

Abstract
Earlier we used a glioma model to identify loci in the mouse genome, which were repeatedly targeted by platelet-derived growth factor (PDGF)-containing Moloney murine leukemia viruses. The gene Prkg2, encoding cyclic guanosine monophosphate (cGMP)-dependent protein kinase II, cGKII, was tagged by retroviral insertions in two brain tumors. The insertions were both situated upstream of the kinase domain and suggested creating a truncated form of the cGKII protein. We transfected different human glioma cell lines with Prkg2 and found an overall reduction in colony formation and cell proliferation compared with controls transfected with truncated Prkg2 (lacking the kinase domain) or empty vector. All glioma cells transfected with the cGKII phosphorylate vasodilator-stimulated phosphoprotein, VASP, after cGMP analog treatment. Glioma cell lines positive for the Sox9 transcription factor showed reduced Sox9 expression when Prkg2 was stably transfected. When cGKII was activated by cGMP analog treatment, Sox9 was phosphorylated, Sox9 protein expression was suppressed and the glioma cell lines displayed loss of cell adhesion, inhibition of Akt phosphorylation and G1 arrest. Sox9 repression by siRNA was similarly shown to reduce glioma cell proliferation. Expression analysis of stem and glial lineage cell markers also suggests that cGKII induces differentiation of glioma cell lines. These findings describe an anti-proliferative role of cGKII in human glioma biology and would further explain the retroviral tagging of the cGKII gene during brain tumor formation in PDGF-induced tumors.
Keywords: glioma, PDGF, cGMP-dependent protein kinase II, Akt, Sox9, VASP
Introduction
The mammalian cyclic guanosine monophosphate (cGMP)-dependent protein kinases, cGKI and cGKII, belong to the family of serine/threonine kinases. The function of the different cGKs has recently been reviewed (Hofmann et al., 2006). In a few cell systems, cGKs have been shown to be involved in the regulation of differentiation or cell proliferation. Activation of the cGKI or cGKII by cGMP and/or cGMP analogs leads to the phosphorylation of vasodilator-stimulated phosphoprotein (VASP) (Smolenski et al., 1998; Gambaryan et al., 2002) on Ser239 and exerts a negative effect on the proliferation of vascular smooth muscle cells (Butt et al., 1994; Chen et al., 2004) and prostatic stromal cells (Cook and Haynes, 2004). Furthermore, cGKII phosphorylates the transcription factor Sox9 and attenuates its nuclear entry (Chikuda et al., 2004), whereupon the inhibitory effect of Sox9 on the differentiation of postmitotic chondrocytes is abrogated. Sox9 is also expressed in neuro-epithelial progenitors in the developing spinal cord and is an important regulator of glial cell maturation (Stolt et al., 2003), but in these cells, the role of cGKII has not been elucidated.
cGKI has been shown to have tumor suppressor properties in colon carcinoma (Deguchi et al., 2004; Hou et al., 2006). Moreover, a recent mutational analysis of human glioblastoma multiforme showed missense mutations in PRKG2 in 2 of 22 tumor samples (Parsons et al., 2008). In an previous study, we used a platelet-derived growth factor (PDGF)-containing Moloney murine leukemia virus (MMLV) to tag genes involved in glioma development (recently reviewed in Johansson Swartling, 2008) and found two proviral integrations situated in exon 9 and intron 9 in the cGKII encoding Prkg2 gene in two tumors (Johansson et al., 2004). Such retroviral tagging is suggested to yield a truncated or disrupted Prkg2 transcript coding for a nonfunctional protein. By contrast, we found a higher expression of Prkg2 in the tumors when comparing pooled RNA from tissue samples from gliomas and normal brains (Johansson et al., 2005) by real-time PCR.
The proto-oncogene Akt is activated in gliomas and many other human cancers mostly due to the loss of the PTEN tumor suppressor (Li et al., 1997; Steck et al., 1997; Wang et al., 1997) and has been found to be a crucial partner to the activated Ras for the induction of glioblastoma-like lesions in mice (Holland et al., 2000). Akt is an important target in glioma therapy (Phillips et al., 2006) and is frequently activated and directs cell survival signals downstream of constitutively activated receptor tyrosine kinases, such as epidermal growth factor receptor or platelet-derived growth factor receptor (PDGFR) (Libermann et al., 1985; Humphrey et al., 1988; Nister et al., 1988; Guha et al., 1995). In this study, we report that cGKII suppresses glioma growth and inhibits Akt phosphorylation. We also found that activated cGKII suppresses Sox9 and that small-interfering RNA (siRNA)-mediated suppression of Sox9 reduces cell proliferation in Sox9-positive glioma cell lines.
Results
Prkg2 inhibits colony formation and proliferation of human glioma cells
In our earlier study (Johansson et al., 2004), two proviral integrations were found in Prkg2 on mouse gliomas, one in reverse direction in exon 9 and one in reverse direction in intron 9 (Figure 1a). If both integrations lead to a disruption of the coding sequence of Prkg2, it suggests that the loss of functional cGKII confers a growth advantage of the glioma cells and thereby contributes to tumor growth. In order to study the effect of Prkg2 on glioma cell growth, we constructed two expression plasmids, one with the full-length cDNA and the other with the truncated variant (ΔPrkg2) containing exons 1–10. Both protein products could be readily detected when transiently expressed in Cos-1 cells (Figure 1b). In a 3-week colony-forming assay using four human glioma cell lines (U-1242MG, U-343MG, U-2987MG and U-Cl2:6MG) transiently transfected with the Prkg2 constructs, we found that fewer colonies were formed in cells transfected with Prkg2 than in those transfected with empty vector (pcDNA3.1) or the truncated form of Prkg2 (Figure 1c).
Figure 1.
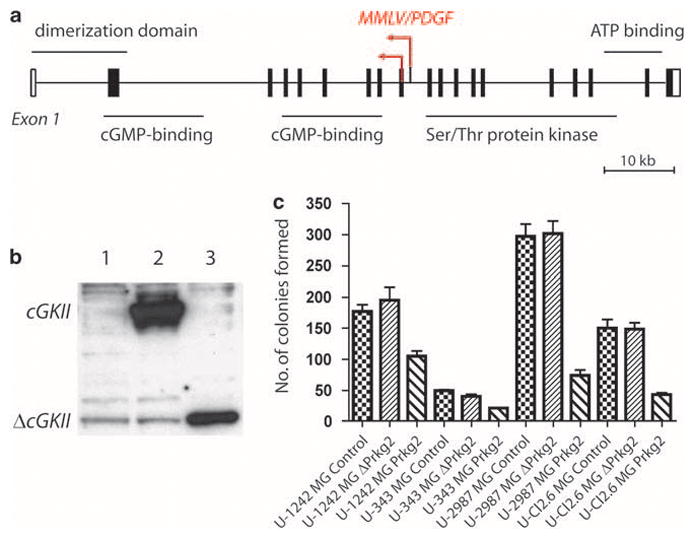
(a) Schematic picture of the two proviral integrations of Moloney murine leukemia virus (MMLV)/platelet-derived growth factor (PDGF) in Prkg2 on mouse chromosome 5. Major structural domains of the protein are presented. Data were obtained from National Center for Biotechnology Information, Mouse 37 assembly and ENSEMBL Release 51, 2008. (b) Transiently transfected Cos-1 cells (after 72 h) overexpressing an empty pcDNA vector (lane 1), full-length cGKII (lane 2, ~85 kDa) or truncated ΔcGKII (lane 3, ~35 kDa). (c) Colony-forming assay showing a number of colonies formed in four different glioma cell lines (U-1242MG, U-343MG, U-2987MG and U-Cl2:6MG) transfected with Prkg2, ΔPrkg2 or empty pcDNA (Control). Colonies were counted after 3 weeks of growth in medium containing 0.8 mg/ml of neomycin. The cells were plated in triplicates and experiments were repeated twice. Results are presented from one representative experiment and significant differences were found for Prkg2 transfections compared with empty vector transfections (P<0.05) with paired t-test for every cell line, but not for ΔPrkg2 and empty vector in U-Cl2:6MG. The U-87MG cell line yielded too few colonies after transfection and was not included.
Glioma cells stably transfected with the full-length Prkg2 were then generated. Two Prkg2-positive clones that were studied (U-2987-P3 and U-2987-P6) were derived from the human glioma cell line U-2987MG, which expresses the stem cell marker Sox2 (Supplementary Figure 1) and another member of the Sox family, Sox9 (Figure 2a). Prkg2-positive clones were also established from U-343MG (U-343-P7, U-343-P8), U-87MG (U-87-P1) and U-1242MG (U-1242-P1) cell lines. These cell lines did not express Sox2 but expressed Sox9 at low and variable levels (Supplementary Figure 1 and Figure 2a). Western blot analysis of the transfected clones showed the expected 86 kDa band of cGKII, whereas the parental cell lines were negative (Figure 2a).
Figure 2.

(a) Western blot of cGKII (only showing the specific 86 kDa band) and Sox9 protein expression in human glioma cell lines (U-2987MG, U-343MG, U-87MG and U-1242MG) and clones transfected with Prkg2 from U-2987MG (U-2987-P3 and U-2987-P6), U-343MG (U-343-P7 and U-343-P8), U-87MG (U-87-P1) and U-1242MG (U-1242-P1) human glioma cell lines (after four passages in culture). (b) Real-time PCR showing relative mRNA expression levels of PRKG2 as well as SOX9 in glioma cell lines compared with a normal human adult brain RNA. The expression levels are also compared with total RNA from normal human fetal brain and two of the Prkg2-transfected cell lines U-2987-P6 and U-87-P1. (c) Western blot showing phosphorylated Sox9 (Ser181) and both phosphorylated VASP residues (Ser 239 and Ser157) in untransfected and Prkg2-transfected glioma cell clones after 1 h of treatment with 8-pCPT-cGMP (250 μM) compared with β-actin loading. (d) Immunostaining of pSox9 (Ser181) in the glioma cells (U-2987MG and U-2987-P6) with or without 4 h of cyclic guanosine monophosphate (cGMP) analog (250 μM) treatment.
Prkg2-transfected glioma cell lines express a functional cGKII that can phosphorylate VASP and Sox9
As shown by quantitative real-time PCR, the expression of endogenous PRKG2 was low in all glioma cell lines compared with normal brain RNA and much lower than in the Prkg2-transfected clones (Figure 2b). All transfected cGKII-positive glioma cell lines expressed a functional cGMP-dependent kinase because treatment with 250 μM of the cGMP analog, 8-pCPT-cGMP, yielded a marked phosphorylation on both Ser239 and Ser157 residues of the endogenous downstream target VASP (Figure 2c). U-2987-P6 cells that had the highest expression of Sox9 also showed a strong phosphorylation of Sox9 (Figure 2c) when treated with the cGMP analog. Treated U-2987-P6 cells showed an accumulation of phosphorylated Sox9 in the cytoplasm compared with untreated cells, in which phosphorylated Sox9 was more restricted to the nucleus (Figure 2d).
Activated cGKII arrests glioma cells in G1 and inhibits Akt phosphorylation
The proliferation rate of cGKII-expressing clones was investigated in a cell counting experiment. U-2987-P6 and U-87-P1 cells were significantly inhibited (unpaired t-test, P<0.01 and P<0.05 respectively) by the cGMP analog as measured by cell counting over a 7-day period (Figure 3a). No significant inhibition of cell proliferation was seen in parental cell lines or clones transfected with empty control vectors after cGMP analog treatment. Flow cytometry of U-2987-P6 cells continuously treated with the cGMP analog for 72 h showed that the cells were arrested in the G1 phase of the cell cycle (Figure 3b). We therefore conclude that activated cGKII provides a growth inhibitory signal in these human glioma cell lines. In both cGKII-expressing clones of U-2987MG glioma cells (U-2987-P3 and U-2987-P6), the addition of 8-pCPT-cGMP led to a pronounced decrease in the level of phosphorylated Akt (Figure 4a and Supplementary Figure 2a). A reduction of Akt phosphorylation was observed 1 h after addition of the cGMP analog, and after 8 h, there was an even more pronounced reduction of pAkt (Figure 4a). The phosphorylation of both Thr308 and Ser473 of Akt was reduced (Figure 4a and Supplementary Figure 2a). The reduction of phosphorylation lasted for at least 24 h after a single treatment and was not preceded by any reduction in the phosphorylation of the Akt kinase PDK1 (Ser241) (Supplementary Figure 2b) or phosphorylation of ERK (Supplementary Figure 2a). No change in pAkt but a slight decrease in total Akt levels was found after both 1 and 8 h of cGMP treatment in the parental cell line U-2987MG. The other Prkg2-expressing clones of U-1242MG and U-343MG did not show any dephosphorylation of Akt (data not shown), whereas the U-87MG clone showed a decrease only after 8 h of treatment (Supplementary Figure 2c).
Figure 3.
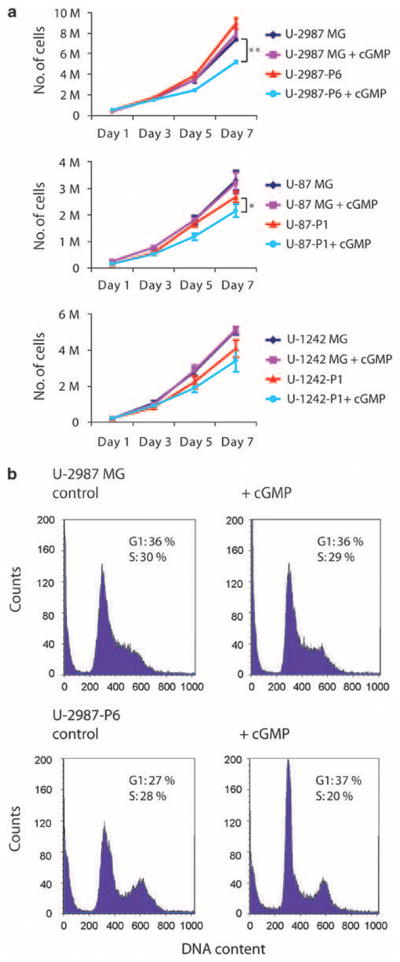
(a) Growth of human glioma cell lines stably transfected with Prkg2 (U-2987-P6, U-87-P1 and U-1242-P1) or empty pcDNA vector (denoted U-2987MG, U-87MG and U-1242MG) when treated with 250 μM of cyclic guanosine monophosphate (cGMP) analog over 7 days. M, million cells; Statistical significance: **P<0.01 and *P<0.05 using Student’s unpaired t-test (error bars indicate standard deviation from triplicates). (b) Flow cytometry of U-2987-MG glioma cells stably transfected with Prkg2 (U-2987-P6) or pcDNA (denoted U-2987MG) and treated with 250 μM of cGMP analog for 72 h.
Figure 4.
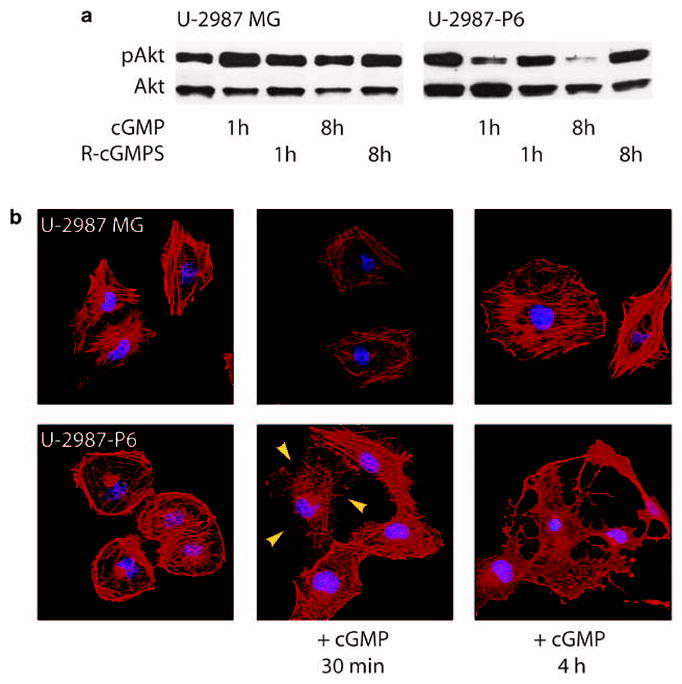
(a) Inhibition of Akt phosphorylation (Thr308) in Prkg2-transfected U-2987MG cell line (U-2987-P6) when stimulated with cyclic guanosine monophosphate (cGMP) analog. (b) Phalloidin staining (red) of U-2987MG and U-2987-P6 glioma cells unstimulated or when stimulated with a cGMP analog for indicated time periods. Yellow arrowheads show the start of arborization and loss of adhesion of the cGKII-positive cells. Blue, DAPI; red, phalloidin coupled to rhodamine; cGMP, 8-pCPT-cGMP (250 μM); R-cGMPS, Rp-pCPT-cGMPS (10 μM).
In the clone with the highest cGKII protein expression (U-2987-P6), the addition of 8-pCPT-cGMP led to a major change in cell morphology, with an apparent collapse of the actin filament network and arborization of the cytoplasm (Figure 4b, Supplementary Figure 3). No sign of cytoplasmic arborization was found in U-343MG, U-87MG or U-1242MG cells expressing cGKII, or in U-2987MG wild-type cells or in U-2987MG cells stably transfected with an empty pcDNA vector after cGMP analog treatment alone (data not shown and Supplementary Figure 3). However, U-87MG cells, stably transfected with Prkg2, became arborized in a similar manner as U-2987-P6 cells when 1mM of the phosphodiesterase inhibitor, 3-isobutyl-1-methylxanthine (IBMX), was added in combination with the cGMP analog (Supplementary Figure 3). By comparison, none of the glioma cell lines or clones arborized when treated with 10 μM of the phosphatidylinositol-3 kinase (PI3K) inhibitor, LY294002, for 4 h (Supplementary Figure 3).
Activation of cGKII is accompanied by the downregulation of Sox9
Compared with the original cell line, SOX9 mRNA expression and the expression of stem cell markers, CD133 and NES, were lower in the Prkg2-transfected cell clone U-2987-P6, as shown by quantitative real-time PCR, whereas the GFAP mRNA levels were unaltered (Figure 5a). Similarly, the mRNA expression of PDGFRa was also lower in the Prkg2-transfected clones (Figure 5a and Supplementary Figure 4a). When U-2987-P6 cells were treated with cGMP analog, the expression of the oligodendrocytic marker, PLP, was greatly diminished (Figure 5a).
Figure 5.
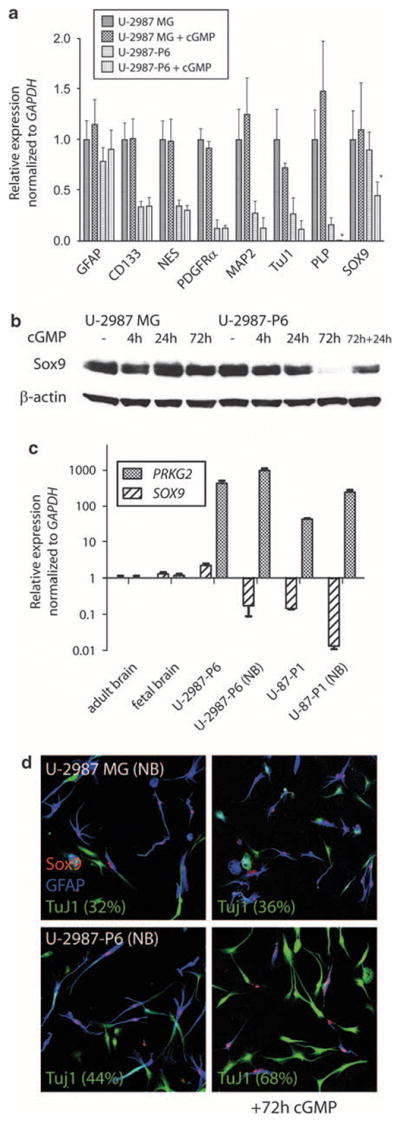
(a) Real-time PCR showing relative expression of the markers of immature cells (CD133, NES), astrocytic (GFAP), oligodendrocytic (PLP) or neuronal (TuJ1, MAP2) lineage for U-2987MG and U-2987-P6 (Prkg2-transfected) glioma cells with or without 72 h of cGMP analog treatment (added every day). Relative expression levels of PDGFRα and SOX9 were also studied. The expression level in U-2987MG cells for each gene was set to 1. Significant different levels: *P<0.05, using Student’s t-test after triplicate measures (after 72 h cGMP analog treatment in U-2987-P6). (b) Western blot showing Sox9 protein levels in U-2987MG and U-2987-P6 after different cGMP analog treatment periods (4, 24 and 72 h) compared with levels of β-actin. For the 72-h treated cells, cGMP analog was added daily. For 72+24 h, cGMP was added daily for the first 72 h, then the cGMP analog was removed for the media for an additional 24 h. (c) Real-time PCR showing relative expression of SOX9 and PRKG2 between the glioma cell lines U-2987MG and U-87MG (and the corresponding Prkg2-transfected clones) when cultured in 10% serum compared with when the cell lines are cultured in neurobasal media (containing EGF and FGF). As compared to controls of total RNA from normal brain (set to 1). (d) Immunostaining of SOX9, TuJ1 and GFAP in glioma cells when differentiating for 72 h in neurobasal media (without growth factors) with or without the cGMP analog (added daily). Relative percentage of TuJ1-expressing cells (after triplicate cell counts) within parentheses under the different conditions are shown.
Moreover, SOX9 expression in U-2987-P6 was significantly reduced upon 4 h of cGMP stimulation (Supplementary Figure 4b). A reduction was also shown for Sox9 protein levels at longer time periods (72 h) of stimulation with cGMP analog (Figure 5b), but not for the Sox family member Sox2 (data not shown). The downregulation of Sox9 was reversible; after the addition of fresh medium without cGMP, the Sox9 protein expression in the U-2987-P6 cells returned to the level of the control (Figure 5b). Staining of U-2987MG and U-2987-P6 for GFAP and TuJ1 showed no obvious differences between the clones when grown in medium containing 10% serum (Supplementary Figure 4c).
Activated cGKII in glioma cells cultured without serum generates more TuJ1-expressing cells
Culturing cGKII-expressing glioma cells in serum-free neurobasal media containing epidermal growth factor (EGF) and fibroblast growth factor (FGF) led to a considerable cell death, although some glioma cells started to grow as floating spheres (Supplementary Figure 5a). RNA prepared from glioma cells cultured for 1 week under these conditions showed lower levels of SOX9 but higher levels of PRKG2 compared with cells grown in serum (Figure 5c and Supplementary Figure 5b). When these glioma spheres were cultured on coverslips coated with poly-L-ornithine and laminin without EGF and FGF, the cGKII-expressing U-2987-P6 clones adhered and formed significantly more TuJ1-positive cells after 72 h of cGMP stimulation compared with both untreated U-2987-P6 and treated or untreated U-2987MG cells (Figure 5d). In contrast to cells grown in 10% serum, Sox9 protein was not expressed in all cells. Co-staining for GFAP, TuJ1 and Sox9 showed that the Sox9-positive cells were almost always positive for GFAP but not for TuJ1 (Figure 5d and Supplementary Figure 5c). Untransfected glioma cells could be passaged for several weeks in neurobasal media while only scattered surviving cells from the transfected clones remained after 3 weeks of culture under these conditions (Supplementary Figure 5a).
Downregulation of Sox9 with specific siRNA inhibits glioma cell proliferation
Both Sox9 and VASP levels could be reduced by their specific siRNAs (Figure 6a). Inhibition of Sox9 by this way significantly (unpaired t-test, P<0.001) reduced cell proliferation (after 7 days) in U-2987MG and U-2987-P6 cells compared with U-1242MG cells that have much lower levels of Sox9 (Figure 6b). Neither control siRNA treatment nor siRNA-induced inhibition of VASP reduced cell proliferation in these glioma cell lines (Figure 6b and data not shown). Treatment of U-2987-P6 cells with cGMP analog again significantly inhibited cell proliferation (Figure 6b). Interestingly, the siRNA downregulation of Sox9 blocked the anti-proliferative effects induced by cGMP treatment in U-2987-P6 cells (Figure 6b).
Figure 6.
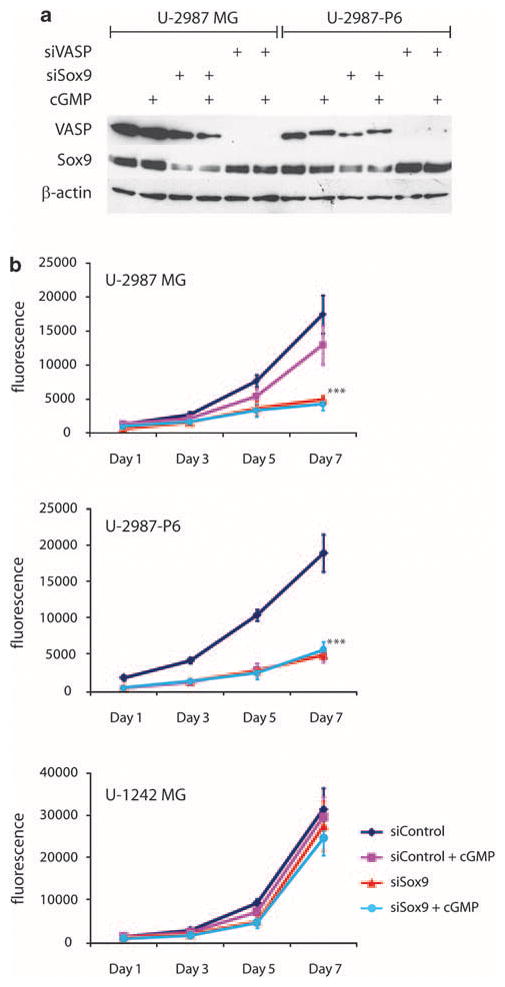
(a) Western blot of U-2987MG and U-2987-P6 after siRNA inhibition of VASP (siVASP) and Sox9 (siSox9) with or without stimulation with 250 μM of 8-pCPT-cGMP for 8 h. (b) Cell proliferation of glioma cell lines (U-2987MG, U-2987-P6 and U-1242MG) transfected with control siRNA-A (siControl) or siRNA directed against Sox9 (siSox9). Fluorescence (measurement of cellular DNA content) proportional to cell number after 1, 3, 5 and 7 days of culturing after siRNA transfection is presented (from quadruplicate CyQUANT measurements). Statistical significance: ***P<0.001, using Student’s unpaired t-test (error bars indicate standard deviation from quadruplicates for siSox9 and siSox9 (+cGMP) compared with siControl for both U-2987MG and U-2987-P6 and also for siControl+cGMP compared with siControl for U-2987-P6).
Discussion
This study shows that expression of cGKII in glioma cells in culture provides a growth inhibitory signal. Cells transduced with a cGKII expression vector showed a marked reduction in colony formation, and in stably transduced cultures, cell proliferation was slowed down and cells accumulated in G1 phase in a cGMP-dependent manner. This notion fits well with the mode of integration of the provirus in the genetic screen where Prkg2 was identified. The integrations were located within the gene, wherein they could be expected to disrupt transcription or give rise to a truncated protein (Johansson et al., 2004), possibly leading to decreased levels of cGKII and selective growth advantage. Inactivation of PRKG2 has also been detected in two leukemia patients, in whom the first exons of PRKG2 were fused to a truncated PDGFR (Walz et al., 2007; Lahortiga et al., 2008). This recombination results in constitutive PDGFR kinase activation in combination with a disruption of the cGKII catalytic domain. It can be noted that truncated catalytic domains of cGKII are able to bind to the full-length protein kinase and to reduce its activity in a dominant-negative manner (Taylor et al., 2002).
Several reports suggest that cGMP is involved in the negative regulation of proliferation (reviewed in Villalobo, 2006). Thus, the growth inhibitory effect of nitric oxide (NO) is in part ascribed to the activation of guanylate cyclase and activation of cGK. Activation of cGKI in colon carcinoma cell lines increases the expression of p21 and p27KIP1 (Cen et al., 2008) However, relatively little is known about the physiological role of cGMP protein kinases in cell growth regulation and signal transduction. A likely explanation why the role of cGKs as growth regulators has been underestimated is that their expression is often rapidly lost when cells are grown in culture (Gudi et al., 1999). This phenomenon may be related to the negative effect of cGKs on cell proliferation; the loss of cGK expression will provide a selective growth advantage, and clones with reduced expression will rapidly expand.
In vivo, cGMP is synthesized in oligodendrocytes in the developing brain, and cGKII is expressed in oligodendrocyte progenitors (Tanaka et al., 1997; Yoshioka et al., 2000). Previous analyses have indicated that mouse brain tumors induced by the PDGFB-encoding MMLV are derived from cells of the oligodendrocyte lineage. In such tumors, Prkg2 was slightly upregulated relative to normal mouse brain (Johansson et al., 2005). This may seem surprising in light of a growth inhibitory effect of cGKII. Because the expression of known markers of oligodendrocyte progenitors, such as Sox10, Ng2, Olig2 and Pdgfra, were upregulated to much higher levels than Prkg2, the relative expression of Prkg2 may actually be lower in the tumor cells than in their progenitors. However, it is not known whether cGKII in these tumors is still functional.
The downstream signals that mediate the growth-suppressive activity to cGKII remain to be identified. Of all cell lines transfected with Prkg2, only clones from U-2987MG cells showed a markedly diminished Akt phosphorylation after exposure to 8-pCPT-cGMP alone, whereas growth suppression was seen in all clones. A reduced Akt phosphorylation is therefore not a sufficient general explanation to the growth suppression. In vascular smooth muscle cells, cGKII-mediated serine phosphorylation of the cytoskeleton-associated protein, VASP, inhibits cell proliferation (Chen et al., 2004). Because we could show an increased phosphorylation of VASP in Prkg2-expressing glioma cells, we could not exclude the possibility that cGKII-catalyzed phophosphorylation of VASP is also involved in the growth inhibitory effects in these cells. In one clone, U-2987-P6, the addition of the cGMP analog led to a reduction in cell adhesion and cytoplasmic arborization. This particular clone was the highest cGKII-expressing one, and also showed the strongest response with regard to VASP phosphorylation. Although VASP phosphorylation has been reported to lead to a detachment of VASP from focal adhesions (Smolenski et al., 2000; Chen et al., 2004), a knockdown of VASP expression by siRNA had no effect on glioma cell morphology (data not shown), suggesting that these cytoskeletal effects were independent of VASP phosphorylation.
Sox9 has been found to be highly expressed in malignant brain tumors, including astrocytic, oligodendroglial and primitive neuro-ectodermal tumors (Kordes and Hagel, 2006). Sox9 downregulation by siRNA inhibits prostate cancer cell proliferation consistent with a block at G0–G1 (Wang et al., 2007); and Sox9 overexpression in prostate cancer cells that express low levels of Sox9 lead to enhanced growth, angiogenesis and invasion in xenograft experiments (Wang et al., 2008). Interestingly, there is a role for Sox9 in PDGF signaling, as Sox9 together with Sox10 has been found to regulate PDGFRα expression in oligodendrocyte progenitors of the spinal cord (Finzsch et al., 2008). Sox9 is also present in the neural crest and neural stem cells and neurospheres (Stolt et al., 2003; Dromard et al., 2007), and is co-expressed with Sox1 and Sox2 in brain regions that harbor neural stem cells (Sottile et al., 2006), but lost upon differentiation and maturation of both oligodendrocytes and neurons (Cheung and Briscoe, 2003; Stolt et al., 2003). We found that Sox9 and PDGFRα-positive U-2987MG glioma cells exhibited lower levels of these proteins in clones transfected with Prkg2, which similar to Sox9 was further reduced on treatment with the cGMP analog. In addition, Sox9 phosphorylation in Prkg2-transfected U-2987-P6 cells displayed identical kinetics similar to the dephosphorylation of Akt, which suggests a possible relationship between these events.
When Sox9 in U-2987MG cells was reduced by siRNA treatment, there was a retardation of cell proliferation that was independent of the continuous cGMP analog treatment. This could suggest that Sox9 is a potential drug target in Sox9-expressing glioma and that activated cGKII inhibits cell proliferation by a Sox9-dependent mechanism. We further showed that Sox9 was downregulated in glioma cells cultured under serum-free conditions. Removal of serum reduced glioma cell proliferation and promoted Prkg2-expressing cells from being GFAP-positive to become more TuJ1-positive after cGMP stimulation. Interestingly, although untransfected glioma cells could be passaged for several weeks in neurobasal media, the Prkg2-transfected glioma cells died off after a couple of weeks in culture. These findings are new for glioma cells but in accordance with previous results on chondrocytes, where cGKII has been shown to attenuate Sox9 entry to the nucleus and thereby cause a switch from proliferation to differentiation (Chikuda et al., 2004). Further analyses of glioma cells isolated from patient samples that have been established in neurobasal media or other serum-free conditions would be necessary to completely understand the anti-proliferative role that cGKII activated by cGMP or inhibition of Sox9 has on these malignant cells.
The observation that NO donors can increase tumor permeability by increasing cGMP levels in a rat brain tumor model suggests a future rationale of increasing cGMP levels to improve drug delivery to brain tumors in patients (Yin et al., 2008). If such therapies also would promote tumor-suppressing effects of protein kinases such as cGKII must be further investigated.
Materials and methods
Antibodies and chemicals
Antibodies against anti-pAkt (Thr308), anti-pAkt (Ser473), anti-Akt, anti-pVASP (Ser157) anti-pVASP (Ser239), anti-pPDK1, anti-pERK, anti-ERK and the anti-PI3k inhibitor, LY294002, were provided by Cell Signaling (Danvers, MA, USA). Antibodies against anti-cGKII (sc-120) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-VASP from BD Laboratories (Franklin Lakes, NJ, USA), anti-Sox9 (AB5535) and anti-β-actin from Millipore Biosciences (Temecula, CA, USA). The anti-GFAP antibody was obtained from Dako (Glostrup, Denmark) and anti-TuJ1 antibody from Covance (Richmond, CA, USA). An anti-Sox9 antibody (AF3075) produced in goat was purchased from R&D Systems (Minneapolis, MN, USA) and anti-pSox9 antibody (Ser181) was purchased from Abcam (Cambridge, MA, USA). Phalloidin and secondary Alexa Fluors antibodies were provided by Molecular Probes (Invitrogen, Carlsbad, CA, USA). The fluorescent-grade substances 8-pCPT-cGMP and Rp-8-pCPT-cGMPS were obtained from Biolog (Bremen, Germany). All other substances used were purchased from Sigma-Aldrich (St Louis, MO, USA).
An antibody directed against anti-cGKII was developed (in collaboration with Carl-Henrik Heldin, Sweden). A peptide in the C-terminal region of human cGKII (734–749) was synthesized. After coupling of cysteine to keyhole limpet hemocyanin (Calbiochem, Darmstadt, Germany), an N2W rabbit was injected with peptide conjugate. The anti-cGKII antibody was eluted with salt after it was bound to its C-terminal peptide attached to the SulfoLink coupling gel according to the manufacturer (Pierce, Rockford, IL, USA). Affinity-purified or unpurified total serum was used in immunostaining. Animal experiments were performed in accordance with the local animal ethics committee (Ethical registration numbers C 47/2 and C 144/5).
Expression vector constructs
The cDNA of rat Prkg2 was kindly provided by Susanne Lohmann (Germany). Full-length Prkg2 and a shorter cDNA (ΔPrkg2) comprising exons 1–10 were cut out with restriction enzymes (New England Biolabs Ipswich, MA, USA) and ligated with T4 DNA ligase (Promega, Madison, WI, USA) into a pcDNA3.1+ plasmid (Invitrogen).
Tumor cell lines
The low-passage brain tumor cell line, U-2987MG, was recently described (was denoted primary culture 18 in Hagerstrand et al., 2006), and all experiments with this cell line were conducted within 10–20 cell passages. The established Cos-1 cell line and human glioma cell lines (U-87MG, U-1242MG, U-Cl2:6MG and U-343MG) have been described elsewhere. Cells were cultured in minimum essential medium Eagle supplemented with 10% fetal bovine serum, 2mM L-glutamine and 100/ml of penicillin and 0.1 mg/ml of streptomycin. For all experiments except for the one conducted in Figure 1, a G418-resistant clone of each cell line transfected with an empty pcDNA3.1 vector (Invitrogen) was used (U-2987MG, pcDNA-Clone 1, U-343MG, pcDNA-Clone 12, U-87MG, pcDNA-clone 4 and U-1242MG, pcDNA-clone 1). These clones could not be distinguished from their parental cell lines and were named as the original lines in the figures.
For serum-free neurobasal conditions, cells were cultured in Neurobasal-A media supplemented with L-glutamine (2mM), 1 × B27 supplement (–A) and N2 supplement (all Gibco, Invitrogen), 20 ng/ml of human EGF (Sigma-Aldrich), and 20 ng/ml of human basic fibroblast growth factor (Peprotech, Rocky Hill, NJ, USA). Cells were plated onto glass coverslips (Fisher Scientific, Pittsburgh, PA, USA) coated each with 50 μg/ml of poly-L-ornithine (Sigma) and 50 μg/ml of mouse laminin (BD Laboratories) for 1 h, and the cells were allowed to differentiate without EGF and FGF. Cells positive for GFAP, TuJ1 or SOX9 were counted (using secondary antibody Alexa Fluor 633 (anti-rabbit), 488 (anti-mouse), 555 (anti-goat), respectively, in blindly selected and photographed areas. The percentage of cells (at least 100 cells counted in each area) positive for GFAP/Sox9, TuJ1, GFAP/Tuj1, Sox9 or GFAP was calculated thrice. The stained cells were counted using a laser setting just beneath the level at which the strongest stained cell was saturated using the LSM Image Browser program (Zeiss, Munich, Germany).
Focus forming and proliferation assays
In the focus-forming assay, 700 000 cells were seeded onto 10 cm cell culture dishes. After 24 h, the cells were transfected with an equal number of plasmids, starting with 5 μg for the empty plasmid, using a FuGENE/DNA ratio of 6:2 according to the manufacturer’s protocol (Roche, Mannheim, Germany). After 72 h in transfection media, cells were seeded onto 3–4 10 cm dishes (depending on the proliferation of the cell line) in minimum essential medium Eagle containing 10% serum and 0.8 mg/ml of G418 sulfate (Calbiochem).
G418-resistant clones were developed and counted after 3 weeks, after staining with Giemsa’s azur eosin methylene blue solution (Merck, Darmstadt, Germany), diluted 1:10 in distilled water. At similar time points, putative Prkg2-expressing clones were picked and propagated in vitro. For the proliferation assay, 400 000 cells per 35mm dish were plated. The number of cells per dish was counted at days 1, 4 and 7 (in triplicates for each time point) using a Coulter counter (Coulter Electronics, Fullerton, CA, USA). The substances, 8-pCPT-cGMP (250 μM) and Rp-8-pCPT-cGMPS (10 μM), were diluted in phosphate-buffered saline (PBS) and used at indicated concentrations.
siRNA and PI3K/Akt inhibition
Sox9 and VASP were inhibited with siRNA directed against corresponding human proteins according to the manufacturer’s protocol (Santa Cruz Biotechnology). A total of 2 × 105 cells were seeded onto six-well plates. After 24 h, 1 μg siRNA was mixed with 8 μl of transfection reagent, incubated for 45 min and transferred to the cells in transfection medium (Santa Cruz Biotechnology). At 48 h after siRNA transfection, the cells were treated with 8-pCPT-cGMP (250 μM) for different time points, photographed or lysed for western blotting. For the cell proliferation assay, 1000 cells per well in a 96-well plate were seeded, incubated for 24 h in the medium without antibiotics, then transfected with 600 ng siRNA against human Sox9 or control siRNA-A (Santa Cruz Biotechnology) using Lipofectamine RNAiMax according to the manufacturer’s protocol (Invitrogen). For cGKII activation, 8-pCPT-cGMP (250 μM) was added 8 h after transfection. The number of cells per dish was counted at days 1, 3, 5 and 7 (in quadruplicates) after transfection using a CyQUANT NF Cell Proliferation Assay Kit and measured according to the manufacturer’s instructions (Invitrogen) in a Synergy2 microplate reader (BioTek, Winooski, VT, USA) at indicated wavelengths. At day 4, the medium was replaced with fresh medium with or without the cGMP analog. When LY294002 was used to inhibit the PI3K/Akt pathway, it was added to cell medium in a concentration of 25 μM.
Real-time PCR
The Power SYBR-Green real-time PCR assay used (Applied Biosystem, Foster City, CA, USA) was carried out as previously described (Johansson et al., 2005). Primers were designed with low penalty and on different exons (when possible), and single amplicons were assured by dissociation curves and the PCR product was displayed on agarose gels after electrophoresis separation. Primer sequences are presented in the Supplementary Table 1. The PRKG2 primers recognized cDNA synthesized from both human (PRKG2) and rat (Prkg2) mRNAs. Human Brain Total RNA and Human Fetal Brain Total RNA used as control RNA were obtained from Clontech.
Flow cytometry
All cells were trypsinzed, collected and washed in PBS. The pellet was resuspended in PBS and stained with propidium iodine as described (Vindelov et al., 1983). Samples were analysed using a FACSort instrument from Becton Dickinson (Erembodegem-Aalst, Belgium).
Tumors and integration sites
The generation of MMLV/PDGFB-induced mouse brain tumors and the cloning of the provirally tagged sequences in the Prkg2 locus have previously been described (Uhrbom et al., 1998; Johansson et al., 2004). According to Ensembl Genome Browser (Release 51, 2008), the two proviral integrations in Prkg2 were located on mouse chromosome 5 in a forward genomic direction starting at the region 99405420 in tumor 14 and starting at the region 99405503 in tumor 21. All animal experiments were performed in accordance with the local animal ethics committee.
Western blotting and immunocytochemistry
Cells were harvested in a lysis buffer (pH 7.4) containing 1% Triton-X 100, 150mM NaCl, 10mM Tris-HCl, 1mM ethyleneglycoltetraacetic acid (EGTA), 1mM EDTA, 0.5% NP-40, 35 ng/ml phenylmethylsulfonyl fluoride, 1.4 μg/ml aprotinin, 1mM Na3VO4, 10mM NaF, 1mM ZnCl2 and 50mM Na2MoO4. Protein concentrations were determined using the BCA Protein Assay (Pierce). Cell lysates were subjected to a NuPAGE 4–12% Bis-Tris gel, and electrophoresis was performed according to the manufacturer’s protocol (Invitrogen). Proteins were transferred to a Hybond ECL, nitrocellulose filter (GE Healthcare, Buckinghamshire, UK), blocked and subjected to antibodies in 5% bovine serum albumin (BSA) (or 5% BSA plus 5% fetal bovine serum for pSox9 immunoblots) in TBS-T (2% Tween-20). Filters were stripped in a solution containing 100mM 2-mercaptoethanol, 2% SDS and 62.5mM Tris-HCl (pH 6.7). Immunoblots were visualized using a Chemoluminiscence Super Signal substrate solution (Pierce) on X-ray films (Hyperfilm ECL, GE Healthcare).
Cells used for immunofluorescence were cultured 24 h directly on cover glasses, washed twice in PBS and fixed with 4% paraformaldehyde in PBS for 10 min. After washing in PBS, the cells were treated with 0.3% Triton X-100 and blocked 1 h in a solution containing 0.05% Tween-20, 4 mg/ml BSA and 5% normal donkey serum (Sigma) in PBS. Primary antibodies were incubated for 1 h in a humidity chamber. After washing with PBS (with 0.05% Tween), the plates were incubated with secondary Alexa antibodies (Molecular Probes, Invitrogen) for 1 h, washed and fixated in Immu-Mount (Thermo Scientific, Pittsburgh, PA, USA). Photographs were obtained using a confocal microscope (Zeiss 510 Confocal Microscope) and processed using the LSM Image Browser program (Zeiss).
Supplementary Material
Acknowledgments
This work was supported by grants from the Swedish Cancer Foundation, the Children’s Cancer Foundation, the Swedish Research Council and the Swedish Foundation for Strategic Research. We acknowledge Lisa Christiansson for skillful technical assistance.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Butt E, Abel K, Krieger M, Palm D, Hoppe V, Hoppe J, et al. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J Biol Chem. 1994;269:14509–14517. [PubMed] [Google Scholar]
- Cen B, Deguchi A, Weinstein IB. Activation of protein kinase G increases the expression of p21CIP1, p27KIP1, and histidine triad protein 1 through Sp1. Cancer Res. 2008;68:5355–5362. doi: 10.1158/0008-5472.CAN-07-6869. [DOI] [PubMed] [Google Scholar]
- Chen L, Daum G, Chitaley K, Coats SA, Bowen-Pope DF, Eigenthaler M, et al. Vasodilator-stimulated phosphoprotein regulates proliferation and growth inhibition by nitric oxide in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:1403–1408. doi: 10.1161/01.ATV.0000134705.39654.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M, Briscoe J. Neural crest development is regulated by the transcription factor Sox9. Development. 2003;130:5681–5693. doi: 10.1242/dev.00808. [DOI] [PubMed] [Google Scholar]
- Chikuda H, Kugimiya F, Hoshi K, Ikeda T, Ogasawara T, Shimoaka T, et al. Cyclic GMP-dependent protein kinase II is a molecular switch from proliferation to hypertrophic differentiation of chondrocytes. Genes Dev. 2004;18:2418–2429. doi: 10.1101/gad.1224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AL, Haynes JM. Protein kinase G II-mediated proliferative effects in human cultured prostatic stromal cells. Cell Signal. 2004;16:253–261. doi: 10.1016/s0898-6568(03)00134-7. [DOI] [PubMed] [Google Scholar]
- Deguchi A, Thompson WJ, Weinstein IB. Activation of protein kinase G is sufficient to induce apoptosis and inhibit cell migration in colon cancer cells. Cancer Res. 2004;64:3966–3973. doi: 10.1158/0008-5472.CAN-03-3740. [DOI] [PubMed] [Google Scholar]
- Dromard C, Bartolami S, Deleyrolle L, Takebayashi H, Ripoll C, Simonneau L, et al. NG2 and Olig2 expression provides evidence for phenotypic deregulation of cultured central nervous system and peripheral nervous system neural precursor cells. Stem Cells. 2007;25:340–353. doi: 10.1634/stemcells.2005-0556. [DOI] [PubMed] [Google Scholar]
- Finzsch M, Stolt CC, Lommes P, Wegner M. Sox9 and Sox10 influence survival and migration of oligodendrocyte precursors in the spinal cord by regulating PDGF receptor alpha expression. Development. 2008;135:637–646. doi: 10.1242/dev.010454. [DOI] [PubMed] [Google Scholar]
- Gambaryan S, Palmetshofer A, Glazova M, Smolenski A, Kristjansson GI, Zimmer M, et al. Inhibition of cGMP-dependent protein kinase II by its own splice isoform. Biochem Biophys Res Commun. 2002;293:1438–1444. doi: 10.1016/S0006-291X(02)00412-6. [DOI] [PubMed] [Google Scholar]
- Gudi T, Hong GK, Vaandrager AB, Lohmann SM, Pilz RB. Nitric oxide and cGMP regulate gene expression in neuronal and glial cells by activating type II cGMP-dependent protein kinase. FASEB J. 1999;13:2143–2152. [PubMed] [Google Scholar]
- Guha A, Dashner K, Black PM, Wagner JA, Stiles CD. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int J Cancer. 1995;60:168–173. doi: 10.1002/ijc.2910600206. [DOI] [PubMed] [Google Scholar]
- Hagerstrand D, Hesselager G, Achterberg S, Wickenberg Bolin U, Kowanetz M, Kastemar M, et al. Characterization of an imatinib-sensitive subset of high-grade human glioma cultures. Oncogene. 2006;25:4913–4922. doi: 10.1038/sj.onc.1209497. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- Hou Y, Gupta N, Schoenlein P, Wong E, Martindale R, Ganapathy V, et al. An anti-tumor role for cGMP-dependent protein kinase. Cancer Lett. 2006;240:60–68. doi: 10.1016/j.canlet.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Humphrey PA, Wong AJ, Vogelstein B, Friedman HS, Werner MH, Bigner DD, et al. Amplification and expression of the epidermal growth factor receptor gene in human glioma xenografts. Cancer Res. 1988;48:2231–2238. [PubMed] [Google Scholar]
- Johansson FK, Brodd J, Eklof C, Ferletta M, Hesselager G, Tiger CF, et al. Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci USA. 2004;101:11334–11337. doi: 10.1073/pnas.0402716101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson FK, Goransson H, Westermark B. Expression analysis of genes involved in brain tumor progression driven by retroviral insertional mutagenesis in mice. Oncogene. 2005;24:3896–3905. doi: 10.1038/sj.onc.1208553. [DOI] [PubMed] [Google Scholar]
- Johansson Swartling F. Identifying candidate genes involved in brain tumor formation. Ups J Med Sci. 2008;113:1–38. doi: 10.3109/2000-1967-215. [DOI] [PubMed] [Google Scholar]
- Kordes U, Hagel C. Expression of SOX9 and SOX10 in central neuroepithelial tumor. J Neurooncol. 2006;80:151–155. doi: 10.1007/s11060-006-9180-7. [DOI] [PubMed] [Google Scholar]
- Lahortiga I, Akin C, Cools J, Wilson TM, Mentens N, Arthur DC, et al. Activity of imatinib in systemic mastocytosis with chronic basophilic leukemia and a PRKG2–PDGFRB fusion. Haematologica. 2008;93:49–56. doi: 10.3324/haematol.11836. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- Nister M, Libermann TA, Betsholtz C, Pettersson M, Claesson-Welsh L, Heldin CH, et al. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res. 1988;48:3910–3918. [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Smolenski A, Burkhardt AM, Eigenthaler M, Butt E, Gambaryan S, Lohmann SM, et al. Functional analysis of cGMP-dependent protein kinases I and II as mediators of NO/cGMP effects. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:134–139. doi: 10.1007/pl00005234. [DOI] [PubMed] [Google Scholar]
- Smolenski A, Poller W, Walter U, Lohmann SM. Regulation of human endothelial cell focal adhesion sites and migration by cGMP-dependent protein kinase I. J Biol Chem. 2000;275:25723–25732. doi: 10.1074/jbc.M909632199. [DOI] [PubMed] [Google Scholar]
- Sottile V, Li M, Scotting PJ. Stem cell marker expression in the Bergmann glia population of the adult mouse brain. Brain Res. 2006;1099:8–17. doi: 10.1016/j.brainres.2006.04.127. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 2003;17:1677–1689. doi: 10.1101/gad.259003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Markerink-van Ittersum M, Steinbusch HW, De Vente J. Nitric oxide-mediated cGMP synthesis in oligodendrocytes in the developing rat brain. Glia. 1997;19:286–297. [PubMed] [Google Scholar]
- Taylor MK, Ahmed R, Begley M, Uhler MD. Autoinhibition and isoform-specific dominant negative inhibition of the type II cGMP-dependent protein kinase. J Biol Chem. 2002;277:37242–37253. doi: 10.1074/jbc.M202060200. [DOI] [PubMed] [Google Scholar]
- Uhrbom L, Hesselager G, Nister M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–5279. [PubMed] [Google Scholar]
- Villalobo A. Nitric oxide and cell proliferation. FEBS J. 2006;273:2329–2344. doi: 10.1111/j.1742-4658.2006.05250.x. [DOI] [PubMed] [Google Scholar]
- Vindelov LL, Christensen IJ, Nissen NI. A detergent-trypsin method for the preparation of nuclei for flow cytometric DNA analysis. Cytometry. 1983;3:323–327. doi: 10.1002/cyto.990030503. [DOI] [PubMed] [Google Scholar]
- Walz C, Metzgeroth G, Haferlach C, Schmitt-Graeff A, Fabarius A, Hagen V, et al. Characterization of three new imatinib-responsive fusion genes in chronic myeloproliferative disorders generated by disruption of the platelet-derived growth factor receptor beta gene. Haematologica. 2007;92:163–169. doi: 10.3324/haematol.10980. [DOI] [PubMed] [Google Scholar]
- Wang H, Leav I, Ibaragi S, Wegner M, Hu GF, Lu ML, et al. SOX9 is expressed in human fetal prostate epithelium and enhances prostate cancer invasion. Cancer Res. 2008;68:1625–1630. doi: 10.1158/0008-5472.CAN-07-5915. [DOI] [PubMed] [Google Scholar]
- Wang H, McKnight NC, Zhang T, Lu ML, Balk SP, Yuan X. SOX9 is expressed in normal prostate basal cells and regulates androgen receptor expression in prostate cancer cells. Cancer Res. 2007;67:528–536. doi: 10.1158/0008-5472.CAN-06-1672. [DOI] [PubMed] [Google Scholar]
- Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, et al. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997;57:4183–4186. [PubMed] [Google Scholar]
- Yin D, Wang X, Konda BM, Ong JM, Hu J, Sacapano MR, et al. Increase in brain tumor permeability in glioma-bearing rats with nitric oxide donors. Clin Cancer Res. 2008;14:4002–4009. doi: 10.1158/1078-0432.CCR-07-1826. [DOI] [PubMed] [Google Scholar]
- Yoshioka A, Yamaya Y, Saiki S, Kanemoto M, Hirose G, Pleasure D. Cyclic GMP/cyclic GMP-dependent protein kinase system prevents excitotoxicity in an immortalized oligodendroglial cell line. J Neurochem. 2000;74:633–640. doi: 10.1046/j.1471-4159.2000.740633.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.