

Abstract
BACKGROUND AND PURPOSE
Cannabinoid CB2 receptor activation by selective agonists has been shown to produce analgesic effects in preclinical models of inflammatory and neuropathic pain. However, mechanisms underlying CB2-mediated analgesic effects remain largely unknown. The present study was conducted to elucidate the CB2 receptor expression in ‘pain relevant’ tissues and the potential sites of action of CB2 agonism in rats.
EXPERIMENTAL APPROACH
Expression of cannabinoid receptor mRNA was evaluated by quantitative RT-PCR in dorsal root ganglia (DRGs), spinal cords, paws and several brain regions of sham, chronic inflammatory pain (CFA) and neuropathic pain (spinal nerve ligation, SNL) rats. The sites of CB2 mediated antinociception were evaluated in vivo following intra-DRG, intrathecal (i.t.) or intraplantar (i.paw) administration of potent CB2-selective agonists A-836339 and AM1241.
KEY RESULTS
CB2 receptor gene expression was significantly up-regulated in DRGs (SNL and CFA), spinal cords (SNL) or paws (CFA) ipsilateral to injury under inflammatory and neuropathic pain conditions. Systemic A-836339 and AM1241 produced dose-dependent efficacy in both inflammatory and neuropathic pain models. Local administration of CB2 agonists also produced significant analgesic effects in SNL (intra-DRG and i.t.) and CFA (intra-DRG) pain models. In contrast to A-836339, i.paw administration of AM-1241 dose-relatedly reversed the CFA-induced thermal hyperalgesia, suggesting that different mechanisms may be contributing to its in vivo properties.
CONCLUSIONS AND IMPLICATIONS
These results demonstrate that both DRG and spinal cord are important sites contributing to CB2 receptor-mediated analgesia and that the changes in CB2 receptor expression play a crucial role for the sites of action in regulating pain perception.
Keywords: cannabinoid, CB2, A-836339, AM1241, inflammatory pain, neuropathic pain
Introduction
The cannabinoid receptors belong to the G-protein coupled receptor (GPCR) super family containing seven transmembrane domains (Matsuda et al., 1990; Munro et al., 1993). Two cannabinoid receptor subtypes have been identified: CB1 and CB2. The CB2 receptor couples through Gi/o proteins to inhibit adenylate cyclase and stimulate MAP kinase activities (Di Marzo et al., 2004). The CB2 receptor shares approximately 44% overall sequence homology with the CB1 receptor and 68% homology within the transmembrane domains Matsuda et al. 1990; Munro et al. 1993).
Although the analgesic properties of non-selective cannabinoid receptor agonists have been known for many years, there is now an increasing body of evidence to support the potential utility of selective cannabinoid CB2 receptor agonists for the treatment of pain (Guindon and Hohmann, 2008). Strong supporting evidence for this hypothesis is provided from knockout studies (Zimmer et al., 1999; Ibrahim et al., 2006), and studies with a handful of CB2-selective agonists, such as HU308 (Hanus et al., 1999), JWH133 (Elmes et al., 2005), AM1241 (Malan et al., 2001, 2003; Ibrahim et al., 2003, 2005), GW405833 (Valenzano et al., 2005; Whiteside et al., 2005, 2007), JWH015 (Romero-Sandoval and Eisenach, 2007), A-796260 (Yao et al., 2008) and GSK554418A (Giblin et al., 2009), which have demonstrated broad-spectrum efficacy in preclinical models of inflammatory, moderate to severe post-operative and neuropathic pain. In this regard, the non-selective cannabinoids such as Δ9-tetrahydrocannabinol (THC) retained analgesic activity in knockout animals lacking the CB1 receptor (Zimmer et al., 1999). Also, a lack of analgesic efficacy for the CB2-selective ligand AM1241 was demonstrated in CB2 knockout mice (Ibrahim et al., 2006), leading to the conclusion that CB2 receptor activation contributes to the analgesic properties of cannabinoids. The discovery of analgesic effects of CB2-selective ligands such as AM1241 also confirmed the potential for use of CB2 receptor agonists in the treatment of pain without causing centrally CB1-mediated side effects such as sedation, loss of motor coordination and hypothermia (Malan et al., 2001; Bingham et al., 2007). Furthermore, studies using the CB2-selective ligand AM1241 have implicated modulation of endogenous opioid systems as the underlying mechanism for CB2 mediated analgesia (Ibrahim et al., 2005), whereas several reports with other selective CB2 agonists A-796260 and GW405833 have failed to demonstrate an opioid dependent mechanism (Whiteside et al., 2005; Yao et al., 2008). These findings suggest that AM1241 may be a unique ligand that is not generally representative of CB2 agonists with respect to mechanism(s) of action.
The CB2 receptor has historically been referred to as the ‘peripheral’ cannabinoid receptor due to its predominant expression on cells of the immune system and the spleen (Galiègue et al., 1995; Di Marzo et al., 2004). In contrast, the CB1 receptor has long been regarded as the ‘central’ cannabinoid receptor for its high level of expression in the brain and other neurological tissues and its mediation of cannabinoid psychotropic effects (Mackie, 2006). However, recent studies have shown up-regulation of the CB2 receptor in CNS tissues such as spinal cord following nerve injury, specifically on non-neuronal cells presumed to be microglia (Zhang et al., 2003; Romero-Sandoval and Eisenach, 2007; Romero-Sandoval et al., 2008). As stated above, analgesia mediated by CB2-selective agonists can offer significant advantages as CB-mediated undesirable side effects are associated with the activation of the CB1 receptor subtype. Yet, the mechanism(s) and site(s) of action underlying CB2-mediated analgesia remain largely unexplained. In the present study, we evaluated CB2 gene expression changes in various tissues obtained from animals under chronic inflammatory (CFA) or neuropathic pain (L5-L6 spinal nerve ligation injury, SNL) conditions. To further support a role for CB2 receptors located in dorsal root ganglia (DRG) and the spinal cord in CB2-mediated analgesia, we investigated the site-specific effects of two CB2-selective agonists A-836339 (Dart et al., 2007; Yao et al., 2009) and AM1241 (Malan et al., 2003) in both inflammatory and neuropathic pain models.
Experimental procedures
Animals, compounds and dosing
Male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 250–300 g at the time of testing were used for all experiments, unless indicated otherwise. The animals were housed in Association for Assessment and Accreditation of Laboratory Animal Care-approved facilities at Abbott Laboratories in a temperature-regulated environment under a controlled 12-h light–dark cycle, with lights on at 0600. Food and water were available ad libitum at all times except during testing. All testing was done following procedures outlined in protocols approved by Abbott Laboratories' Institutional Animal Care and Use Committee and followed the Guidelines on Ethical Standards for Investigations of Experimental Pain in Conscious Animals laid down by the International Association for the Study of Pain (Zimmermann, 1983).
A-836339 was synthesized at Abbott Laboratories (Dart et al., 2007). AM1241 is available through Sigma-Aldrich Chemical Co (catalogue #A6478, St. Louis, MO). Rimonabant (also known as SR141716A, a CB1 receptor selective antagonist) and SR144528 (a CB2 receptor selective antagonist) were also prepared at Abbott Laboratories according to literature methods (Barth et al., 1995, 1997). Gabapentin was purchased from ChemPacific (Baltimore, MD). Complete Freund's adjuvant (CFA) was obtained from Sigma-Aldrich Chemical Co.
A-836339 and AM1241 (dissolved in 5% DMSO/95% PEG-400, v/v) and gabapentin (prepared in water) were administered intraperitoneally (i.p.) at a volume of 2 mL·kg−1 30 min before behavioural testing. A-836339 and AM1241 were dissolved in 10% DMSO/90% hydroxyl-β-cyclodextrin (30%, w/w) in water (v/v) for intra-DRG or i.t. (administration 30 min before behavioural testing) at an injection volume of 10 µL and for i.paw administration (50 µL). For the antagonist blockade studies, rimonabant or SR144528 was dissolved in 5% DMSO/95% PEG-400 (v/v, 1 mL·kg−1) was i.p. administered 15 min before CB2 agonist administration.
RNA isolation and real-time quantitative polymerase chain reaction (qRT-PCR) for CB receptor mRNA gene profiling
Tissues of interest, that is, paws, spinal cords, DRGs and brain regions (hippocampus, thalamus, sensory cortex and brain stem) were collected individually from animals 48 h post CFA injection or 14 days post-L5/L6 spinal nerve ligation surgery after the rats were humanely killed (with CO2). Tissues from sham operated animals were used as controls. Total RNA samples were prepared using Trizol® reagent (Invitrogen, Carlsbad, CA, USA) following the vendor's protocol. RNA samples were treated with approximately 30 Kunitz units of DNase I (Qiagen, Valencia, CA, USA) for 15 min at room temperature to remove genomic DNA contamination. For CB2 detection, custom forward (5′ GCA GCG TGA CCA TGA CCT T-3′) and reverse (5′-AGG TAT CGG TCA ACA GCA GTC AG-3′) primers (accession #NM020543) were used with a probe (5′-ACG GCC TCT GTG GGC AGC CTG-3′) conjugated at the 5′ end with 6-carboxyfluorescein (FAM) and at the 3′ end with Black Hole Quencher™ 1. TaqMan Gene Expression Assays were used for detection of CB1 (Rn00562880_m1) and HPRT1 (Rn01527840_m1) (Applied Biosystems, Foster City, CA, USA). RT-PCR reactions were prepared in a total reaction volume of 25 µL using the SuperScript III Platinum One-Step Quantitative RT-PCR System (Invitrogen, Carlsbad, CA, USA) and analysed using the 7300 Real-Time PCR System (Applied Biosystems). The relative levels of CB1 and CB2 expression were normalized to the expression of HPRT1. CB2 receptor protein levels were not conducted in this study because a robust commercially available CB2 antibody was not available, according to the data observed in our laboratory.
In vivo pain models
Rat complete Freund's adjuvant (CFA) – induced chronic inflammatory pain
Chronic inflammatory mechanical allodynia was induced by injection of 150 µL of a 50% emulsion of CFA in phosphate buffered saline (PBS) into the intra-plantar surface (palmar site) of the right hind paw in rats; control animals received only PBS treatment. Thermal hyperalgesia was assessed 48 h post CFA injection. On the day of testing, A-836339 or AM1241 was injected 30 min (i.p., i.t., intra-DRG or i.paw) before testing for thermal hyperalgesic effects.
Thermal hyperalgesia was determined using a commercially available thermal paw stimulator (UARDG, University of California, San Diego, CA, USA) as described by Hargreaves et al. (1988). Rats were placed into individual plastic cubicles mounted on a glass surface maintained at 30°C, and allowed a 20 min habituation period. A thermal stimulus, in the form of radiant heat emitted from a focused projection bulb, was then applied to the plantar surface of each hind paw. The stimulus current was maintained at 4.50 ± 0.05 amp, and the maximum time of exposure was set at 20.48 s to limit possible tissue damage. The latency to a brisk withdrawal of the hind paw from the thermal stimulus was recorded automatically using photodiode motion sensors. The right and left hind paws of each rat were tested in three sequential trials at approximately 5 min intervals. Paw withdrawal latency (PWL) was calculated as the mean of the two shortest latencies. PWL were measured 30 min post-A-836339 or AM1241 administration in both the CFA-inflamed and un-injected paws.
Rat SNL model of neuropathic pain
As previously described in detail by Kim and Chung (1992), rats were placed under isoflurane anaesthesia and a 1.5 cm incision was made dorsal to the lumbosacral plexus. The paraspinal muscles (left side) were separated from the spinous processes, the L5 and L6 spinal nerves isolated, and tightly ligated with 5-0 silk suture distal to the dorsal root ganglion. Care was taken to avoid ligating the L4 spinal nerve. Following spinal nerve ligation, a minimum of 7 days of recovery and no more than 2 weeks was allowed prior to the behavioural testing (mechanical allodynia). Only rats with threshold scores ≤4.5×g were considered allodynic and utilized in pharmacological experiments.
Mechanical allodynia was measured using calibrated von Frey filaments (Stoelting, Wood Dale, IL). Paw withdrawal threshold (PWT) was determined by using the Dixon's up–down method (Chaplan et al., 1994). Rats were placed into inverted individual plastic containers (20 × 12.5 × 20 cm) on top of a suspended wire mesh with a 1 cm2 grid to provide access to the ventral side of the hind paws, and acclimated to the test chambers for 20 min. The von Frey filaments were presented perpendicularly to the plantar surface of the selected hind paw, and then held in this position for approximately 8 s with enough force to cause a slight bend in the filament. Positive responses included an abrupt withdrawal of the hind paw from the stimulus, or flinching behaviour immediately following removal of the stimulus. A 50% withdrawal threshold was determined using an up–down procedure (Dixon, 1980). The strength of the maximum filament used for von Frey testing was 15.0×g. A per cent maximal possible effect (% MPE) of testing compound was calculated according to the formula: [(compound – treated threshold) – (vehicle – treated threshold)]/[(maximum threshold) – (vehicle-treated threshold)] × 100%, where the maximum threshold was equal to 15×g.
Rat chronic constriction injury (CCI) model of neuropathic pain
As previously described in detail by the method of Bennett and Xie (1988), the right common sciatic nerve was isolated at mid-thigh level, and loosely ligated by four chromic gut (5-0) ties separated by an interval of 1 mm. All animals were left to recover for at least 2 weeks and no more than 3 weeks prior to testing of mechanical allodynia.
Mechanical testing was measured using calibrated von Frey filaments as the procedures described above. Only rats with a baseline threshold score of less than 4.5×g were used in this study, and animals demonstrating motor deficit were excluded.
Rat intrathecal catheterization
A group of rats were implanted with i.t. catheters, as previously described (Yaksh and Rudy, 1976), to investigate potential spinal sites of action of A-836339 and AM1241 in this model. Rats were placed under isoflurane anaesthesia and mounted onto an intrathecal stereotaxic instrument by placing the animal into blunt ear bars, which held the animal's head firmly. An incision was made vertically from the dorsal surface of the occipital bone to the base of the skull (2 cm). Tissue was then displaced using a blunt probe so that the atlanto-occipital membrane at the base of the skull was clearly seen. A custom-made intrathecal PE-5 catheter (Marsil Enterprises, San Diego, CA, USA) was inserted through the atlanto-occipital membrane via a small hole in the cisterna magnum. The catheter was then advanced 8.5 cm caudally such that the tip ended in the spinal subarachnoid space around the lumbar enlargement (L4-L6). The catheter was then secured to the musculature at the incision site. The incision was closed with surgical wound clips. The catheter was filled with sterile physiological saline and the end of the catheter was heat-sealed. Animals with catheters were allowed 1 week of recovery from surgery before behavioural testing. For i.t. injection, a Hamilton syringe (50 µL) was connected to the external portion of the catheter and 10 µL of drug solution was slowly injected into the catheter over a period of 1 min. The catheter was subsequently flushed with 10 µL of sterile water and the behavioural testing was conducted 30 min post-CB2 agonist administration. After the behavioural testing was completed, cannula placement was confirmed by the infusion of 0.5% Evans blue dye in saline solution (10 µL) and subsequent dissection. In the pilot studies, we had demonstrated that intrathecal catheterization procedures did not change the paw withdrawal baseline of either CFA-inflamed or SNL paws.
Rat intra-DRG catheterization
A group of rats were also implanted with intrathecal catheters as previously described (Rueter et al., 2003) to investigate a potential DRG site of action of A-836339 and AM1241. Under isoflurane anaesthesia, an incision was made on the dorsal portion of the hip and the muscle was blunt dissected to reveal the spinal processes. The left L5 DRG was exposed by removing the posterior articular process of the L5 vertebra. The catheter constructed of PE20 tubing was implanted with the tip positioned about 1 to 2 mm dorsal to the exposed L5 DRG. A small piece of absorbable gelatin sponge (Gelfoam®, Pharmacia & Upjohn Co, Division of Pfizer Inc, Kalamazoo, MI, USA) was packed between the DRG and the tip of the PE tubing to prevent the catheter from damaging the ganglion. The catheter was sutured to the muscle and fascia, then run subcutaneously and exteriorized between the shoulder blades. Saline was infused into the catheter, and the catheter was heat-sealed. For intra-DRG injection, a Hamilton syringe (50 µL) was connected to the external portion of the catheter and 10 µL of drug solution was slowly injected into the catheter over a period of 1 min. The catheter was subsequently flushed with 10 µL of sterile water and behavioural testing was conducted 30 min post-CB2 agonist administration. At the end of each experiment, the area of initial operation was re-exposed and the status of the tubing was examined. An injection of 10 µL of Evans blue dye (0.5%) showed that the tubing permitted unobstructed, free passage of injected material in all cases. In the pilot studies, we had demonstrated that intra-DRG catheterization procedures did not alter the paw withdrawal baseline of either CFA-inflamed or SNL paws.
Data analysis
The statistical analysis was carried out using GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA). The values were represented as mean ± S.E.M. All in vivo behavioural studies to determine the sites of actions were conducted in a randomized blinded fashion. Statistical significance of group means difference was measured by one-way analysis of variance (anova), followed by Bonferroni's post hoc analysis. In all cases P < 0.05 was assumed as the level for statistical significance. ED50 values (Effective Dose, 50%) (GraphPad Prism) were also calculated by linear regression analysis and reported with the 95% confidence interval (95%CI). The drug/molecular target nomenclature (e.g. receptors, ion channels and so on) used in the present study conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Results
Changes in CB mRNA expression in the CFA model of inflammatory pain
To determine if induction of an inflammatory pain state altered expression of CB2 receptors in tissues associated with pain responses, expression of CB2 mRNA was analysed in the L3-L5 spinal cords and DRGs, paws and several brain regions including hippocampus, sensory cortex, thalamus and brain stem 48 h after CFA injection, using qRT-PCR. The levels of CB2 mRNA were significantly up-regulated in ipsilateral DRGs (Figure 1A) and paws (Figure 1C) as compared with the sham controls, whereas the expression of the CB2 mRNA in spinal cord (Figure 1B) and hippocampus, thalamus, cortex and brain stem was not altered (Table 1). Interestingly, the contralateral DRGs also showed increased levels of CB2 mRNA expression as compared with sham controls (Figure 1A). The expression of the CB1 mRNA in these animals was not altered (Table 2).
Figure 1.
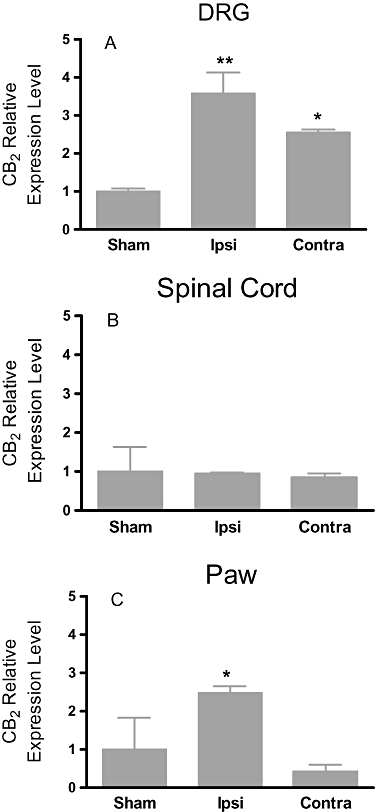
Expression of CB2 mRNA in the CFA model of inflammatory pain in rats. CB2 gene expression up-regulation observed in ipsilateral paw and DRG of CFA rats, but not in spinal cord. The relative levels of CB2 expression were normalized to the expression of HPRT1. Data expressed as mean ± SEM **P < 0.01 as compared with sham rats (n = 5).
Table 1.
Expression of CB2 mRNA in the CFA model of inflammatory pain in rats. CB2 gene expressions detected in supra-spinal tissues hippocampus, thalamus, cortex and brainstem were not altered. Data expressed as mean ± SEM (n = 3–5)
| CB2 relative expression levelsa | ||
|---|---|---|
| Tissues | Sham | CFA |
| Hippocampus | 1.83 ± 0.87 | 2.01 ± 0.38 |
| Thalamus | 1.14 ± 0.29 | 1.04 ± 0.09 |
| Sensory cortex | 1.61 ± 0.68 | 1.57 ± 0.36 |
| Brainstem | 0.98 ± 0.05 | 0.88 ± 0.08 |
The relative levels of CB2 expression were normalized to the expression of HPRT1.
Table 2.
Expression of CB1 mRNA in the CFA model of inflammatory pain and the SNL model of neuropathic pain in rats. CB1 gene expressions observed in ipsilateral tissues (paw, DRG and spinal cord) and supra-spinal tissues (hippocampus, thalamus, cortex and brainstem) were not altered. Data expressed as mean ± SEM (n = 3–5)
| Tissues | CB1 relative expression levelsa | |
|---|---|---|
| Sham | CFA | |
| DRG | 1.00 ± 0.11 | 1.31 ± 0.01 |
| Spinal cord | 0.99 ± 0.29 | 0.99 ± 0.29 |
| Paw | 1.00 ± 0.50 | 0.25 ± 0.10 |
| Hippocampus | 1.01 ± 0.38 | 2.14 ± 0.96 |
| Thalamus | 1.00 ± 0.47 | 0.82 ± 0.27 |
| Sensory cortex | 1.00 ± 0.16 | 1.11 ± 0.42 |
| Brainstem | 1.04 ± 0.58 | 1.47 ± 0.63 |
| Sham | SNL | |
| DRG | 1.33 ± 0.10 | 1.31 ± 0.03 |
| Spinal cord | 1.25 ± 0.44 | 0.94 ± 0.15 |
| Paw | 0.82 ± 0.30 | 0.40 ± 0.10 |
| Hippocampus | 2.09 ± 0.76 | 2.07 ± 1.37 |
| Thalamus | 0.50 ± 0.18 | 1.12 ± 0.47 |
| Sensory cortex | 1.54 ± 1.05 | 1.23 ± 0.20 |
| Brainstem | 1.83 ± 0.04 | 2.04 ± 0.65 |
The relative levels of CB1 expression were normalized to the expression of HPRT1.
Changes in CB mRNA expression in the SNL model of neuropathic pain
To elucidate the possible changes in CB2 receptors in the SNL model of neuropathic pain model, we also examined CB2 mRNA levels in the tissues as described above for the CFA model. The tissues were collected 2 weeks after ligation of the L5-L6 spinal nerve. Ipsilateral L5-L6 DRGs had a significantly higher level of CB2 mRNA as compared with the contralateral side and sham controls (Figure 2A). The contralateral DRGs also showed increased levels of CB2 receptor expression as compared with sham controls (Figure 2A). A significant increase (68% as compared with sham control) of CB2 mRNA expression in the ipsilateral spinal cord was also observed (Figure 2B). In contrast, expression of the CB2 mRNA in supraspinal tissues, hippocampus, thalamus, cortex and brain stem was not altered as compared with sham groups (Figure 2C–F). The expression of CB2 mRNA was also not changed in paw tissues derived from SNL as compared with sham rats (data not shown). No difference in the expression of the CB1 mRNA in these tissues was detected (Table 2).
Figure 2.
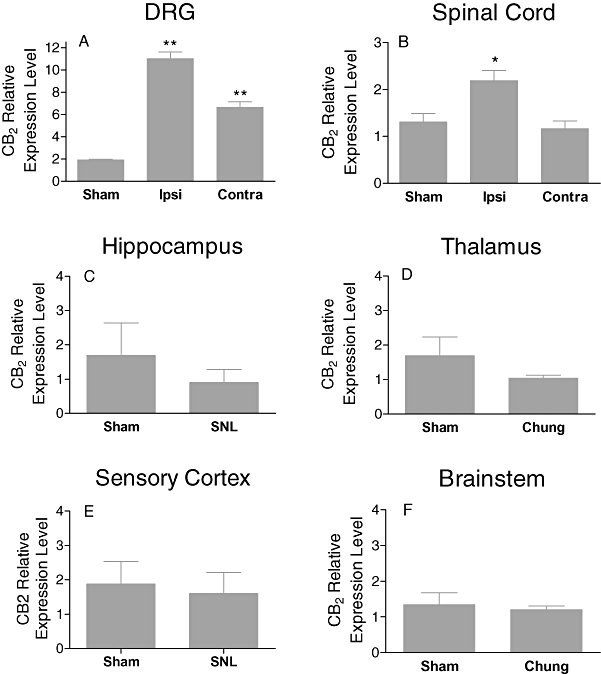
Upregulation of CB2 gene expression in the SNL model of chronic neuropathic pain in rats. In spinal cord, a significant increase in CB2 mRNA versus sham operated was observed. In DRGs, 11 fold increases in CB2 message versus sham operated rats was observed. CB2 gene expression was not upregulated in sensory cortex, hippocampus, thalamus or brainstem. The relative levels of CB2 expression were normalized to the expression of HPRT1. Data expressed as mean ± SEM. *P < 0.05; **P < 0.01 as compared with sham rats (n = 5).
Effects of A-836339 on CFA-induced chronic inflammatory thermal hyperalgesia
A-836339 elicited significant anti-hyperalgesic effects in CFA-induced inflammatory pain in rats. Administration of CFA produced a significant decrease in PWL, from 11.6 ± 0.5 to 5.8 ± 0.3 s, demonstrating inflammation-induced thermal hypersensitivity. A-836339 (1, 3, 10 µmol·kg−1, i.p.) significantly reversed CFA-induced decrease in PWL to control levels in a dose-related fashion, resulting in an 80% effect at the highest dose tested with an ED50 value of 1.8 µmol·kg−1 (95% CI = 1.5–2.2) (Table 3). A-836339 at 10 µmol·kg−1 had no effect on PWL of the contralateral non-inflamed paw (10.1 ± 0.3 s), indicative of a specific anti-hyperalgesic effect. Systemic administration of SR144528 (10 µmol·kg−1, i.p.), a CB2 receptor selective antagonist, completely reversed A-836339-evoked anti-hyperalgesic effect. In contrast, rimonabant (10 µmol·kg−1, i.p.), a CB1 receptor selective antagonist did not significantly block the anti-hyperalgesic effect of A-836339 (Table 3). These data demonstrate that the effects of A-836339 are mediated through activation of CB2 receptors. However, the effects of A-836339 in the CFA model were not reversed by an opioid receptor antagonist naloxone. A-836339 alone (10 µmol·kg−1, i.p.) produced a significant anti-hyperalgesic effect (58%, P < 0.01 vs. vehicle). Pretreatment with naloxone (10 mg·kg−1 i.p.) 20 min prior to administration of A-836339 did not block the anti-hyperalgesic effect of A-836339 (71%, P < 0.01 vs. vehicle, P > 0.05 vs. A-836339 alone).
Table 3.
Efficacy of A-836339 in the CFA-induced inflammatory pain model. Administration of CFA produced a significant decrease in paw withdrawal latencies (PWL) in the ipsilateral but not contralateral paws, significantly diminished from 11.6 ± 0.5 to 5.8 ± 0.3 s. A-836339 exhibited dose-dependent reversal of the decreased PWL and the effects were blocked by antagonists selective at CB2 (SR144528), but not at CB1 (rimonabant) receptors
| A-836339 | ||
|---|---|---|
| Treatment | µmol·kg−1 i.p. | Percent reversal (%) |
| A-836339 | 1 | 31 ± 4**, n = 12 |
| 3 | 68 ± 7**, n = 12 | |
| 10 | 80 ± 5**, n = 12 | |
| + CB2 antagonist SR144528 (10 µmol·kg−1) | ||
| A-836339 alone | 10 | 91 ± 4**, n = 6 |
| A836339 + SR144528 | 10 | 16 ± 5++, n = 6 |
| SR144528 alone | 5 ± 3, n = 6 | |
| + CB1 antagonist rimonabant (10 µmol·kg−1) | ||
| A-836339 alone | 10 | 97 ± 3**, n = 6 |
| A836339 + rimonabant | 10 | 87 ± 4, n = 6 |
| Rimonabant alone | 0 ± 5, n = 6 | |
Antagonist was administered i.p.15 min before A-836339 injection (10 µmol·kg−1). Data are expressed as mean ± SEM
P < 0.01 versus vehicle treated group
P < 0.01 versus A-836339 alone.
To test potential sites of action, A-836339 at 100 nmol·rat−1 (= 0.3 µmol·kg−1) was administered directly into the L4-L6 spinal levels or L5 DRG in rats with chronically implanted i.t. or intra-DRG catheters. Intra-DRG administration of A-836339 significantly reversed CFA-induced hyperalgesic effect (65%, P < 0.01 vs. vehicle, n = 8) (Figure 3A). In contrast, i.t administration of A-836339 at the same dose did not significantly produce reversal of CFA-induced decrease in PWL (14%, P > 0.05 vs. vehicle, n = 8) (Figure 3A). A-836339 was also directly administered (50 µL/i.paw) into the CFA-inflamed or non-injured hindpaws to examine whether the CB2 activation at local paw site contributes to systemic efficacy of the compound. Ipsilateral i.paw administration (palmar site) of A-836339 did not produce any reversal of thermal hyperalgesia. A weak effect (27%, P < 0.05 vs. vehicle) was observed at the highest dose 300 nmol/i.paw. However, similar effects (33%, P < 0.05 vs. vehicle) were also observed with the contralateral i.paw application at this dose (Figure 3B).
Figure 3.
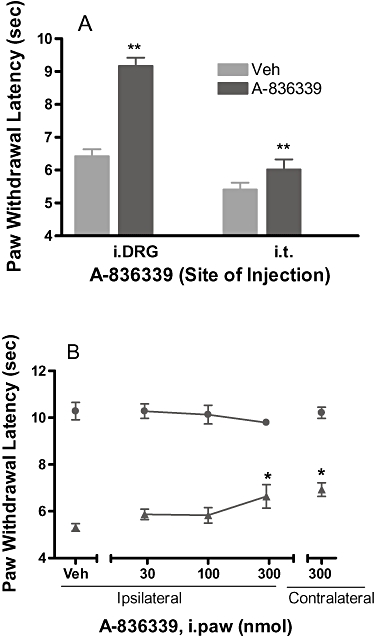
Local site of action of CB2 agonist A-836339 on the CFA model of inflammatory pain in rats. (A) Effects of A-836339 on thermal hyperalgesia following i.DRG or i.t. administration (100 nmol·rat−1). Responses of only the ipsilateral paws of the treated animals were shown. Responses of the respective contralateral paws of all treatment groups are similar to that of the vehicle treated contralateral paws (not shown). (B) Effects of A-836339 on thermal hyperalgesia (▴ ipsilateral paw, • contralateral paw) following ipsilateral or contralateral injection (i.paw) into the intra-plantar surface of the hindpaw. Data represent mean ± SEM (n = 6–8). *P < 0.05; **P < 0.01 as compared with vehicle-treated animals.
Effects of A-836339 in chronic models of neuropathic pain
Administration of A-836339 also produced a significant reversal of nerve injury-induced tactile hypersensitivity in the rat SNL model of neuropathic pain. A reduction in PWTs was observed ipsilateral to the nerve injury (3.1 ± 0.2×g), demonstrating the development of mechanical allodynia. Systemic A-836339 treatment attenuated SNL-induced mechanical allodynia in a dose-related manner with an ED50 of 14.5 µmol·kg−1 i.p. (95% CI: 11–19) and a 67% reduction (P < 0.01 vs. vehicle) at the highest dose tested (30 µmol·kg−1) (Figure 4A). Under the same conditions, i.p. administration of gabapentin (500 µmol/kg−1), a clinical-use analgesic for neuropathic pain, was used as a positive control and produced a statistically significant reversal (53%, P < 0.01) (Figure 4A).
Figure 4.
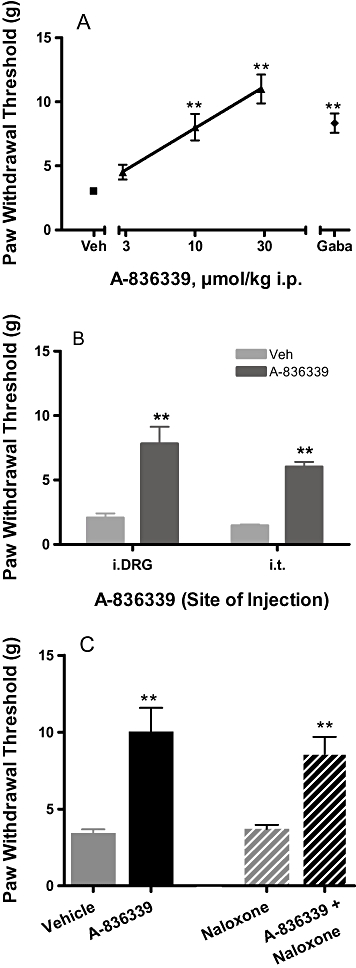
Effects of CB2 agonist A-836339 on mechanical allodynia in the SNL model of neuropathic pain in rats. (A) A-836339 (▴) dose-dependently attenuated mechanical allodynia. Two weeks following spinal nerve injury, A-836339 was injected 30 min before testing. Gabapentin (♦, gaba, 500 µmol·kg−1 i.p.) was included as a positive control. Data expressed as mean ± SEM (n = 12). *P < 0.05; **P < 0.01 as compared with vehicle-treated animals ( , veh). (B) Effects of A-836339 on mechanical allodynia in the SNL model of neuropathic pain following iDRG and i.t. administration (100 nmol·rat−1). Data represent mean ± SEM (n = 8). **P < 0.01 as compared with vehicle-treated animals. (C) Lack of naloxone blockade of A-836339 (30 µmol·kg−1 i.p.) reversal of mechanical allodynia. Data represent mean ± SEM (n = 6). **P < 0.01 as compared with vehicle-treated animals. Responses of only the ipsilateral paws of the treated animals were shown. Responses of the respective contralateral paws of all treatment groups are similar to that of the vehicle treated contralateral paws (not shown).
, veh). (B) Effects of A-836339 on mechanical allodynia in the SNL model of neuropathic pain following iDRG and i.t. administration (100 nmol·rat−1). Data represent mean ± SEM (n = 8). **P < 0.01 as compared with vehicle-treated animals. (C) Lack of naloxone blockade of A-836339 (30 µmol·kg−1 i.p.) reversal of mechanical allodynia. Data represent mean ± SEM (n = 6). **P < 0.01 as compared with vehicle-treated animals. Responses of only the ipsilateral paws of the treated animals were shown. Responses of the respective contralateral paws of all treatment groups are similar to that of the vehicle treated contralateral paws (not shown).
Separate studies were conducted to determine the potential sites of action of CB2 agonism induced anti-allodynic effects. A-836339 (100 nmol·rat−1 = 0.3 µmol·kg−1) was administered directly into the L4-L6 spinal levels or L5 DRG in rats with chronically implanted i.t. or intra-DRG catheters. Intra-DRG administration of A-836339 significantly attenuated mechanical allodynia (45%, P < 0.01, n = 8) compared with vehicle treated animals assessed 30 min after dosing (Figure 5B). Similarly, i.t administration of A-836339 at the same dose also significantly produced reversal of SNL-induced decrease in PWT (33%, P < 0.01 vs. vehicle, n = 8) (Figure 4B). Pretreatment with naloxone (10 mg·kg−1 i.p.) 20 min prior to administration of A-836339 (30 µmol·kg−1, i.p.) did not reverse or attenuate the anti-allodynic effects of A-836339 (69%, P < 0.01 vs. vehicle, n = 6) (Figure 4C).
Figure 5.
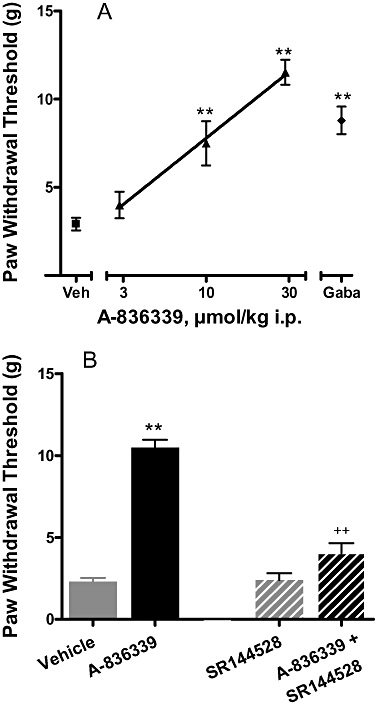
Effects of CB2 agonist A-836339 on mechanical allodynia in the CCI model of neuropathic pain in rats. (A) A-836339 (▴) dose-dependently attenuated mechanical allodynia. Two weeks following spinal nerve injury, A-836339 was injected 30 min before testing. Gabapentin (♦, gaba, 500 µmol·kg−1 i.p.) was included as a positive control. Data expressed as mean ± SEM (n = 12). *P < 0.05, **P < 0.01 as compared with vehicle-treated animals ( , veh). (B) Antagonism of the effect of A-836339 (30 µmol·kg−1, i.p.) by SR144528 (10 µmol·kg−1, i.p.). Data represent mean ± SEM (n = 6). **P < 0.01 as compared with vehicle-treated animals, ++P < 0.01 as compared with A-836339 alone. Responses of only the ipsilateral paws of the treated animals were shown. Responses of the respective contralateral paws of all treatment groups are similar to that of the vehicle treated contralateral paws (not shown).
, veh). (B) Antagonism of the effect of A-836339 (30 µmol·kg−1, i.p.) by SR144528 (10 µmol·kg−1, i.p.). Data represent mean ± SEM (n = 6). **P < 0.01 as compared with vehicle-treated animals, ++P < 0.01 as compared with A-836339 alone. Responses of only the ipsilateral paws of the treated animals were shown. Responses of the respective contralateral paws of all treatment groups are similar to that of the vehicle treated contralateral paws (not shown).
In rats, CCI of the sciatic nerve produced a decrease in PWT to mechanical stimulation with von Frey monofilaments 2 weeks following surgery (PWT = 2.2 ± 0.2×g, Figure 5A), demonstrating the development of mechanical allodynia. Administration of A-836339 attenuated CCI-induced mechanical allodynia in a dose-related manner (n = 12) and produced a 71% effect (P < 0.01 vs. vehicle) at the highest dose (30 µmol·kg−1 i.p.) tested. In the same study, intraperitoneal administration of gabapentin (500 µmol·kg−1) also produced a statistically significant reversal (49%, P < 0.01 vs. vehicle group) of mechanical allodynia. Systemic administration of SR144528 (10 µmol·kg−1, i.p.), a CB2 receptor selective antagonist, completely reversed A-836339-evoked anti-allodynic effect (Figure 5B). A-836339 alone (30 µmol·kg−1, i.p.) produced a significant reversal of allodynia (64% at 30 min, P < 0.01 vs. vehicle, n = 6) and the effects were significantly blocked by the pretreatment with SR144528 (10 µmol·kg−1, i.p.) 15 min prior to administration of A-836339 (14%, P < 0.01 vs. A-836339 alone, n = 6). These results demonstrated that the analgesic effects of A-836339 in the neuropathic pain model were also mediated through CB2 receptor activation.
Effects of AM1241 in inflammatory and neuropathic pain models
To further support a role for CB2 receptors located in DRG and the spinal cord in CB2-mediated analgesia, we also evaluated the effects of CB2 selective reference agonist AM1241 following intra-DRG and i.t. administration. In the CFA-induced inflammatory pain model, acute systemic administration of AM1241 dose-dependently reversed thermal hyperalgesia by 22, 55 and 78% at 2, 6 and 20 µmol·kg−1, i.p., respectively (n = 6) (Figure 6A). AM1241 at 20 µmol·kg−1 dose had no effect on PWL of the contralateral non-inflamed paw (10.4 ± 0.4 s), indicative of a specific anti-hyperalgesic effect in this model. i.t. administration of AM1241 (100 nmol·rat−1 = 0.2 µmol·kg−1) directly into the L4-L6 spinal levels produced a weak anti-hyperalgesic effect (29%, P < 0.01, vs. vehicle). However, a near full efficacy (76%, P < 0.01 vs. vehicle) was observed when the compound was administered into L5 DRG in rats with chronically implanted catheters (Figure 6B). Consistent with literature findings (Malan et al., 2001), we also demonstrated that ipsilateral paw injection (palmar site) of AM1241 dose-relatedly reversed thermal hyperalgesia with a 62% effect (P < 0.01, vs. vehicle) at 6 µmol·kg−1 (Figure 6C). In contrast, an injection of 6 µmol·kg−1 into the contralateral paw only produced a marginal effect (18%), which was significantly different from the effect upon ipsialateral injection (P < 0.01). AM1241 (6 µmol·kg−1) was more efficacious in producing antinociception when administered i.p. than when administered i.paw contralaterally (Figure 6A and C, P < 0.01 55% vs. 18%). This is possibly because the systemic absorption and distribution of the compound is much more efficient from the peritoneal cavity than from paw tissue.
Figure 6.
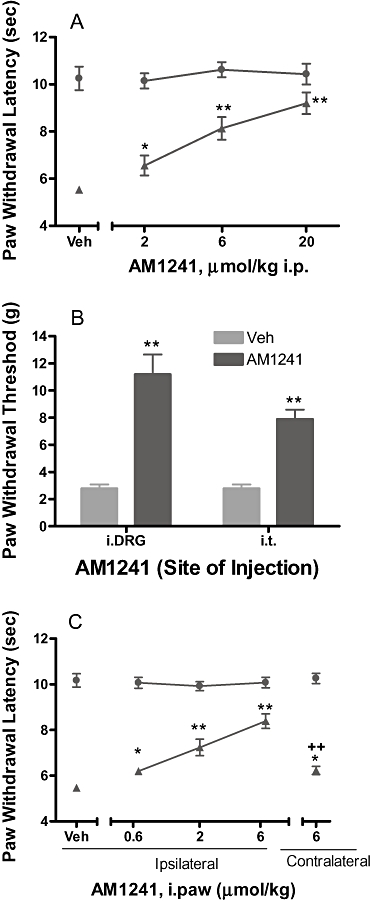
Effects of CB2 agonist AM1241 on the CFA model of inflammatory pain in rats. (A) Effects of AM1241 on thermal hyperalgesia (▴ ipsilateral paw, • contralateral paw) following systemic i.p. administration. (B) Effects of AM1241 on thermal hyperalgesia following i.DRG or i.t. administration (100 nmol·rat−1). Responses of only the ipsilateral paws of the treated animals were shown. Responses of the respective contralateral paws of all treatment groups are similar to that of the vehicle treated contralateral paws (not shown). (C) Effects of AM1241 on thermal hyperalgesia (▴ ipsilateral paw, • contralateral paw) following the hindpaw ipsilateral or contralateral injection (i.paw). Data represent mean ± SEM (n = 6–8). *P < 0.05; **P < 0.01 as compared with vehicle-treated animals; ++P < 0.01 as compared with ipsilateral paw injection.
In the SNL neuropathic pain model, AM1241 significantly reversed mechanical allodynia by 23, 48 and 58%, at 3, 10 and 30 µmol·kg−1, i.p., respectively (n = 6), as compared with the vehicle controls (Figure 7A). Intra-DRG administration of AM1241 (100 nmol = 0.2 µmol·kg−1) attenuated mechanical allodynia (69% P < 0.01 vs. vehicle, n = 8) compared with vehicle treated animals. AM1241 also produced significant effect upon i.t administration (42%, P < 0.01 vs. vehicle, n = 8) (Figure 7B). However, the effects of AM1241 in the SNL model were not sensitive to naloxone blockade (Figure 7C). AM1241 alone (30 µmol·kg−1, i.p.) produced a significant reversal of allodynia (56%, P < 0.01 vs. vehicle, n = 6). Pretreatment with naloxone (10 mg·kg−1) 20 min prior to administration of AM1241 (30 µmol·kg−1, i.p.) did not reverse or attenuate the anti-allodynic effects of AM1241 (55%, P < 0.01 vs. vehicle, n = 6) (Figure 7C). These results are in contrast to the full reversal of the anti-hyperalgesic effects of AM1241 by naloxone under an identical treatment protocol in the CFA model of chronic inflammatory pain (Yao et al., 2008).
Figure 7.
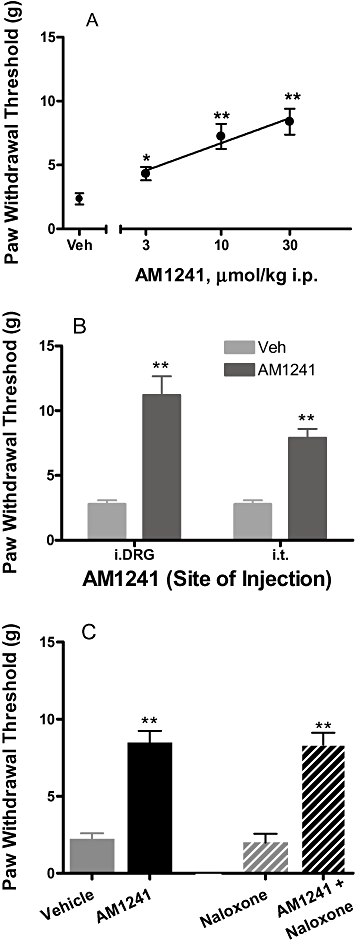
Effects of CB2 agonist AM1241 on mechanical allodynia in the SNL model of neuropathic pain in rats. (A) AM1241 dose-dependently attenuated mechanical allodynia. One to two weeks following spinal nerve injury, A-836339 was injected 30 min before testing. Data expressed as mean ± SEM (n = 6). *P < 0.05, **P < 0.01 as compared with vehicle-treated animals. (B) Effects of AM1241 on mechanical allodynia following i.DRG and i.t. administration. Data represent mean ± SEM (n = 7–8). *P < 0.05; **P < 0.01 as compared with vehicle-treated animals. (C) Lack of naloxone blockade of AM1241 (30 µmol·kg−1 i.p.) reversal of mechanical allodynia. Data represent mean ± SEM (n = 6). **P < 0.01 vs. vehicle-treated animals. Responses of only the ipsilateral paws of the treated animals were shown. Responses of the respective contralateral paws of all treatment groups are similar to that of the vehicle treated contralateral paws (not shown).
Discussion and conclusions
The present study investigated the potential sites of action for CB2 receptor activation-induced analgesic effects in preclinical models of inflammatory and neuropathic pain, using a potent and selective CB2 agonist A-836339 (Dart et al., 2007) and a literature CB2 agonist AM1241 (Malan et al., 2003). A-836339 was potent and efficacious in inflammatory and neuropathic pain models following systemic administration. The analgesic effects of A-836339 were CB2 receptor mediated as they were blocked by a selective CB2 antagonist but not by a selective CB1 antagonist. We had previously reported that A-836339 exhibits high binding affinities at the human and rat CB2 receptors (Ki = 0.4 and 0.8 nM, respectively) and had high selectivity over the CB1 receptor (>200) (Yao et al., 2009). Unlike AM1241 (Ibrahim et al., 2005), the antinociceptive effects evoked by A-836339 do not involve the µ-opioid receptor, a finding similar to those previously reported for A-796260 (Yao et al., 2008) and GW405833 (Whiteside et al., 2005).
Our data also demonstrate that both the DRGs and the spinal cord are important sites contributing to CB2 receptor-mediated analgesia, and that increased CB2 gene expression in DRG in animal models of inflammatory and neuropathic pain plays a significant role for the sites of action in regulating pain perception. To our knowledge, this is the first time a DRG site of action of CB2 agonism has been demonstrated in the preclinical pain models of inflammatory and neuropathic pain following the intra-DRG injection of CB2 agonists. Interestingly, in an in vitro setting, Sagar et al. (2005) has previously reported effects of a CB2 agonist JWH133 on calcium responses of DRG neurons from SNL rats. CB2 mRNA expression was significantly up-regulated in the ipsilateral DRG following L5-L6 spinal nerve injury in rats and similar expression profiles were observed in tissues from CFA-treated animals. CB2 gene expression also appeared to be increased in the spinal cord of neuropathic animals, whereas no significant changes were observed in the supraspinal brain regions. The finding of CB2 mRNA up-regulation in the spinal cords derived from neuropathic (SNL) and not from the inflammatory (CFA) pain model could mean in a broad sense that neuropathic pain is associated with a more central component, whereas inflammatory pain is more peripheral. These results were also in line with the weak anti-hyperalgesic effects of A-836339 and AM-1241 observed in the CFA model of inflammatory pain following the i.t. administration (Figures 3A and 6B). The expression of CB1 was not significantly changed in the tissues examined, consistent with that reported by Zhang et al. (2003). The increase of CB2 and CB1 receptors had been reported in ipsilateral paw skin, L3-L4 DRG or spinal cord derived from neuropathic rats and mice following the saphenous nerve partial ligation (Walczak et al., 2005, 2006), inconsistent with some but not all of our observations in CFA inflammatory and SNL neuropathic pain conditions. The CB2 expression was also up-regulated in contralateral DRGs in both CFA inflammatory and SNL neuropathic pain models. The reason for these findings is currently not clear. Whilst the pathophysiology behind this symmetry is unexplained, there are well documented evidences that indicate peripheral-nerve lesions can affect the contralateral non-lesioned neurons. These contralateral effects are qualitatively similar to those occurring at the ipsilateral side, but are usually smaller in magnitude and have a briefer time course (Koltzenburg et al., 1999). Nonetheless, neither A-836339 nor AM1241 had any effect on PWL of the contralateral non-inflamed paws in the present study, indicative of a specific anti-hyperalgesic effect of the compounds.
To further support a role for the CB2 receptors located in DRG and the spinal cord in CB2-mediated analgesia, we demonstrated the analgesic efficacy of the CB2-selective agonists A-836339 and AM1241 following intra-DRG or i.t. administration in rats with chronic inflammation and neuropathic pain. The doses are well below those required to produce comparable efficacy upon systemic administration and, though the concentration of CB2 agonist at the receptor level in DRG and spinal cords is unknown, it would be expected that local (i.DRG or i.t.) administration of drugs does not result in the systemic exposure and, subsequently, accessing the spinal cord or DRG. Nevertheless, our results further emphasize that both the DRG and spinal cord levels are important sites for CB2 mediated analgesia in chronic neuropathic and inflammatory pain. Tonic activity of the CB receptor at spinal cords and skin tissues has been reported previously in different models (Richardson et al., 1997; Calignano et al., 1998; Lever and Malcangio, 2002). It would be expected that the up-regulation of CB2 receptors would be accompanied by increased tonic activation and CB2 antagonists would be pro-nociceptive. However, the present results had demonstrated that the analgesic effect produced by A-836339 was reversed by systemic administration of the CB2 antagonist SR1144528, which, by itself, did not produce hyperalgesia in CFA model (Table 3). Studies designed to further demonstrate the blockade of CB2 antagonists locally administered (i.t. or intra-DRG) on systemic CB2 agonism-mediated effects would be needed to address this question.
The mechanism of CB2 receptor-mediated antinociception has not been readily explained. CB2 receptors are not normally present in the spinal cord or brain or peripheral neurons because the receptor expression in these tissues is below the detection limit of available technique (Howlett, 1995; Pertwee, 1997). The effects of CB2 agonists were assumed to arise as a result of activation of receptors on peripheral immune and inflammatory cells and, under some pathological conditions, on microglia (Carlisle et al., 2002; Walter et al., 2003; Núñez et al., 2004; Cabral and Marciano-Cabral, 2005; Benito et al., 2008). The findings that CB2 receptor expression is up-regulated in the spinal cords and DRG tissues obtained from rats under inflammatory or neuropathic pain conditions in the present study suggest that they might mediate some of the analgesic effects of systemically administered CB2 agonists. Several studies have demonstrated a novel functional role of spinal CB2 receptors in modulating nociceptive processing in neuropathic, but not sham-operated, rats (Sagar et al., 2005), supporting their presence in the spinal cord of neuropathic rats (Zhang et al., 2003; Wotherspoon et al., 2005; Beltramo et al., 2006).
The CB2 receptor has also been identified in microglial cultures of neonatal rat spinal cord (Guo et al., 2007). In a rat L5 spinal nerve transaction model, CB2 expression is up-regulated in spinal microglia and the CB2 agonist JWH-015 (i.t.) reverses hypersensitivity following nerve injury, which can be blocked by AM630 (CB2 antagonist) but not AM281 (CB1 receptor antagonist, i.t.) (Romero-Sandoval and Eisenach, 2007). Appearance of CB2 receptor expression, though the specific response is not robust, also coincides with the activation of spinal microglial and astrocytic cells following either peripheral nerve injury or paw incision (Romero-Sandoval et al., 2007, 2008). The same authors also showed spinal cord as the site of action in the skin incisional model of post-operative pain (at 24 hr post surgery). Microglial and astrocytic activation is well known to play an important role in the initiation and maintenance of hypersensitivity in neuropathic pain (Watkins et al., 2001; Guo et al., 2007). Therefore, we speculate that CB2 agonism-inhibited glial activation would be, at least in part, the cause of analgesic effects induced by A-836339 and AM1241.
In the present study, we also demonstrated a novel finding that CB2 gene expression was significantly up-regulated in the ipsilateral paw tissues in a model of inflammatory (CFA) pain. CB2 receptor is highly expressed in the immune cells (Galiègue et al., 1995; Di Marzo et al., 2004) and increases in CB2 mRNA levels in the CFA-inflamed paw tissues would be expected because of the immune cell infiltration. Interestingly, A-836339 did not exhibit any local, peripheral effect following ipsilateral i.paw injection up to a dose of 100 nmol/i.paw in the CFA model. Although the modest analgesic activity was produced at 300 nmol/i.paw, similar effects were also observed with the contralateral i.paw administration, suggesting that the effect of i.paw A-836339 at that dose may be systemic rather than local. The reason for this is currently unexplained. In contrast, our data demonstrated the local site of action following i.paw injection of AM1241 in the CFA model, as an injection of 6 µmol·kg−1 into the contralateral paw only produced a marginal effect (18%), which was significantly different from the effect upon ipsialateral injection (62%, P < 0.01 vs. contralateral i.paw) (Figure 6C). The results are consistent with the literature findings, that CB2 agonist AM1241 suppressed the carrageenan or capsaicin-evoked thermal and mechanical hyperalgesia and allodynia in rats after local administration to the ipsilateral paw but was inactive after administration to the contralateral paw (Hohmann et al., 2004; Gutierrez et al., 2007). Similarly, it has also been reported that AM1241, administered locally in the paw, is sufficient to suppress C-fibre–evoked responses and windup at the level of the spinal dorsal horn and the AM1241-induced suppression of electrically evoked responses is blocked by the CB2 antagonist but not by the CB1 antagonist intraplantar, administered to the carrageenan-injected paw (Nackley et al., 2004).
The antinociceptive effects evoked by A-836339 do not involve the µ-opioid receptor in inflammatory (CFA) as well as neuropathic (SNL) pain as the effects are not sensitive to the pre-treatment of naloxone, a finding similar to those previously reported for other CB2 agonists A-796260 (Yao et al., 2008) and GW405833 (Whiteside et al., 2005). Interestingly, the blockade effect of AM1241 by naloxone is only observed in the CFA model of inflammatory pain (Yao et al., 2008) but not in the chronic (SNL) model of neuropathic pain in rats (Figure 7C). The reason for the difference between two models is currently unknown. Whether CFA injection up-regulates endogenous opioid levels in the periphery remains to be determined. In naïve rats, CB2 immunolabelling was detected on β-endorphin-containing keratinocytes in stratum granulosum throughout the epidermis of the hind paw and the antinociceptive effects of AM1241 were prevented in rats when naloxone or antiserum to β-endorphin was injected in the hindpaw where the noxious thermal stimulus was applied (Ibrahim et al., 2005). Therefore, the µ-opioid receptor dependency of CB2-mediated analgesic effect may be only true for specific compounds like AM1241 for specific efficacy models. A-836339 is shown to exhibit relatively few off-target interactions (Yao et al., 2009), which is in contrast to the CB2-selective ligand AM1241 that exhibits significant radioligand binding affinity to a large number of additional GPCR and ion channel targets (Yao et al., 2008). Therefore, AM1241 may interact with additional targets that may contribute to the antinociceptive efficacy through the regulation of the opioid receptor pathway. Taken together, our data have provided evidence that A-836339 could serve as a useful tool for further characterization of CB2 receptor pharmacology with respect to site(s) or mechanism(s) of action. It would also be interesting to see if there is pharmacological interaction between CB2 agonists and clinical-use analgesic drugs in the preclinical models of pain.
In summary, we have demonstrated a functional inhibitory effect of intrathecal or intra-DRG administration of the CB2-selective agonists A-836339 and AM1241. The data complement the findings that CB2 receptor mRNA is up-regulated in the spinal cord and DRG tissues obtained from rats under inflammatory or neuropathic pain conditions, but not sham-operated animals, suggesting that CB2 agonists may elicit their analgesic effects by acting not only at peripheral DRG sites but also at central levels of the spinal cord, making CB2 an attractive target for chronic pain treatment.
Acknowledgments
The authors would like to thank Dr Michael W. Decker for his valuable input on the manuscript.
Glossary
Abbreviations
- A-836339
2,2,3,3-tetramethyl-cyclopropanecarboxylic acid [3-(2-methoxy-ethyl)-4,5-dimethyl-3H-thiazol-(2Z)-ylidene]-amide
- AM1241
2-iodo-5-nitro-phenyl)-[1-(1-methyl-piperidin-2-ylmethyl)-1H-indol-3-yl]-methanone
- CB
cannabinoid, DRG, dorsal root ganglia
- CCI
chronic constriction injury
- CFA
complete Freund's adjuvant
- i.t.
intrathecal
- qRT-PCR
real-time quantitative polymerase chain reaction
- rimonabant
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride
- spinal nerve ligation, SNL
spinal nerve ligation
- SR144528 (SR2)
5-(4-chloro-3-methyl-phenyl)-1-(4-methyl-benzyl)-1H-pyrazole-3-carboxylic acid [(1S,2S,4R)-1,3,3-trimethyl-bicyclo(2.2.1)hept-2-yl]-amide
Conflict of interest
All authors are employees of Abbott Laboratories who funded the research.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edn) 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. (2008 revision) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth F, Casellas P, Congy C, Martinez S, Rinaldi M, Anne-Archard G. Preparation of N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl) 4-methylpyrazole-3-carboxamide as a cannabinoid receptor antagonist. European Patent Appl. 1995 EP 656354. [Google Scholar]
- Barth F, Casellas P, Millan J, Oustric D, Rinaldi M, Sarran M. 3-Pyrazolecarboxamide derivatives having cannabinoid receptor. PCT Patent Appl. 1997 WO 9721682. [Google Scholar]
- Beltramo M, Bernardini N, Bertorelli R, Campanella M, Nicolussi E, Fredduzzi S, et al. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur J Neurosci. 2006;23:1530–1538. doi: 10.1111/j.1460-9568.2006.04684.x. [DOI] [PubMed] [Google Scholar]
- Benito C, Tolón RM, Pazos MR, Núñez E, Castillo AI, Romero J. Cannabinoid CB2 receptors in human brain inflammation. Br J Pharmacol. 2008;153:277–285. doi: 10.1038/sj.bjp.0707505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bingham B, Jones PG, Uveges AJ, Kotnis S, Lu P, Smith VA, et al. Species-specific in vitro pharmacological effects of the cannabinoid receptor 2 (CB2) selective ligand AM1241 and its resolved enantiomers. Br J Pharmacol. 2007;151:1061–1070. doi: 10.1038/sj.bjp.0707303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral GA, Marciano-Cabral F. Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J Leukoc Biol. 2005;78:1192–1197. doi: 10.1189/jlb.0405216. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Carlisle SJ, Marciano-Cabral F, Staab A, Ludwick C, Cabral GA. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int Immunopharmacol. 2002;2:69–82. doi: 10.1016/s1567-5769(01)00147-3. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Dart MJ, Carroll WA, Florjancic AS, Frost JM, Gallagher ME, Kolasa T, et al. Novel iminothiazole compounds as cannabinoid receptor ligands and their preparation, pharmaceutical compositions and use in the treatment of diseases. 2007. PCT International Application World Patent WO 2007140385.
- Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Elmes SJ, Winyard LA, Medhurst SJ, Clayton NM, Wilson AW, Kendall DA, et al. Activation of CB1 and CB2 receptors attenuates the induction and maintenance of inflammatory pain in the rat. Pain. 2005;118:327–335. doi: 10.1016/j.pain.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- Giblin GM, Billinton A, Briggs M, Brown AJ, Chessell IP, Clayton NM, et al. Discovery of 1-[4-(3-chlorophenylamino)-1-methyl-1H-pyrrolo[3,2-c]pyridin-7-yl]-1-morpholin-4-ylmethanone (GSK554418A), a brain penetrant 5-azaindole CB2 agonist for the treatment of chronic pain. J Med Chem. 2009;52:5785–5788. doi: 10.1021/jm9009857. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez T, Farthing JN, Zvonok AM, Makriyannis A, Hohmann AG. Activation of peripheral cannabinoid CB1 and CB2 receptors suppresses the maintenance of inflammatory nociception: a comparative analysis. Br J Pharmacol. 2007;150:153–163. doi: 10.1038/sj.bjp.0706984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, et al. HU-308: a specific agonist for CB2, a peripheral cannabinoid receptor. Proc Natl Acad Sci U S A. 1999;96:14228–14233. doi: 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A. Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. J Pharmacol Exp Ther. 2004;308:446–453. doi: 10.1124/jpet.103.060079. [DOI] [PubMed] [Google Scholar]
- Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, Jr, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci U S A. 2003;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, Jr, et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci U S A. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, et al. CB2 cannabinoid receptor mediation of antinociception. Pain. 2006;122:36–42. doi: 10.1016/j.pain.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Wall PD, McMahon SB. Does the right side know what the left is doing? Trends Neurosci. 1999;22:122–127. doi: 10.1016/s0166-2236(98)01302-2. [DOI] [PubMed] [Google Scholar]
- Lever IJ, Malcangio M. CB1 receptor antagonist SR141716A increases capsaicin-evoked release of Substance P from the adult mouse spinal cord. Br J Pharmacol. 2002;135:21–24. doi: 10.1038/sj.bjp.0704506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T, et al. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Lai J, Vanderah TW, Makriyannis A, Porreca F. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Curr Opin Pharmacol. 2003;3:62–67. doi: 10.1016/s1471-4892(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Zvonok AM, Makriyannis A, Hohmann AG. Activation of cannabinoid CB2 receptors suppresses C-fiber responses and windup in spinal wide dynamic range neurons in the absence and presence of inflammation. J Neurophysiol. 2004;92:3562–3574. doi: 10.1152/jn.00886.2003. [DOI] [PubMed] [Google Scholar]
- Núñez E, Benito C, Pazos MR, Barbachano A, Fajardo O, González S, et al. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse. 2004;53:208–213. doi: 10.1002/syn.20050. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Aanonsen L, Hargreaves KM. SR 141716A, a cannabinoid receptor antagonist, produces hyperalgesia in untreated mice. Eur J Pharmacol. 1997;319:R3–R4. doi: 10.1016/s0014-2999(96)00952-1. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval A, Eisenach JC. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology. 2007;106:787–794. doi: 10.1097/01.anes.0000264765.33673.6c. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval A, Nutile-McMenemy N, DeLeo JA. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology. 2008;108:722–734. doi: 10.1097/ALN.0b013e318167af74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter LE, Kohlhaas KL, Curzon P, Surowy CS, Meyer MD. Peripheral and central sites of action for A-85380 in the spinal nerve ligation model of neuropathic pain. Pain. 2003;103:269–276. doi: 10.1016/s0304-3959(02)00455-4. [DOI] [PubMed] [Google Scholar]
- Sagar DR, Kelly S, Millns PJ, O'Shaughnessey CT, Kendall DA, Chapman V. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur J Neurosci. 2005;22:371–379. doi: 10.1111/j.1460-9568.2005.04206.x. [DOI] [PubMed] [Google Scholar]
- Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology. 2005;48:658–672. doi: 10.1016/j.neuropharm.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Walczak JS, Pichette V, Leblond F, Desbiens K, Beaulieu P. Behavioral, pharmacological and molecular characterization of the saphenous nerve partial ligation: a new model of neuropathic pain. Neuroscience. 2005;132:1093–1102. doi: 10.1016/j.neuroscience.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Walczak JS, Pichette V, Leblond F, Desbiens K, Beaulieu P. Characterization of chronic constriction of the saphenous nerve, a model of neuropathic pain in mice showing rapid molecular and electrophysiological changes. J Neurosci Res. 2006;83:1310–1322. doi: 10.1002/jnr.20821. [DOI] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, et al. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. 2003;23:1398–1405. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Gottshall SL, Boulet JM, Chaffer SM, Harrison JE, Pearson MS, et al. A role for cannabinoid receptors, but not endogenous opioids, in the antinociceptive activity of the CB2-selective agonist, GW405833. Eur J Pharmacol. 2005;528:65–72. doi: 10.1016/j.ejphar.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Lee GP, Valenzano KJ. The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists. Curr Med Chem. 2007;14:917–936. doi: 10.2174/092986707780363023. [DOI] [PubMed] [Google Scholar]
- Wotherspoon G, Fox A, McIntyre P, Colley S, Bevan S, Winter J. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience. 2005;135:235–245. doi: 10.1016/j.neuroscience.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Yao BB, Hsieh GC, Frost JM, Fan Y, Garrison TR, Daza AV, et al. In vitro and in vivo characterization of A-796260: a selective cannabinoid CB2 receptor agonist exhibiting analgesic activity in rodent pain models. Br J Pharmacol. 2008;153:390–401. doi: 10.1038/sj.bjp.0707568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao BB, Hsieh GC, Daza AV, Fan Y, Grayson G, Garrison TR, et al. Characterization of a cannabinoid CB2 receptor-selective agonist, A-836339 [2,2,3,3-tetramethyl-cyclopropane carboxylic acid [3-(2-methoxy-ethyl)-4,5-dimethyl-3H-thiazol-(2Z)-ylidene]-amide], using in vitro pharmacological assays, in vivo pain models, and pharmacological magnetic resonance imaging. J Pharmacol Exp Ther. 2009;328:141–151. doi: 10.1124/jpet.108.145011. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O'Donnell D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci. 2003;17:2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.