoint after completion of DNA repair [36]. By contrast, in HBV X-expressing cells PLK1 mediates checkpoint adaptation by attenuating both the DNA damage checkpoint and DNA repair [37]. Specifically, the HBV X protein activates PLK1 in the G2 phase in non-transformed hepatocytes [37]; in turn, activated PLK1 enables propagation of DNA damage to dividing X-expressing cells by concurrently suppressing DNA repair and the DNA damage checkpoint thereby resulting in the generation of polyploidy [37]. Thus, these cell-based studies of the HBV X protein have identified PLK1 as a mechanistically meaningful gene, likely initiating hepatocyte transformation in X-expressing cells. Elevated PLK1 protein has been observed in human liver tumors relative to normal peritumoral tissue [38], [39], including human liver tumors from HBV-HCC patients (F. Lu and O. Andrisani, unpublished data). Therefore, the combination of cell-based [33], [34], [37] and clinical studies [38], [39] identify PLK1, a member of the proliferation gene signature [18], [24], [27], as a mechanistically significant gene having the potential to be clinically relevant for diagnosis and therapy of HCC. Currently, PLK1 inhibitors are in clinical trials for various types of human cancers [35], [40], [41]. Therefore, PLK1 could serve as therapy target for HCC. We propose the proliferation signature and PLK1 are promising diagnostic and prognostic markers for determination of poor prognosis HCC.
2. Hepatic progenitor-specific and EpCAM-positive gene signature in HCC pathogenesis
As described earlier, section 1.1, Lee et al. [21] identified the hepatoblast HB-specific subgroup of HCCs by comparative analyses of the gene expression profile of fetal hepatoblasts vs. hepatocyte-originating HCCs. Employing a different microarray strategy, Kim et al. [42] from Xin Wei Wang’s laboratory identified the involvement of hepatic progenitors in HCC pathogenesis. Specifically, a 30 gene signature was identified that becomes significantly altered in patients with preneoplastic chronic liver disease (hepatitis, fibrosis and cirrhosis). A principal gene, displaying the largest fold increase in expression was TACSTD1 encoding the epithelial cell adhesion molecule EpCAM, a stem cell antigen, exhibited elevated expression in tumors of epithelial origin [43]. Earlier observations by De Boer et al. [44] demonstrated that adult hepatocytes are EpCAM-negative, while the bile duct epithelium is EpCAM-positive. Interestingly, expression of EpCAM was observed during fetal liver development, liver regeneration, and liver repair associated with cirrhosis. More recent studies have shown that indeed EpCAM is a marker of hepatic progenitors, suggesting that EPCAM-positive HCCs are of hepatic progenitor cell origin. Hepatic progenitors include both hepatic stem cells (HpSCs) and the committed hepatoblasts [45], [46], [47]. Table II shows markers that characterize hepatic stem cells, hepatoblasts, and differentiated hepatocytes. EpCAM-positive and AFP-negative cells characterize hepatic stem cells located in the canals of Herring in the adult liver; EpCAM-positive and AFP-positive characterize hepatoblasts. The hepatoblast cell number is reduced with age, except during cirrhosis and HCC [48]. Alpha fetal protein (AFP) is a liver specific gene, expressed exclusively in embryonic liver [48]. AFP is a known prognostic indicator for HCC that is found elevated in 60% of HCC patients [49].
Table II.
Markers of Hepatic progenitors and Cancer initiating cells
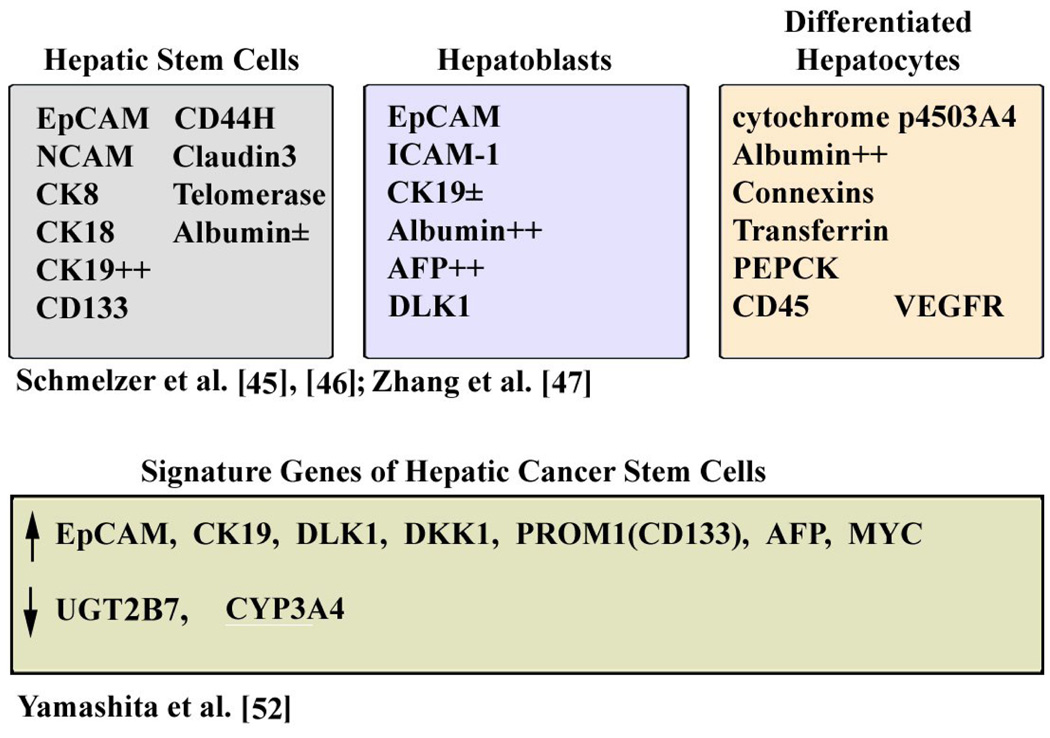 |
Significantly, studies by Yamashita et al. [25] demonstrated that enhanced expression of EpCAM and EpCAM co-expressed genes is prognostic of subtypes of HCCs. Microarray studies and immunohistochemistry classified HCCs into EpCAM-positive and EpCAM-negative subtypes, with high accuracy. The EpCAM-positive gene signature is comprised of 70 genes and exhibits increased expression of marker genes characterizing hepatic progenitors. These include cytokeratin 19 (CK19), c-kit and Wnt signaling-induced genes DKK1 [50] and BAMBI [51]. Also, the EpCAM-positive gene signature exhibited down-regulated expression of the hepatocyte-specific genes, including UGT2B7 (Uridine diphosphate glycosyl transferase 2) and APOC1 (Apolipoprotein C1). The gene expression of BAMBI and DKK1 positively correlated with EpCAM expression, whereas, expression of the hepatocyte-specific genes UGT2B7 and APOC1 was inversely linked to EpCAM expression [25]. Furthermore, the EpCAM-positive subtype was further subgrouped to AFP-positive and AFP-negative tumors. HCCs that were EpCAM-positive but AFP-negative co-clustered with higher levels of CK19 and CK7 expression indicating that they resemble HpSCs. On the other hand, HCCs that were positive for both EpCAM and AFP markers abundantly co-expressed DKK1, BAMBI, and the hepatoblast markers DLK1 (Delta like-1) and PROM1 (Prominin or CD133 antigen). Thus, these subtypes of HCC tumors express markers characteristic of specific stages of hepatic cell lineages (Table II). Recent studies have demonstrated that EpCAM-positive hepatocellular carcinoma cells are tumor initiating cells or Cancer Stem Cells (CSCs) [52]. EpCAM-positive cells isolated by FACS from human HCCs self-renewed, differentiated in vitro and formed large tumors in NOD/SCID mice [52]. These are considered features of cancer initiating stem cells. Key genes up-regulated in hepatic CSCs (EpCAM-positive) isolated from human HCC tumors include: TACSTD1, CK19, DLK1, DKK1, PROM1, AFP, and MYC. Down-regulated genes include liver-specific genes UGT2B7 and CYP3A4 expressed in higher levels in EpCAM-negative cells [52] and Table II.
2.1 Role of EpCAM in cancer and stem cell signaling
Elevated EpCAM expression has been identified in many epithelial cell-derived human cancers and cancer initiating cells [53]. Elevated EpCAM expression is linked to poor prognosis for various types of human cancer, including HCC [25]. Moreover, EpCAM is highly expressed in undifferentiated human embryonic stem cells [54]. EpCAM enhances cell proliferation by increasing expression of c-myc, cyclinA, and cyclinE [55]. Regarding the mechanism by which EpCAM affects proliferation and cancer development, recent studies [56] have shown that EpCAM participates in intracellular signaling by undergoing intramembranous proteolysis via the action of the TNF-α converting enzyme (TACE) and presenilin-2(PS-2). Proteolysis of EpCAM results in release of its intracellular domain, EpICD. In turn, the released EpICD associates with components of the Wnt pathway, β-catenin and LEF-1, and regulates gene transcription, inducing proliferation and tumorigenesis in mice. Therefore, the study by Maetzel et al. [56] provides a mechanistic link between expression of EpCAM, proliferation of stem cells, and cancer development by cancer initiating cells after aberrant EpCAM re-expression. This conclusion raises the question of mechanism(s) regulating expression of EpCAM.
2.2 Expression of EpCAM : Regulation by the Polycomb (PRC2) chromatin modifying complex
In human embryonic stem cells (hESCs), expression of EpCAM is regulated by epigenetic histone modification mechanisms that involve dynamic changes in trimethylation of lysine 27 of histone 3, H3K27me3 [54]. H3K27me3 silences gene transcription. Upon hESCs differentiation, the EpCAM promoter is transcriptionally silenced via the repressive H3K27me3 modification, mediated by the interplay of SUZ12 and JMJD3 [54]. SUZ12 is an essential component of the chromatin modifying Polycomb Repressive Complex (PRC2). PRC2 is a histone methyltransferase comprised of EZH2, EED, and SUZ12. PRC2 proteins bind to specific regions of DNA and direct repressive posttranslational modification of H3K27me3, mediating epigenetic regulation of gene expression in development, differentiation, and maintaining cell fate [57], [58]. Conversely, JMJD3 is a histone demethylase of H3K27me3 [59]. Whether deregulation of this interplay between SUZ12 and JMJD3 results in EpCAM re-expression in cancer initiating cells and cancer pathogenesis remains to be determined. Likewise, the molecular mechanism(s) regulating the activity or protein levels of the PRC2 complex and JMJD3, during cancer development, remain to be understood. A recent review by Bracken and Helin [60] addresses how deregulation of polycomb group proteins could contribute to tumorigenesis.
3. Gene targets of the PRC2 complex relevant to HCC pathogenesis
In human embryonic fibroblasts, Bracken et al. [61] identified more than 1000 genes that are transcriptionally silenced by the PRC2 complex, including TACSTD1/EpCAM. We reasoned that these data [61] provide important mechanistic insights regarding genes and pathways that can be deregulated by the PRC2 complex and contribute to HCC pathogenesis. Accordingly, in Table III, we provide a list of select PRC2 target genes identified in human embryonic fibroblasts [61] which are relevant to HCC. These include: 1) TACSTD (EpCAM) and IGFII; these two genes were shown by independent studies to be targets of epigenetic regulation by the PRC2 complex [54], [62]. The significance of EpCAM overexpression in HCCs has been presented in section 2. Regarding IGFII, it is upregulated in poor prognosis human HCCs such as the G1 subtype identified by Boyault et al. [22], which over-express both the proliferation and fetal gene signatures. Another study [63] also identified IGFII over-expression in human HCCs; these investigators also noted an inverse relationship between IGFII expression and expression of interferon (IFN) regulated genes, although the mechanism for this exclusive relationship is not understood [63]. 2) DKK1, 2 [50] and BAMBI [51] have been found to be up-regulated in human HCC samples [18, 25, 52] and hepatoblastomas [26]. Their elevated expression positively correlated with EpCAM expression and was linked to poor prognosis [25, 52]. 3) Myc, ATF3, HesX1, and KLF1 are up-regulated in human HCCs, as shown by Lee et al. [21] and Yamashita et al. [25]. 4) Wnt 1, 2, 3, 6, 10, 11, and LEF-1 are components of the Wnt signaling pathway and may contribute to the activation of Wnt signaling as observed by Yamashita et al. [25]. 5) RPA-2, MCM-5, CCNA1, and CCND2 are members of the proliferation gene signature discussed in section 1.1, and 6) TERT (telomerase) is the hallmark of cancer cells [64] as well as a feature determining stem cell fate [45], [65].
Table III.
PRC2 target genes of relevance to HCC
 |
The overlap of the PRC2 target genes (Table III) with the EpCAM-positive, hepatic cancer stem cell signature (Table II) and the proliferation signature (Table I) suggests that these two gene signatures identified in human HCCs are connected via a common mechanism. We propose that this mechanism involves loss of gene silencing via H3K27me3, leading to deregulated expression of genes, including EpCAM, IGFII, BAMBI, DKK1, 2, and enhanced expression of the proliferation cluster including c-myc, MCM-5, CCNA1 and CCND2 genes. Whether changes in the pattern of H3K27me3 occur during HCC pathogenesis is unknown. Thus, it will be important to determine changes in the pattern of H3K27me3 in human HCCs and, in turn, to determine mechanisms leading to loss/down-regulation of PRC2 components and/or enhanced demethylation of H3K27me3. Recent studies by Helin’s group demonstrated enhanced expression of JMJD3 demethylase in response to senescence induced by expression of the oncogene BRAF [66]. This study raises the possibility that upregulation of JMJD3 may also be involved in HCC pathogenesis. Earlier studies [59] demonstrated histone demethylase JMJD3 is quickly induced by NF-κB in primary mouse macrophages in response to inflammatory stimuli. Since human HCC is usually preceded by chronic liver inflammation and cirrhosis, it will be very interesting to determine the expression level of JMJD3 demethylase in chronic liver disease such as cirrhosis, and link its role to HCC pathogenesis. Conversely, down-regulation of PRC2 components may turn out to be another likely mechanism. Our studies (Wang et al., submitted) have identified SUZ12 as a gene whose protein levels are down-regulated in an in vitro HBV pX-mediated cellular transformation model [34], in HCC cell lines, and in human HCCs (F. Lu and Andrisani unpublished observations), while PLK1 protein levels increase. This inverse/exclusive relationship between protein levels of PLK1 and SUZ12 suggests that a regulatory mechanism links PLK1, a proliferation cluster gene, to down-regulation of SUZ12, a likely regulator of the hepatic cancer initiating/stem cell gene cluster.
4. Concluding remarks
In this review we have highlighted the significance of two gene signatures identified in human HCCs. These are the proliferation cluster gene signature and the EpCAM-positive, hepatic cancer stem cell gene signature. We have summarized results that strongly suggest these two gene signatures are linked mechanistically, via H3K27me3 demethylation. More work is needed to understand the molecular mechanisms deregulating H3K27me3 demethylation during oncogenic transformation and HCC pathogenesis, employing liver cancer animal models and human HCC tissues. The mechanistic connection between the proliferation cluster and the hepatic cancer stem cell gene signatures renders these gene signatures promising as clinical prognosticators for HCC.
Acknowledgments
The authors thank Drs. MA Buendia and RL Hullinger for critical reading of the manuscript. This work has been supported by NIH grants DK044533 and CA135192 to OMA. LS supported by the Bilsland’s graduate student fellowship of Purdue University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1.Parkin DM. International variation. Oncogene. 2004;23:6329–6340. doi: 10.1038/sj.onc.1207726. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 3.Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2:1129–1133. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- 4.Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215–219. doi: 10.1016/s1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 5.Cougot D, Neuveut C, Buendia MA. HBV-induced hepatocarcinogenesis. J Clin Virol. 2005;34 Suppl 1:S75–S78. doi: 10.1016/s1386-6532(05)80014-9. [DOI] [PubMed] [Google Scholar]
- 6.Llovet JM, Burroughs A, Bruix J. Hepatocellular Carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 7.Zoulim F, Poynard T, Degos F, Slama A, El Hasnoui A, P. Blin F, et al. A prospective study of the evolution of lamivudine resistance mutations in patients with chronic hepatitis B treated with lamivudine. J Viral Hepatitis. 2006;13:278–288. doi: 10.1111/j.1365-2893.2005.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 9.Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, et al. Design and Endpoints of Clinical Trials in Hepatocellular Carcinoma. J Natl Cancer Inst. 2008;100:698–711. doi: 10.1093/jnci/djn134. [DOI] [PubMed] [Google Scholar]
- 10.Llovet JM, Ricci S, Mazzafero V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 11.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mostertz W, Stevenson M, Acharya C, Chan I, Walters K, Lamlertthon W, Barry W, Crawford J, Nevins J, Potti A. Age- and sex-specific genomic profiles in non-small-cell lung cancer. JAMA. 2010;303:535–543. doi: 10.1001/jama.2010.80. [DOI] [PubMed] [Google Scholar]
- 13.Mojica W, Hawthorn L. Normal colon epithelium: a data set for analysis of gene expression and alternative splicing events in colon disease. BMC Genomics. 2010;11:5. doi: 10.1186/1471-2164-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gevaert O, Daemen A, De Moor B, Libbrecht L. A taxonomy of epithelial human cancer and their metastases. BMC Med Genomics. 2009;2:69. doi: 10.1186/1755-8794-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathe EA, Nguyen GH, Bowman ED, Zhao Y, Budhu A, et al. MicroRNA expression in squamous cell carcinoma and adenocarcinomaof the esophagus: associations with survival. Clin Cancer Res. 2009;15:6192–6200. doi: 10.1158/1078-0432.CCR-09-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoshida Y, Nijman SMB, Kobayashi M, Chan JA, Brunet J-P, et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iizuka N, Hamamoto Y, Tsunedomi R, Oka M. Translational microarray systems for outcome prediction of hepatocellular carcinoma. Cancer Sci. 2008;99:659–665. doi: 10.1111/j.1349-7006.2008.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Cheung ST, So S, Fan ST, Barry C, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–1939. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J-S, Chu I-S, Heo J, Calvisi DF, Sun Z, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–676. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- 20.Pagano M, Benmaamar R. When protein destruction runs amok, malignancy is on the loose. Cancer Cell. 2003;4:251–256. doi: 10.1016/s1535-6108(03)00243-5. [DOI] [PubMed] [Google Scholar]
- 21.Lee J-S, Heo J, Libbrecht L, Chu I-S, Kaposi-Novak P, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nature Med. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 22.Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 23.Yu D, Thomas-Tikhonenko A. A non-transgenic mouse model for B-cell lymphoma: in vivo infection of p53-null bone marrow progenitors by a Myc retrovirus is sufficient for tumorigenesis. Oncogene. 2001;21:1922–1927. doi: 10.1038/sj.onc.1205244. [DOI] [PubMed] [Google Scholar]
- 24.Yu D, Cozma D, Park A, Thomas-Tikhonenko A. Functional Validation of Genes Implicated in Lymphomagenesis:An in Vivo Selection Assay Using a Myc-induced B-Cell Tumor. Ann Ny Acad Sci. 2005;1059:145–159. doi: 10.1196/annals.1339.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, et al. EpCAM and α-Fetoprotein Expression Defines Novel Prognostic Subtypes of Hepatocellular Carcinoma. Cancer Res. 2008;68:1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 26.Cairo S, Armengol C, De Reyniès A, Wei Y, Thomas E, Renard CA, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14:471–484. doi: 10.1016/j.ccr.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nature Genetics. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 28.Teradillos O, Billet O, Renard CA, Levy R, Molina T, Briand P, et al. The hepatitis B virus X gene potentiatesc-myc-induced liver oncogenesis in transgenic mice. Oncogene. 1997;14:395–404. doi: 10.1038/sj.onc.1200850. [DOI] [PubMed] [Google Scholar]
- 29.Madden CR, Finegold MJ, Slagle BL. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J Virol. 2001;75:3851–3858. doi: 10.1128/JVI.75.8.3851-3858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrisani OM, Barnabas S. The transcriptional function of the hepatitis B virus X protein and its role in hepatocarcinogenesis (Review) Inter J Oncol. 1999;15:373–379. doi: 10.3892/ijo.15.2.373. [DOI] [PubMed] [Google Scholar]
- 31.Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725–12734. doi: 10.1128/JVI.78.23.12725-12734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepat. 2010;52:594–604. doi: 10.1016/j.jhep.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 33.Rakotomalala L, Studach L, Wang W-H, Gregori G, Hullinger RL, Andrisani OM. Hepatitis B virus X protein increases the Cdt1 to geminin ratio inducing DNA re-replication and polyploidy. J Biol Chem. 2008;283:28729–28740. doi: 10.1074/jbc.M802751200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Studach L, Rakotomalala L, Wang W-H, Hullinger RL, Cairo S, Buendia MA, Andrisani OM. Polo-like kinase1 inhibition suppresses Hepatitis B virus X protein-induced transformation, in an in vitro model of liver cancer progression. Hepatology. 2009;50:414–423. doi: 10.1002/hep.22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nature reviews/Drug discovery. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 36.van Vught MA, Bras A, Madema RH. Polo-like kinase 1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. 2004;15:799–811. doi: 10.1016/j.molcel.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 37.Studach L, Wang W-H, Weber G, Tang J, Hullinger RL, Malbrue R, et al. Polo-like Kinase 1 activated by Hepatitis B Virus X protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. J Biol Chem. 2010;285 doi: 10.1074/jbc.M109.093963. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He ZL, Zheng H, Lin H, Miao XY, Zhong DW. Overexpression of polo-like kinase1 in hepatocellular carcinoma patients. World J Gastroenterology. 2009;15:4177–4182. doi: 10.3748/wjg.15.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pellegrino R, Calvisi DF, Ladu S, Ehemann V, Staniscia T, Evert M, et al. Oncogenic and Tumor Suppressive Roles of Polo-Like Kinases in Human Hepatocellular Carcinoma. Hepatology. 2010;51:857–868. doi: 10.1002/hep.23467. [DOI] [PubMed] [Google Scholar]
- 40.Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 41.Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 42.Kim WK, Ye Q, Forgues M, Budhu A, Sime J, et al. Cancer-associated Molecular Signature in the Tissue Samples of Patients with Cirrhosis. Hepatology. 2004;39:518–527. doi: 10.1002/hep.20053. [DOI] [PubMed] [Google Scholar]
- 43.Balzar M, Winter MJ, de Boer CJ, Litvinov SV. The biology of the 17-1A antigen (Ep-CAM) J Mol Med. 1999;77:699–712. doi: 10.1007/s001099900038. [DOI] [PubMed] [Google Scholar]
- 44.de Boer CJ, van Krieken JH, Janssen-van Rhijm CM, Litvinov SV. Expression of Ep-CAM in normal, regenerating, metaplastic, and neoplastic liver. J Pathol. 1999;188:201–206. doi: 10.1002/(SICI)1096-9896(199906)188:2<201::AID-PATH339>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 45.Schmelzer E, Wauthier E, Reid LM. The phenotypes of pluripotent human hepatic progenitors. STEM CELLS. 2006;24:1852–1858. doi: 10.1634/stemcells.2006-0036. [DOI] [PubMed] [Google Scholar]
- 46.Schmelzer E, Zhang L, Bruce A, Wauthier E, Ludlow J, et al. Human hepatic stem cells from fetal postnatal donors. J Exp Med. 2007;204:1973–1987. doi: 10.1084/jem.20061603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Theise N, Chua M, Reid LM. The Stem Cell Niche of Human Livers: Symmetry between Development and Regeneration. Hepatology. 2008;48:1598–1607. doi: 10.1002/hep.22516. [DOI] [PubMed] [Google Scholar]
- 48.Takayasu K, Arii S, Ikai I, et al. Prospective cohort study of transarterial chemoembolization for unresectable hepatocellular carcinoma in 8510 patients. Gastroenterology. 2006;131:461–469. doi: 10.1053/j.gastro.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 49.Taketa K. α-Fetoprotein: reevaluation in hepatology. Hepatology. 1990;12:1420–1432. doi: 10.1002/hep.1840120625. [DOI] [PubMed] [Google Scholar]
- 50.Chamorro MN, Schwartz DR, Vonica A, Brivanlou AH, Cho KR, Varmus HE. FGF-20 and DKK1 are transcriptional targets of β-catenin and FGF-20 implicated in cancer development. EMBO J. 2005;24:73–84. doi: 10.1038/sj.emboj.7600460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sekiya T, Adach S, Kohu K, et al. Identification of BMP and activin membrane-bound inhibitor (BAMBI), an inhibitor of TGF- β signaling as a target of the β-catenin pathwayin colorectal tumor cells. J Biol Chem. 2004;279:6840–6846. doi: 10.1074/jbc.M310876200. [DOI] [PubMed] [Google Scholar]
- 52.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, et al. EpCAM-positive hepatocellular carcinoma cells are tumor initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009;69:5627–5629. doi: 10.1158/0008-5472.CAN-09-0654. [DOI] [PubMed] [Google Scholar]
- 54.Lu T-Y, Lu R-M, Liao M-Y, Yu J, Chung C-H, et al. Epithelial Cell Adhesion Molecule Regulation Is Associated with the Maintenance of the Undifferentiated Phenotype of Human Embryonic Stem Cells. J Biol Chem. 2010;285:8719–8732. doi: 10.1074/jbc.M109.077081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munz M, Kieu C, Mack B, Schmitt B, Zeidler R, Gires O. The carcinoma-associated antigen EpCAM upregulates c-myc and induces cell proliferation. Oncogene. 2004;23:5748–5758. doi: 10.1038/sj.onc.1207610. [DOI] [PubMed] [Google Scholar]
- 56.Maetzel D, Denzel S, Mack B, Canis M, Went P, et al. Nuclear signaling by tumour-associated antigen EpCAM. Nature Cell Biol. 2009;11:162–171. doi: 10.1038/ncb1824. [DOI] [PubMed] [Google Scholar]
- 57.Schuttengrubber B, Chourrout D, Vervoot M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 58.Passini D, Bracken AP, Hansen JB, Capillo M, Helin K. The Polycomb Group Protein Suz12 Is Required for Embryonic Stem Cell Differentiation. Mol Cell Biol. 2007;27:3769–3779. doi: 10.1128/MCB.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 60.Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer. 2009;9:773–784. doi: 10.1038/nrc2736. [DOI] [PubMed] [Google Scholar]
- 61.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;209:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li T, Hu JF, Qiu X, Ling J, Chen H, Wang S, Hou A, Vu TH, Hoffman AR. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol Cell Biol. 2008;28:6473–6482. doi: 10.1128/MCB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Breuhahn K, Vreden S, Haddad R, Beckebaum S, Strippel D, et al. Molecular Profiling of Hepatocellular Carcinoma Defines Mutually Exclusive Interferon Regulation and Insulin-Like-Growth Factor II Overexpression. Cancer Res. 2004;64:6058–6064. doi: 10.1158/0008-5472.CAN-04-0292. [DOI] [PubMed] [Google Scholar]
- 64.Shay JW, Zou Y, Hiyama E, Wright WE. Telomerase and cancer. Hum Molec Genet. 2001;10:677–685. doi: 10.1093/hmg/10.7.677. [DOI] [PubMed] [Google Scholar]
- 65.Agarwal S, Loh Y-H, McLoughlin EM, Huang J, Park I-H, Miller JD, Huo H, Okuka M, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–296. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Agger K, Cloos PAC, Rudkjaer L, Williams K, Andersen G, Christensen J, Helin K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene and stress-induced senescence. Genes Dev. 2009;23:1171–1176. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]