

Abstract
BACKGROUND AND PURPOSE
The 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphocholine (edelfosine) is an ether-linked phospholipid with promising anti-cancer properties but some side effects that preclude its full clinical therapeutic exploitation. We hypothesized that this lipid could interact with plasma membrane ion channels and modulate their function.
EXPERIMENTAL APPROACH
Using cell migration-proliferation assays, patch clamp, spectrofluorimetry and 125I-Apamin binding experiments, we studied the effects of edelfosine on the migration of breast cancer MDA-MB-435s cells, mediated by the small conductance Ca2+-activated K+ channel, SK3/KCa2.3.
KEY RESULTS
Edelfosine (1 µM) caused plasma membrane depolarization by substantially inhibiting activity of SK3/KCa2.3 channels, which we had previously demonstrated to play an important role in cancer cell migration. Edelfosine did not inhibit 125I-Apamin binding to this SKCa channel; rather, it reduced the calcium sensitivity of SK3/KCa2.3 channel and dramatically decreased intracellular Ca2+ concentration, probably by insertion in the plasma membrane, as suggested by proteinase K experiments. Edelfosine reduced cell migration to the same extent as known SKCa channel blockers. In contrast, K+ channel openers prevented edelfosine-induced anti-migratory effects. SK3 protein knockdown decreased cell migration and totally abolished the effect of edelfosine on MDA-MB-435s cell migration. In contrast, transient expression of SK3/KCa2.3 protein in a SK3/KCa2.3-deficient cell line increased cell migration and made these cells responsive to edelfosine.
CONCLUSIONS AND IMPLICATIONS
Our data clearly establish edelfosine as an inhibitor of cancer cell migration by acting on SK3/KCa2.3 channels and provide insights into the future development of a new class of migration-targeted, anti-cancer agents.
Keywords: alkyl lipid, edelfosine, SK3/KCa2.3, KCNN3, migration
Introduction
The 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphocholine (edelfosine, also known as ET18-OCH3), is the prototype of a class of anti-tumour ether lipids, the alkyl lysophopholipids (APLs) (Gajate and Mollinedo, 2002). Edelfosine has been reported to have anti-tumourogenic activity by acting through different physiological pathways. This ether lipid was found to stop tumour cell proliferation by inhibiting cell division, leading to the accumulation of cells in phases G2/M or G0/G1 of the cell cycle (Roos and Berdel, 1986; Engebraaten et al., 1991; Boggs et al., 1995; Lohmeyer and Workman, 1995; Principe and Braquet, 1995; Pushkareva et al., 1999). It also induced Ca2+-independent apoptosis of tumour cells at 10 µM (normal cells are resistant to edelfosine at this concentration) through the activation of Fas/CD95 death receptors (Mollinedo et al., 2004), and has anti-angiogenic (Candal et al., 1994; Vogler et al., 1998) as well as anti-invasive effects (Storme et al., 1985; Van Blitterswijk et al., 1987; Bolscher et al., 1988; Slaton et al., 1994; Haugland et al., 1999; Steelant et al., 2001). Some authors have proposed that the anti-invasive effect of edelfosine could be due to the inhibition of tumour cell migration (Slaton et al., 1994; Haugland et al., 1999), but this remains a highly contentious issue (Storme et al., 1985). The disadvantages associated with the use of APLs have been that their effective concentrations are generally cytotoxic. Due to their detergent-like character, they intercalate into cellular membranes, and at high concentration, they exert cytotoxic effects by solubilizing cell membranes and causing cell lysis (Wiese et al., 2000).
Unlike most anti-neoplastic drugs, the anti-tumour effects of ether-lipids are distinct because they do not directly interact with DNA. Instead, due to their phospholipid nature, they are incorporated into cell membranes where they can affect proteins (Gajate and Mollinedo, 2002). To date, a potential action of edelfosine on ion channels has not been considered. Accumulating evidence indicates that Ca2+-activated K+ channels (KCa) are involved in the control of proliferation (Wang, 2004) and migration of cancer cells (Schwab et al., 1999; Bordey et al., 2000; Kraft et al., 2003). Recently, we reported that the small conductance Ca2+-activated K+ channel (SKCa) SK3/KCa2.3 (channel nomenclature follows Alexander et al., 2009) is expressed in a highly metastasizing breast cancer cell line, MDA-MB-435s, and in melanoma cells where it promotes cell migration with no significant effect on cell proliferation (Potier et al., 2006; Chantome et al., 2009). The expression of SK3/KCa2.3 channels hyperpolarizes plasma membrane and leads to an increase of Ca2+ entry through voltage-independent Ca2+ channels and therefore to a rise in intracellular Ca2+ concentration [Ca2+]i (Potier et al., 2006).
In this study, we have demonstrated that edelfosine inhibited SK3/KCa2.3-mediated MDA-MB-435s cell migration by reducing its Ca2+ sensitivity, leading to cell depolarization and subsequent reduction of [Ca2+]i. Edelfosine had no effect on IKCa channels or on the migration of non-cancerous breast epithelial cells that do not express SK3/KCa2.3 channels. Thus, we propose that those SK3/KCa2.3 channels, which mediate the inhibitory effects of edelfosine on cell migration, could be used as a functional molecular target for the development of new edelfosine-like APL, with preserved inhibitory effects on SK3/KCa2.3 channels, without toxicity on non-tumour cells.
Methods
Cell culture
The human breast cancer cell lines MDA-MB-435s and the immortalized normal breast epithelial cell lines MCF-10A and 184A1 were cultured as already described (Potier et al., 2006). The human embryonic kidney cells, HEK293, were grown in Dulbecco's modified Eagle medium with 10% fetal bovine serum in a humidified atmosphere of 5% CO2 in 37°C. All cell lines were obtained from the American Type Culture Collection (ATCC, LGC Promochem, Molsheim, France).
In vitro cell proliferation and cell migration
Cell proliferation was determined using the tetrazolium salt reduction method, as described (Roger et al., 2004). For migration, cells were seeded on 24-well plates and grown for 48 h. Drugs were then added for 24 h at concentrations that had no effect on cell proliferation. Cell migration was analysed in 24-well plates containing 8 µm pore size cell culture inserts (Becton Dickinson, Le Pont de Claix, France), as described (Roger et al., 2004).
Cell cycle analysis
Cell cycle analysis was performed as described (Barascu et al., 2006). Briefly, analyses were performed using a flow cytometer equipped with a 488 nm argon laser. The red DNA fluorescence signal was analysed as total (area) versus peak signal and data were recorded for at least 10 000 events.
Cytotoxic assays
To distinguish between a cytostatic and a cytotoxic effect, two cytotoxic assays were used. In the first one, cell viability after 24 h of treatment was assessed by the Trypan blue exclusion method. In the second method, cells were incubated with edelfosine at 1, 3, 10 and 30 µM for 8 h, and then washed three times with fresh culture medium. The remaining viable cells were allowed to grow for 6 days before being quantified using the MTT assay as described previously.
125I-Apamin binding
125I-Apamin binding experiments were carried out by CEREP company (Celle L'evescault, France). Membrane homogenates of cerebral cortex (120 µg protein) are incubated for 60 min at 4°C with 7 pM [125I]apamin (Kd) in the absence or presence of edelfosine in a buffer containing 50 mM Tris-HCl (pH 7.4), 5.4 mM KCl and 0.1% BSA. Non-specific binding is determined in the presence of 100 nM apamin. Following incubation, the samples are filtered rapidly under vacuum through glass fibre filters (GF/B, Packard, Meriden, CT, USA) pre-soaked with 0.3% PEI and rinsed several times with ice-cold 50 mM Tris-HCl using a 96-sample cell harvester (Unifilter, Packard). The filters are dried then counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard). The standard reference compound is apamin, which is tested in each experiment at several concentrations to obtain a competition curve from which its IC50 is calculated (13 pM). The results are expressed as a percent of control specific binding [ (measured specific binding/control specific binding) × 100] obtained in the presence of edelfosine.
Electrophysiology and intracellular Ca2+ measurements
Whole-cell K+ currents were recorded as already described (Potier et al., 2006; Chantome et al., 2009). Briefly, for electrophysiology experiments, cells were seeded into 35 mm Petri dishes at 2500 cells/cm2. Patch pipettes were pulled from non-heparinized hematocrit tubes to a resistance of 3–6 MΩ. Whole-cell macroscopic K+ currents were generated by stepwise 8 mV depolarizing pulses from a constant holding potential of −70 up to +58 mV. Signals were filtered at 1 kHz and digitized at 10 kHz. The acute effects of edelfosine were tested on HEK293 cells (that expressed recombinant SK3/KCa2.3 and IKCa channels) using a ramp protocol from +60 to −100 mV, with a holding potential of 0 mV (500 ms duration; 4 s intervals) to inactivate endogenous K+ currents. Statistical analysis was performed at 0 mV in order to minimize endogenous HEK293 Cl- currents (ECl= 0 mV).
Intracellular Ca2+ ([Ca2+]i) measurements were made using the fluorescent dye Fura-2. Cells were cultured at 3.5 × 104 cells per dish in glass bottom dishes (WillCo Wells, Amsterdam, the Netherlands). Cells were loaded for 75 min at rom temperature in physiological saline solution (PSS; see Solutions) containing 5 µM Fura-2 AM (the membrane-permeant acetoxymthylester form of Fura-2). Cells were then rinsed four times with 1 mL PSS and allowed to de-esterify for at least 15 min at room temperature. The dish was then placed on the stage of a Nikon Eclipse TE2000-S inverted epi-illumination microscope (Nikon, Champigny sur Marne, France). Excitation light at the two excitation wavelength maxima of fura-2 (340/380 nm) was chopped by a monochromator (Cairn Optoscan, Kent, UK). The excitation protocol was a 50 ms excitation at each wavelength every 2 s. Fluorescence emission at 510 ± 20 nm was detected by photomultiplier tube (PMT) placed on the side of the microscope. The analogue signal of the PMT was digitized by a digidata 1322-A converter (Axon Instruments, Foster City, CA, USA) at a sampling frequency of 2 kHz and futher analysed using Clampfit 8.1 (Axon Instruments). [Ca2+]i was calculated as described previously using in situ calibration (Gannier et al., 1994). A Kd of 150 nM was used for these calculations, according to the information supplied for this batch of fura-2 (Invitrogen Life Technologies, Cergy Pontoise, France).
Signals were captured using 1322-A Digidata converter (Axon Instruments) and pClamp 8.1 software (Axon Instruments). The analyses were performed using Clampfit 8.1 and Origin 7.0 softwares (Microcal Software, Northampton, MA, USA).
Western blot experiments
Western blot experiments were performed as already described (Potier et al., 2006). Briefly, proteins were electrotransferred onto polyvinylidene fluoride membranes which were incubated with an anti- SK3/KCa2.3 antibody directed against amino acids 2–21 (Sigma-Aldrich, St Quentin Fallavier, France) followed by incubation with a horseradish peroxidase-conjugated anti-rabbit IgG (Jackson Immuno-Research Laboratories, West Grove, PA, USA). Anti-GAPDH (monoclonal) and anti-actin (Sigma-Aldrich) antibodies were used for Western blot loading controls.
Proteinase K digestion
Proteinase K digestion was performed as described previously for SK3/KCa2.3 protein (Syme et al., 2003). Briefly, MDA-MB-435s cells were incubated with 10 mM HEPES, 150 mM NaCl, and 2 mM CaCl2 (pH 7.4) with or without 200 µg·mL−1 proteinase K (Sigma-Aldrich) at 37°C for 30 min. Proteinase K digestion was quenched by adding ice-cold phosphate-buffered saline containing 6 mM phenylmethylsulfonyl fluoride and 25 mM EDTA. Lysates were prepared and analysed by Western blot as described previously.
Constructions, transient and stable transfections
Two SK3/KCa2.3 specific siRNAs were designed as already described (Potier et al., 2006). Transfection into MDA-MB-435s was performed as previously described (Chajes et al., 2006). The plasmid containing full-length rat KCNN3 cDNA (KCNN3-pTracer-CMV2) and the empty vector (pTracer-CMV2) (generous gifts from Dr S. Lidofsky, USA) were transfected into MCF-7 and 184A1 cells using lipofectamine 2000 (Invitrogen Life Technologies) as already described (Potier et al., 2006). Rat KCNN3 cDNA was subcloned in a lentivector as already described (Chantome et al., 2009) and HEK293 cells were transduced at multiplicities of infection of 2 in the presence of polybrene (4 µg·mL−1, Sigma-Aldrich) to obtain stable expression of the SK3 channels. Human HA-tagged KCNN4 cDNA cloned into pCDNA3.1(+) vector (Syme et al., 2003) was kindly provided by Dr D.C. Devor (University of Pittsburgh, USA). The stable expression of the IKCa channels in HEK293 cells was generated by subjecting cells to antibiotic selection (1 mg·mL−1 G418) 48 h post-transfection, during 20 days
Solutions and drugs
The PSS had the following composition (in mM): NaCl 140, MgCl2 1, KCl 4, CaCl2 2, D-glucose 11.1 and HEPES 10, adjusted to pH 7.4 with NaOH 1 M. The pipette solutions were named based on their calculated free Ca2+ concentration as already described (Potier et al., 2006; Chantome et al., 2009). pCa X indicates a free Ca2+ concentration of 10−X M. The pipette solution for the whole-cell recordings contained (in mM): K-glutamate 125, KCl 20, MgCl2 1, Mg-ATP 1, HEPES 10, and pH was adjusted to 7.2 with KOH and various concentrations of CaCl2 and EGTA were added to obtain calculated pCa6 (8.7 mM CaCl2 and 10 mM EGTA), pCa6.4 (0.7 mM CaCl2 and 1 mM EGTA) or pCa7 (0.37 mM CaCl2 and 1 mM EGTA). The concentrations of free calcium were estimated using WebMaxC (http://www.stanford.edu/cpatton/webmaxcS.htm).
Tetraethylammonium (TEA), 4-aminopyridine (4-AP), NS 1619 [1-(2′-hydroxy-5′-trifluoro methylphenyl)-5-trifluoromethyl-2(3H)benzimidazolone)], apamin, BMS 204-352, iberiotoxin, clotrimazole and edelfosine were added to the PSS or culture media at the concentrations indicated in the figure legends. Except for BMS-204352 which was kindly given by Dominique Cahard (UMR 6014 CNRS, University of Rouen), all drugs and chemicals were purchased from Sigma-Aldrich.
Statistics
Unless otherwise indicated, data were expressed as mean ± standard error of the mean (n = number of cells). Comparisons between two means were made using Mann–Whitney or paired t-tests as appropriate. For comparison between more than two means, we used Kruskal–Wallis one-way analysis of variance followed by Dunn's test. Differences were considered significant when P < 0.05.
Results
Edelfosine inhibits cell migration through modulation of K+ channel activity
To determine the highest concentration of edelfosine devoid of cytotoxic and cytostatic effects, we established a dose-response curve to a range of concentrations (1–30 µM). As shown in Figure 1, edelfosine did not impede MDA-MB-435s cell proliferation at concentrations up to 1 µM. At concentrations of edelfosine higher than 1 µM, cell proliferation decreased in a dose and time-dependent manner with an IC50 of 5.0 ± 1.0 and 2.6 ± 0.3 µM, after 24 and 48 h respectively (Figure 1A). Toxicity tests (see Methods) showed no effect on cell viability up to 3 µM edelfosine, with a drastic effect on cell viability at 10 µM (Figure 1B). Edelfosine did not induce apoptosis at concentrations up to 10 µM (data not shown). As already described for other cell lines, the accumulation of cells in G2/M phase of the cell cycle was observed only when the concentrations of edelfosine used were 3 µM or higher (Figure 1C). Based on the dose-response curve thus established, we decided for the remainder of the study to use 1 µM edelfosine in cell migration assays.
Figure 1.
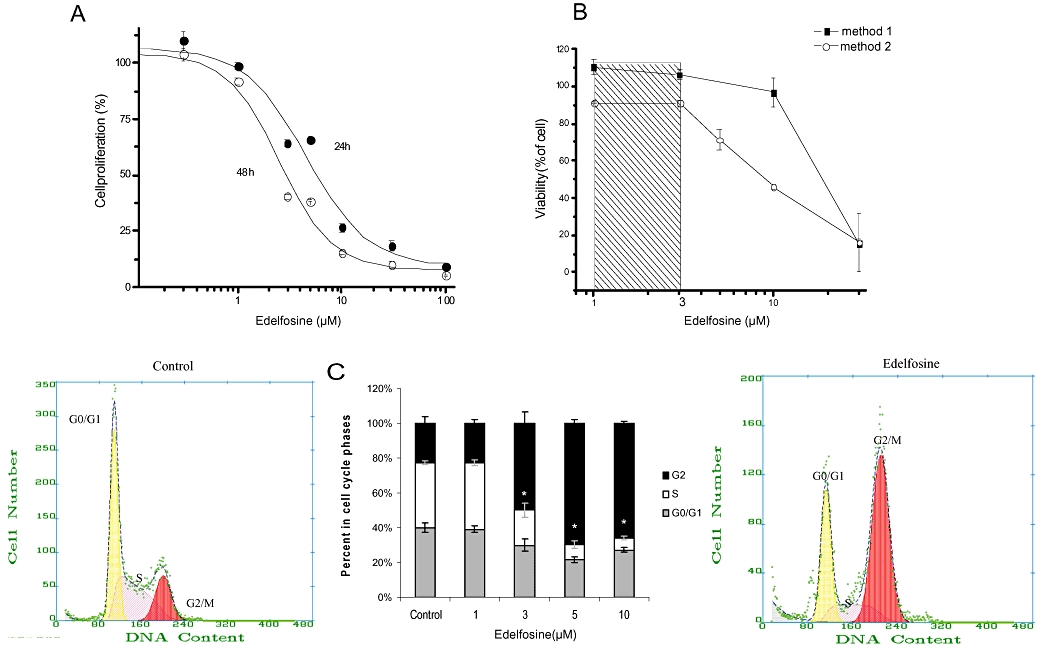
Effect of edelfosine on MDA-MB-435s cell survival, proliferation, toxicity and on cell cycle parameters. (A) Dose and time-dependent effects of edelfosine on cell survival and proliferation. (B) Dose-dependent toxicity of edelfosine. For Trypan blue experiments (method 1), cells were treated for 24 h with increasing concentration of edelfosine. For the other cytotoxic test (method 2), cells were treated with edelfosine for 8 h. Then, edelfosine was removed for 6 days and cells were quantified using MTT assay. (C) Histograms showing the effect of increasing concentrations of edelfosine on cell cycle phases. Cells were incubated for 24 h with edelfosine at different concentrations and their distribution in the cell cycle was assessed by flow cytofluorimetry. Results from two separate experiments performed in triplicate are expressed as mean ± SEM. *Significantly different from control at P < 0.05.
As shown in Figure 2, cell migration was decreased by almost 50% following treatment with 1 µM edelfosine. In the presence of apamin, a well-known blocker of SK2/KCa2.2 and SK3/KCa2.3 channels, edelfosine had no additional effect on cell migration (Figure 2A), suggesting that edelfosine is mediating its effects on cell migration through the inhibition of SK3/SK2 channels. Similarly, 4-AP and TEA, two potassium channel blockers that were found to block SKCa channels in MDA-MB-435s cells (Potier et al., 2006) caused no additional inhibition of cell migration in the presence of edelfosine (Figure 2B). We already found that apamin, TEA and 4-AP depolarized the plasma membrane of MDA-MB-435s cells (Potier et al., 2006). In order to test if edelfosine reduced cell migration by similarly depolarizing plasma cell membranes, we tested the effect of NS-1619 and BMS-204352, two well-known openers of big conductance calcium-activated K+ channels (BKCa) that were found to hyperpolarize plasma membrane cells (Holland et al., 1996; Gribkoff et al., 2001; Roger et al., 2004; Gessner et al., 2005). Figure 2C and D show that in contrast to the K+ channel blockers described previously, BKCa channel openers prevented the effect of edelfosine on cell migration. Note that these openers had no effect on cell migration when they are applied in the absence of edelfosine (Figure 2C,D). These results suggested that the effect of edelfosine on cell migration was prevented by the hyperpolarization induced by openers of BKCa channels.
Figure 2.
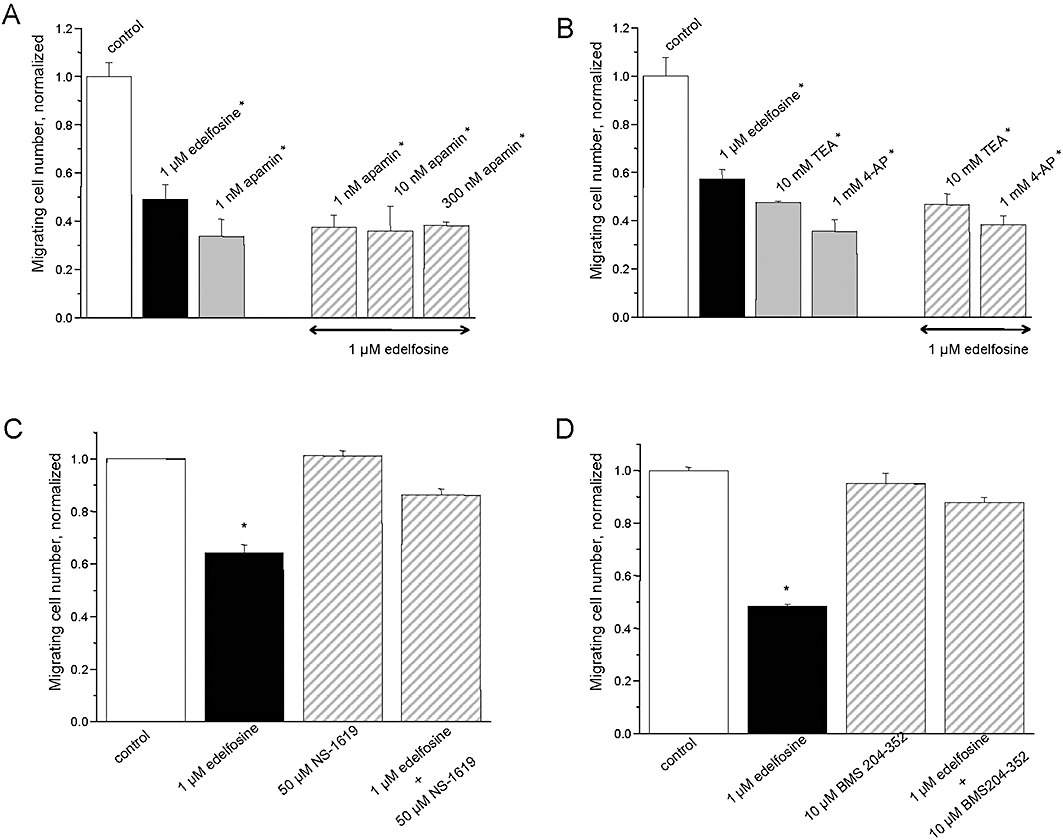
Effects of edelfosine on MDA-MB-435s cell migration. Histograms showing the effect of apamin (A), tetraethylammonium (TEA), 4-aminopyridine (4-AP) (B) and of NS-1619 (C), BMS-204352 (D) with and without 1 µM edelfosine for 24 h. The normalized cell number corresponds to the ratio of the total number of migrating cells in the presence of drug/total number of migrating cells in control experiments. Results from two separate experiments performed in triplicate are expressed as mean ± SEM. *Significantly different from control at P < 0.05.
Edelfosine decreases SKCa channel activity involved in membrane potential regulation
We performed whole-cell patch-clamp recordings to provide strong evidence that edelfosine decreases MDA-MB-435s cells migration by impairing SKCa channel activity. Using 10 mV voltage steps, we calculated cell capacitance as a measurement of cell size. No statistical difference was observed between control cells (33.2 ± 1.8 pF; n = 28) and edelfosine-treated cells (33.2 ± 1.4 pF; n = 18). Figure 3A shows typical examples of whole-cell outward K+ currents recorded in control, untreated MDA-MB435s cells and those treated with 1 µM edelfosine for 24 h. Compared with control cells, edelfosine caused a large reduction of the total whole-cell K+ currents (68% reduction at +58 mV; Figure 3B). The current density in edelfosine-treated cells was markedly lower than in control cells. Over the physiological range of resting membrane potential (i.e. −50 to −30 mV), edelfosine significantly decreased outward K+ currents (Figure 3B) and, as expected, depolarized the membrane potential of MDA-MB-435s cells from −44 ± 3 to −26 ± 4 mV. We recently demonstrated that apamin-sensitive currents were the main K+ currents regulating the membrane potential of MDA-MB-435s cells (Potier et al., 2006). Therefore, we examined the effects of apamin on edelfosine-insensitive currents. Figure 3C shows that apamin, used at the high concentration of 100 nM, had no significant effect on the remaining K+ outward current recorded from edelfosine-treated cells.
Figure 3.
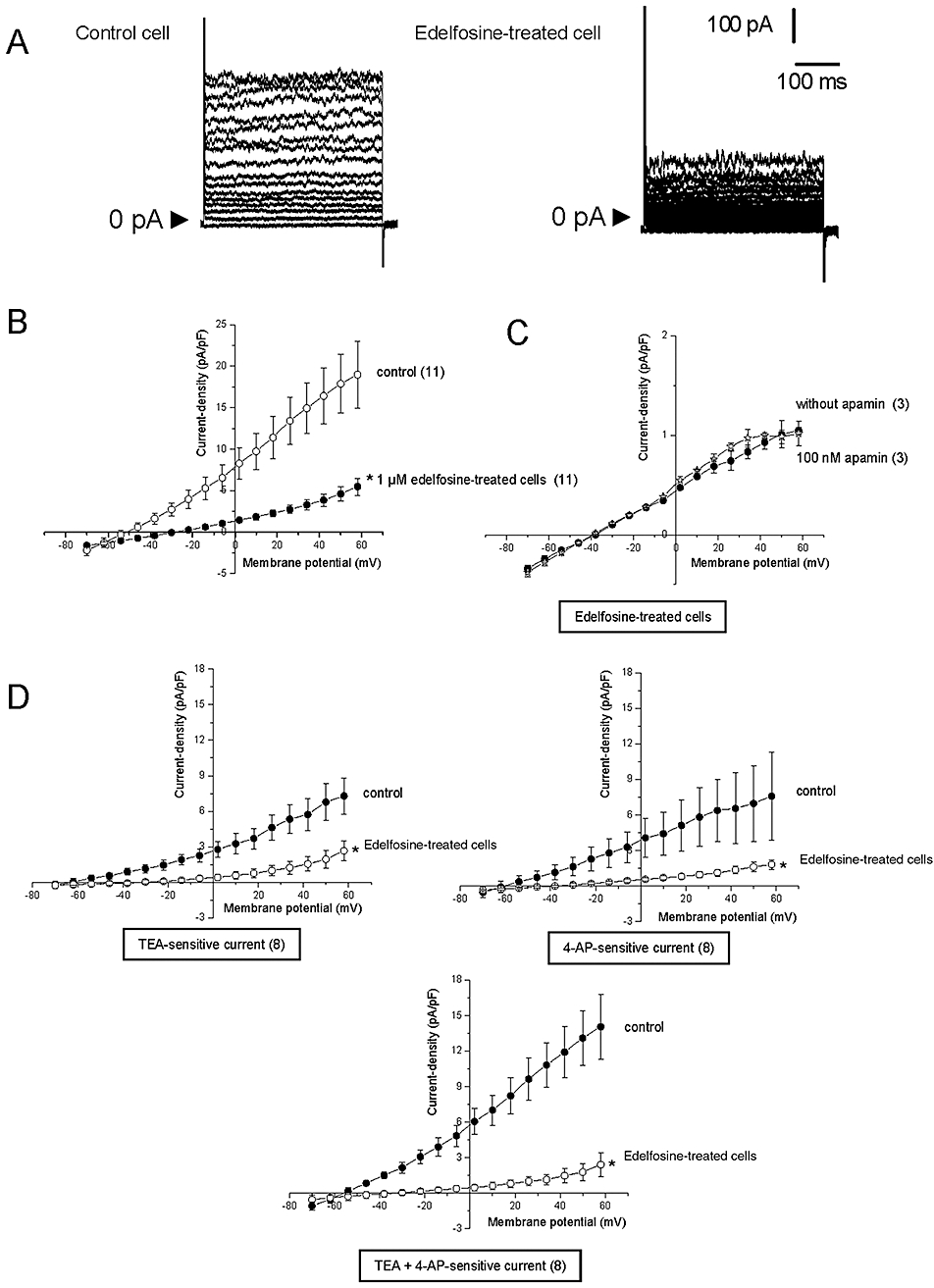
Effects of edelfosine on SKCa currents of MDA-MB-435s cells. (A) Example of whole-cell K+ currents recorded in one cell grown in a normal medium (control) or after treatment with 1 µM edelfosine for 24 h. Currents were generated by stepwise 8 mV depolarizing pulses (400 ms duration; 5 s intervals) from a constant holding potential of −70 up to +58 mV. (B) Current density-voltage relationships obtained in control and in edelfosine-treated cells. The current density–voltage relation was obtained by dividing the averaged steady-state currents elicited between −70 and +58 mV by the respective cell capacitance. (C) Current density–voltage relations obtained in edelfosine-treated cells, with and without 100 nM apamin. (D) Tetraethylammonium/4-aminopyridine (TEA/4-AP)-sensitive current density–voltage relationships during voltage steps in untreated and in 24 h edelfosine-treated cells. The magnitude of TEA, 4-AP and TEA + 4-AP-sensitive currents were obtained by subtracting the outward current recorded in the presence of these drugs from the net outward current observed in normal physiological saline solution. These drug-sensitive currents were compared in control condition and following 24 h treatment with 1 µM edelfosine. Results represent the mean ± SEM *Significantly different from control at P < 0.05. The numbers in brackets indicate the number of cells.
The nature of the protein inhibited by edelfosine was further examined using TEA and 4-AP (Figure 3D). We have demonstrated that 10 mM TEA and 5 mM 4-AP decreased the amplitude of the SK3/KCa2.3 currents on MDA-MB-435s cells (Potier et al., 2006). Therefore, we conducted additional experiments to assess the effects of these two inhibitors either individually or together on K+ currents in edelfosine-treated and -untreated cells. To determine the magnitude of TEA and 4-AP-sensitive currents in edelfosine-treated (for 24 h) and -untreated cells, we first recorded whole cell currents in the absence or presence of 10 mM TEA and/or 5 mM 4-AP. Then, TEA and/or 4-AP-sensitive currents were obtained by subtracting the net outward current observed in PSS solution from the outward current recorded in the presence of TEA and/or-AP. As shown in Figure 3D, edelfosine clearly and dramatically decreased TEA- and 4-AP-sensitive currents, suggesting that edelfosine reduced the activity of SK3/KCa2.3 channels in MDA-MB-435s.
Edelfosine decreases SK3/KCa2.3 channel activity but not the activity of IKCa channel
The effect of edelfosine was directly and acutely tested on recombinant SK3/KCa2.3 channels expressed in HEK293 cells. Figure 4A shows the typical SK3/KCa2.3 currents elicited by voltage ramps (from +60 to −100 mV), with a holding potential of 0 mV to inactive endogenous K+ currents. Superfusion with 1 µM edelfosine reduced the current and shifted the current reversal potential toward more positive potentials consistent with an effect of edelfosine on SK3/KCa2.3 channel conductance. Superfusion of the same cell with apamin completely blocked the current, confirming the SK3/KCa2.3 nature of the channel (Figure 3A). This experiment was repeated four times and 1 µM edelfosine reduced the amplitude of apamin-sensitive current by 79.1 ± 3.7%. The endogenous HEK293 K+ current was not significantly affected by 1 µM edelfosine (data not shown). The time course for SK3/KCa2.3 channel inhibition by edelfosine was further analysed at a membrane potential of 0 mV in order to minimize endogenous HEK293 Cl- currents (ECl= 0 mV). Figure 4A shows that 1 µM edelfosine reversibly decreased the amplitude of SK3/KCa2.3 current and total inhibition was observed after 5 min application. Finally, the application of apamin blocked the current. The selectivity of edelfosine toward IKCa channel was tested. Figure 4B shows representative IKCa currents recorded in HEK293 cells expressing IKCa channels in control condition and after application of 1 µM edelfosine. The experimental protocol was similar to the one used in Figure 4A. Edeldosine at 1 µM only slightly and not significantly reduced the amplitude of IKCa current to 7.00 ± 0.03% (n = 3; Figure 4B). After washout, 1 µM clotrimazole was applied and reversibly decreased the outward current, demonstrating that it was mediated by IKCa channels. Note that 10 µM clotrimazole totally inhibited this current (data not shown).
Figure 4.
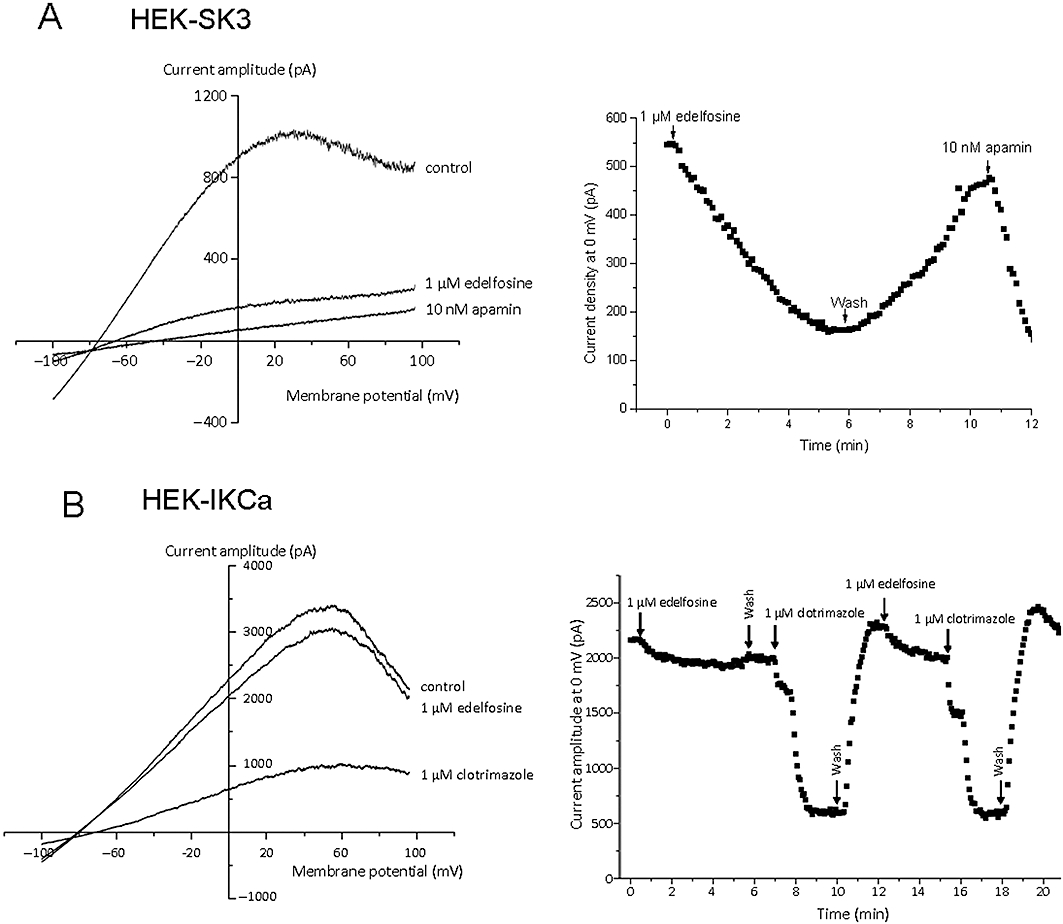
Effect of edelfosine on recombinant SK3/KCa2.3 channel and IKCa channel. (A) Left: Example of recombinant SK3 whole-cell K+ currents, recorded in one cell, obtained in control condition or after acute application of 1 µM edelfosine or 10 nM apamin (pCa = 6). Right: SK3 current obtained at 0 mV from voltage ramps (left) as a function of time. The cell was exposed to 1 µM edelfosine or 10 nM apamin as indicated by the arrows. (B) Left: Example of recombinant IKCa whole-cell K+ currents, recorded in one cell, obtained in control conditions or after acute application of 1 µM edelfosine or 1 µM clotrimazole (pCa = 6). Right: IKCa current obtained at 0 mV from voltage ramps (left) as a function of time. The cell was exposed to 1 µM edelfosine or 1 µM clotrimazole as indicated by the arrows.
Edelfosine decreases cancer cell migration by acting on SK3/KCa2.3 channel activity
We previously demonstrated that SK3/KCa2.3channels was the SKCa channel involved in MDA-MB-435s migration (Potier et al., 2006). To determine whether SK3/KCa2.3 channel inhibition is mediating the effect of edelfosine, SK3/KCa2.3 channel expression was knocked down by transfecting MDA-MB-435s cells with either two sets of siRNA targeted against SK3/KCa2.3 gene used individually or with scrambled siRNA used as a negative control. Knocking down SK3/KCa2.3 channels not only reduced the number of MDA-MB-435s migrating cells, but also totally suppressed the inhibitory effect of edelfosine on cell migration (Figure 5A). To further validate the requirement of SK3/KCa2.3 channels for the effect of edelfosine, we asked whether ‘forced’ SK3/KCa2.3 expression in cells lacking these channels would lead to a gain of sensitivity to edelfosine, in terms of cell migration. We addressed this question by transfecting rat rSK3/KCa2.3 cDNA in the SK3-negative 184A1 cell line. Note that edelfosine had no effect on wild-type untransfected 184A1 cell migration (Figure 5A). In contrast, the ectopic expression of SK3/KCa2.3 protein in 184A1 cells led to plasma membrane hyperpolarization (not shown) and increased cell migration rate by 30% (Potier et al., 2006), while edelfosine treatment decreased migration in SK3-expressing 184A1 cells (Figure 5A).
Figure 5.
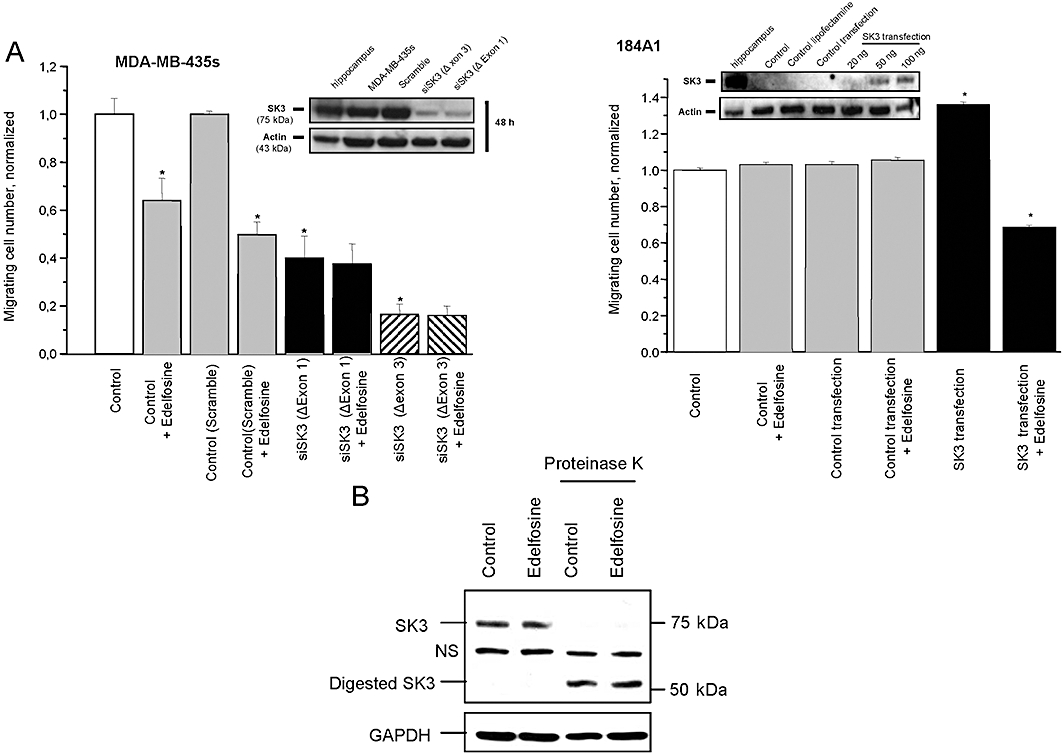
SK3/KCa2.3 gene transcript destruction suppresses edelfosine effect on MDA-MB-435s cells and SK3/KCa2.3 gene expression renders 184A1 cells sensitive to edelfosine. (A) Left: Histograms showing the effect of siRNA transfection on cell migration, after 24 h, with or without 1 µM edelfosine. Results from two separate experiments performed in triplicate are expressed as mean ± SEM. The normalized cell number corresponds to the ratio of total number of migrating cells in the presence of edelfosine (1 µM)/total number of migrating cells in control experiments. The insets are Western blot patterns showing the silencing effect on the expression of SK3/KCa2.3 protein of the two siRNAs designed against SK3/KCa2.3 mRNA. Cells were transfected with siRNA-lipofectamine complexes for 48 h. Right: Histograms showing the number of migrating cells after transient transfection of SK3/KCa2.3-pTracer-CMV2 plasmid or empty vector in non-cancerous (184A1) breast epithelial cells with or without 1 µM edelfosine treatment. The insets are Western blot patterns showing the expression of the SK3/KCa2.3protein channel after transient transfection of SK3/KCa2.3-pTracer-CMV2 plasmid or empty vector in 184A1 cells. Results from two separate experiments performed in triplicate are expressed as mean ± SEM. (B) Western blot of SK3/KCa2.3 protein following incubation in absence or presence of proteinase K. Proteinase K digestion was performed on cells treated or not with 1 µM edelfosine for 24 h. NS, non-specific. *Significantly different from control at P < 0.05.
To determine if the decrease in current density observed following edelfosine treatment (Figure 3) was due to a decrease in the expression of SK3/KCa2.3 protein at the plasma membrane, we assessed cell-surface protein expression using a proteinase K cleavage assay as described previously (Syme et al., 2003). In the absence of proteinase K, SK3/KCa2.3 ran at an apparent molecular mass of 75 kDa, consistent with the full-length protein (Figure 5B). Edelfosine treatment did not change the expression level or size of the SK3/KCa2.3 protein. Indeed, the SK3/KCa2.3 antibody revealed a band with similar intensity and size both in control untreated- and edelfosine treated-cells. Following proteinase K treatment, almost all of this 75 kDa-band was converted to a product with an apparent molecular mass of 50 kDa both in control untreated- and edelfosine treated-cells. This demonstrates that the majority of the SK3/KCa2.3 proteins are expressed at the cell surface and that edelfosine did not change the SK3/KCa2.3 protein expression and location.
In addition to the inhibition of SK3/KCa2.3 channel activity, edelfosine reduces Ca2+ sensitivity of SK3/KCa2.3 channels and decreases [Ca2+]i
Edelfosine was tested in the presence of two different Ca2+ concentrations (pCa7 and 6.4 indicate the free Ca2+ concentration in the pipette solution), and Figure 6A shows the average inhibition plotted for this two pCa solutions. Iberiotoxin (100 nM) was added to pharmacologically block BKCa currents, and apamin was used to isolate SK3/KCa2.3-mediated current [SK1 is not expressed in MDA-MB-435s and SK2 is not functional (Potier et al., 2006) ]. Edelfosine caused an almost full inhibition of the SK3/KCa2.3-mediated current at pCa7 (100 nM), whereas the inhibition was substantially smaller at pCa 6.4 (400 nM). Thus, the inhibition by edelfosine decreased with increasing [Ca2+]i. As the edelfosine effect was dependent on [Ca2+]i, we investigated if edelfosine could bind Ca2+ ions. Chelation of Ca2+ has been already reported with bisphsophonates (Lamson et al., 1984; Boulenc et al., 1995). Using a Ca2+-sensitive electrode, we measured pCa values in solutions containing increasing concentrations of edelfosine and of an alendronate derivative (Figure 6B). These pCa values were compared with values obtained with EGTA, a well-known selective Ca2+ chelator (Figure 6B). The results are illustrated in Figure 6B and show that only EGTA at 0.1 mM and alendronate at 2 mM modified the control pCa value (0.1 mM). This suggested that edelfosine, even at 2 mM (a concentration 20 times higher than the control [Ca2+]i), cannot complex Ca2+ ions.
Figure 6.
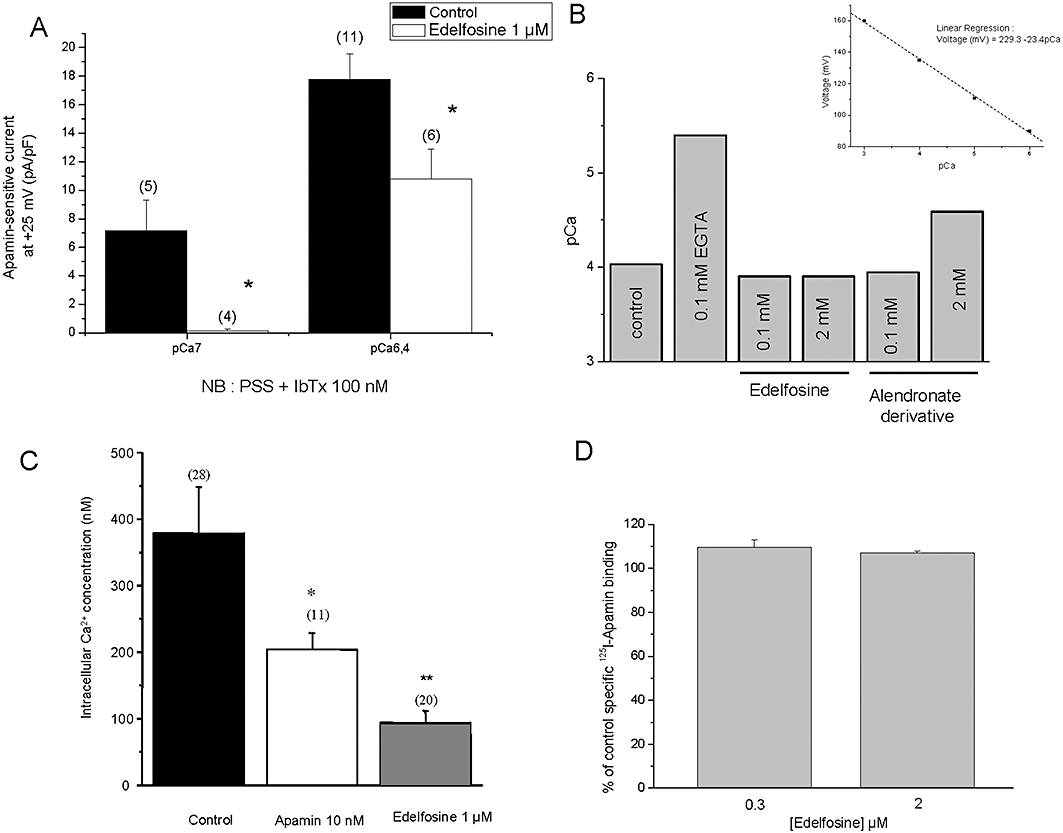
Mechanisms of action of edelfosine on SK3/KCa2.3-dependent cell migration. (A) Apamin-sensitive currents recorded at +25 mV (holding potential =−70 mV). Whole-cell recordings of MDA-MB-435s cells were obtained at two pCa pipette solutions, pCa 7 and pCa 6.4 and in the presence of 100 nM iberiotoxin in order to completely block BKCa channels. Apamin-sensitive currents, defined as the difference between outward currents recorded in drug-free bath solution and after superfusion with 10 nM apamin, were recorded in physiological condition (control) and after 24 h treatment with 1 µM of edelfosine. *Significantly different from control at P < 0.05. The number of investigated cells is indicated in brackets. (B) Comparative effect of EGTA, edelfosine and alendronate derivative to complex calcium and to reduce pCa. Measurements of pCa values in a pCa4 solution (containing in mM: HEPES 10; EGTA 1, MgCl2 1, KCl, 150, CaCl2, 1.1, pH 7.2 KOH) as control solution and with increasing concentrations of edelfosine and an alendronate derivative. The pCa values were measured by a Ca2+-sensitive electrode. The inset is the voltage-pCa relationship obtained with the Ca2+-sensitive electrode and used to determine the pCa. Reported results were obtained in a single experiment and that was repeated two times. (C) Intracellular Ca2+ concentration [Ca2+]i was measured in MDA-MB-453s cells in physiological condition (control) and following 24 h treatment with 10 nM of apamin and 1 µM of edelfosine. [Ca2+]i was measured using the fluorescent dye Fura-2. *P < 0.05; **P < 0.01. The number of investigated cells is indicated in brackets. (D) Histogram showing the % of control specific binding of 125I-Apamin (7 pM) to SKCa channel obtained from membrane homogenates of cerebral cortex by 0.3 and 2 µM edelfosine.
Because SK3/KCa2.3 channels can regulate intracellular Ca2+ levels (Potier et al., 2006), we measured [Ca2+]i in control-untreated cells and in cells treated with 1 µM edelfosine. We found that edelfosine considerably decreased the [Ca2+]i from 379 ± 30 nM (control cells; n = 28) to 69 ± 9 nM (edelfosine-treated cells; n = 20) (Figure 6C). To investigate whether edelfosine interacts with the apamin binding site, 125I-Apamin binding studies were performed. Figure 6D shows that edelfosine did not inhibit 125I-Apamin binding to membrane expressing SKCa channels, suggesting that this lipid acts at a site that is distinct from the apamin binding site. Clearly, further studies are needed to identify the precise binding site of edelfosine on SK3 channels.
Discussion and conclusions
This study demonstrates for the first time that edelfosine decreases the activity of SK3/KCa2.3, an ion channel involved in cancer cell migration (Potier et al., 2006; Chantome et al., 2009).
As already reported in other cancer cells, we found that, in MDA-MB-435s cells, concentrations of edelfosine equal or higher than 3 µM applied during 24 h reduced cancer cell proliferation and led to the accumulation of cells in the G2/M phase of the cell cycle (Roos and Berdel, 1986; Engebraaten et al., 1991; Boggs et al., 1995; Lohmeyer and Workman, 1995; Principe and Braquet, 1995; Pushkareva et al., 1999). In contrast to leukaemic or some solid tumour cells other than breast cancer cells (Mollinedo et al., 2004; Nieto-Miguel et al., 2006; 2007;), edelfosine was not found to induce apoptosis. This discrepancy is likely to be due to the intrinsic differences between cell types and its elucidation will require additional studies.
It has already been demonstrated that edelfosine decreases migration of tumour cells, but its exact mechanisms of action remained unknown (Slaton et al., 1994; Vogler et al., 1998; Haugland et al., 1999). Recently, we identified SK3/KCa2.3 channels as a new mediator of breast and melanoma cancer cell migration (Potier et al., 2006; Chantome et al., 2009). In the present study, we found that edelfosine reduced the migration of MDA-MB-435s cells by decreasing the activity of SK3/KCa2.3 channels and limiting Ca2+ entry into cells. This is a consequence of the membrane depolarization induced by edelfosine and subsequent decrease of Ca2+entry due to the reduction of driving force for Ca2+ (Figure 7). Indeed, the effect of edelfosine on cell migration was prevented by the addition of BKCa channel openers (NS-1019 and BMS 204-352), which are known to hyperpolarize the membrane of breast epithelial cancer cells (Roger et al., 2004). This demonstrates that membrane depolarization of MDA-MB-435s is the critical modification induced by edelfosine. In addition, the inhibition by edelfosine decreased with increasing [Ca2+]i, suggesting that this ether lipid reduced the calcium sensitivity of SK3/KCa2.3 channels. In contrast to well-established pore blockers, such as apamin, edelfosine acts as an inhibitory lipid interacting at a site distinct from the apamin binding site in SKCa channels. Interestingly, edelfosine was found to have no effect on IKCa channel, suggesting a selectivity of edelfosine towards the other members of the KCa channels. Because edelfosine contains a phosphate group, we tested the hypothesis that edelfosine would act by complexing calcium. This compound was not found to complex calcium even for a concentration 20 times higher than the calcium-free concentration. Thus, this cannot explain the effect of 1 µM edelfosine on SK3/KCa2.3 channels recorded with pCa7 or 6.4. Furthermore, edelfosine has no effect on IKCa channels, strongly suggesting that another mechanism, more selective, is involved in the reduction of SK3 channel activity. Interestingly, the alendronate derivative at 2 mM was found to complex calcium. This is in agreement with the results obtained using tiludronate (another bisphosphonate), showing that the concentration of this compound needed to complex 50% of the free calcium ions was found to be 20–40 times higher than EGTA (Boulenc et al., 1995).
Figure 7.
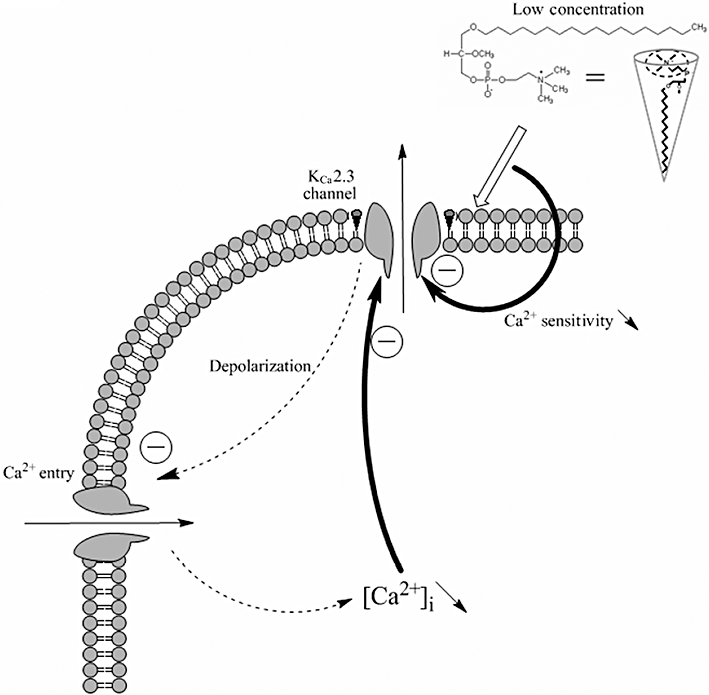
Proposed model to explain how edelfosine decreases SK3/KCa2.3-dependent cancer cells migration. When SK3/KCa2.3 channels are expressed and active in plasma membranes, they hyperpolarize the membrane. This leads to an increase of Ca2+ entry through voltage-independent Ca2+ channels and raised [Ca2+]i, promoting cell migration. This model proposes that edelfosine incorporates within plasma membrane and blocks SK3/KCa2.3 channel-induced hyperpolarization. This leads to a membrane depolarization and to a decrease of the driving force for Ca2+ and then to decreased SK3/KCa2.3 channel activity. Additionally, edelfosine reduced the Ca2+ sensitivity of SK3/KCa2.3 channels, leading to a further reduction of SK3/KCa2.3 channel activity. Consequently, [Ca2+]i is reduced and cell migration decreased. The inset represents the cone shape form proposed for edelfosine [modified from (Patel et al., 2001)].
Edelfosine, at much higher concentrations than those used in our study (20 µM vs. 1 µM), was found to inhibit the invasion of MO4 cells without affecting cell migration (Storme et al., 1985). The absence of edelfosine effect on MO4 cell migration could be explained by the absence of SK3/KCa2.3-mediated cell migration mechanism in MO4 cells. Alternatively, the high concentration of edelfosine used by these authors which inhibited MO4 cell proliferation (Storme et al., 1985) and induced apoptosis, might hide the effects on MO4 cell migration.
Because of its phospholipid structure, edelfosine may produce some of its effects on SK3/KCa2.3 channel after being incorporated into cellular membranes. Indeed, edelfosine is readily incorporated into lipid monolayers and bilayers (Busto et al., 2007), in cellular membranes of MCF-7 cells (estimated by a reduction of the total fatty acids/phosphorus ratio) (Besson et al., 1996), and in other tumour cells, as discussed by Mollinedo et al. (2004). Furthermore, edelfosine was found to change the fatty acid composition of membrane phospholipids (Besson et al., 1996) and the proportion of phosphatidylcholine and sphingomyelin of membrane phospholipids (P. Besson, unpubl. data). This can explain how this compound modifies plasma membrane lipid composition resulting in selective association and displacement of proteins with lipid raft scaffolds (Gajate et al., 2004; Zaremberg et al., 2005; Gajate and Mollinedo, 2007). This alteration in plasma lipid membranes could in turn affect the activity of lipid-sensitive channels, possibly by increasing the membrane fluidity (Hac-Wydro and Dynarowicz-Latka, 2010). This could explain why edelfosine reduced SK3/KCa2.3 channel activity without changing SK3/KCa2.3 protein expression and location at the plasma membrane, as documented by our results using proteinase K.
Different mechanisms for edelfosine uptake have been suggested, including direct adsorption at the plasma membrane, lipid flip-flop and endocytosis (Mollinedo et al., 2004). In this case, edelfosine seems to act through lipid raft reorganization, probably by the displacement of an essential protein from lipid rafts (Gajate et al., 2004; 2009; Gajate and Mollinedo, 2007; Zaremberg et al., 2005). Because of its conic shape (see Figure 6), edelfosine could affect the activity of SK3/KCa2.3 channels by changing the physical properties of the membrane. This was already observed with mechano-gated 2P domain K+ channels (TREK/TRAAK channels). Indeed, the conic shape of neutral lysophospholipids, such as lysophosphatidylcholine, as well as platelet activating factor (independently of its receptor) tends to favour a convex deformation of the plasma membrane, which leads to TREK-1/TRAAK channels opening (Maingret et al., 2000; Patel et al., 2001). Recently, Brainard et al. (2005) demonstrated that KCa channels localize to caveolae close to the cytoskeleton in order to form an actin-KCa channels-caveolin microdomain complex. According to those findings, it would be interesting to examine whether or not edelfosine integrates at a specific plasma membrane location close to SK3/KCa2.3 channels.
Besides a role in cell proliferation, K+ channels seem to be crucial for other mechanisms such as apoptosis (see Burg et al., 2006) or migration/invasion (see Schwab et al., 2007). Surprisingly, although cancer cell migration studies related to ion channels are few, it has emerged that ion channels belonging to the KCa channel sub-family are the channels able to promote cancer cell migration. The role of BKCa channels in cancer cell migration is not fully understood: activation of BKCa channels reduced, or was not involved, in cancer cell migration (Kraft et al., 2003; Roger et al., 2004). Besides our results, Schwab et al. (2007) have shown that the activity of IKCa is required for optimal cell migration. Currently, there is an increasing list of recent patents related to the use of K+ channel modulators in the anti-cancer therapy (Villalonga et al., 2007). To date, only IKCa blockers have been proposed for the treatment of prostate, pancreatic and endometrial cancer, based on their ability to inhibit in vitro cell proliferation. The involvement of SK3/KCa2.3 channel as a new K+ channel promoting cell migration and the identification of edelfosine as a new inhibitor of SK3/KCa2.3 channels may help to develop new SKCa channels blockers and a new class of migration-targeted anti-cancer agents.
Despite the emergence of edelfosine as a very effective and promising anti-tumour agent, its clinical therapeutic use has been hampered by several side effects, including gastrointestinal, lung, liver, renal and haemolytic toxicities (Gajate and Mollinedo, 2002). In particular, haemolysis has been considered a major side effect of edelfosine when given i.v. To alleviate these adverse effects, there is a need to develop new analogues of edelfosine, having greater or identical anti-tumour activity but with less toxicity. Our identification of the SK3/KCa2.3 channel as a molecular target accounting for the effects of edelfosine on cell migration, a pivotal process in tumour expansion and metastasis, opens up the way to consider the synthesis of molecular analogues. The fact that edelfosine decreases SK3/KCa2.3 channel activity and the migration of MDA-MB-43s cells, provides a unique opportunity to identify analogues having activity as SK3/KCa2.3 inhibitors but no toxic actions.
Acknowledgments
This work was funded by ‘Ligue contre le Cancer - Région Centre’, ‘Fondation Carrefour’, ‘INSERM’ and ‘Cancéropôle Grand Ouest’. M. Potier, A. Chantome, A. Girault and S. Roger held fellowships from, respectively, the ‘Ligue contre le Cancer’, ‘Agence National de la Recherche’, ‘Région du Centre’ and the ‘Ministère de la Recherche et de la Technologie’. We thank Dr M. Trebak for the helpful comments in writing the manuscript; Dr S. Lidofsky and Dr D.C. Devor for, respectively, the kind gift of KCNN3 and KCNN4 plasmids; and D. Cahard for the kind gift of BMS-204352. We thank Aurélia Barascu, Veronique Chajès and Lysiane Boulay for their technical assistance and Catherine Leroy for secretarial support.
Glossary
Abbreviations
- 4-AP
4-aminopyridine
- APL
alkyl lysophospholipids
- BKCa
big conductance Ca2+-activated K+ channels
- edelfosine
1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphocholine
- IKCa
intermediate conductance Ca2+-activated K+ channels
- KCa
Ca2+-activated K+ channels
- SKCa
small conductance Ca2+-activated K+ channels
- TEA
tetraethylammonium
Conflicts of interest
None
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) Br J Pharmacol. (4th edn) 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barascu A, Besson P, Le Floch O, Bougnoux P, Jourdan ML. CDK1-cyclin B1 mediates the inhibition of proliferation induced by omega-3 fatty acids in MDA-MB-231 breast cancer cells. Int J Biochem Cell Biol. 2006;38:196–208. doi: 10.1016/j.biocel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Besson P, Gore J, Vincent E, Hoinard C, Bougnoux P. Inhibition of Na+/H+ exchanger activity by an alkyl-lysophospholipid analogue in a human breast cancer cell line. Biochem Pharmacol. 1996;51:1153–1158. doi: 10.1016/0006-2952(96)00029-9. [DOI] [PubMed] [Google Scholar]
- Boggs KP, Rock CO, Jackowski S. Lysophosphatidylcholine attenuates the cytotoxic effects of the antineoplastic phospholipid 1-O-octadecyl-2-O-methyl-rac-glycero-3- phosphocholine. J Biol Chem. 1995;270:11612–11618. doi: 10.1074/jbc.270.19.11612. [DOI] [PubMed] [Google Scholar]
- Bolscher JG, Schallier DC, Van Rooy H, Storme GA, Smets LA. Modification of cell surface carbohydrates and invasive behavior by an alkyl lysophospholipid. Cancer Res. 1988;48:977–982. [PubMed] [Google Scholar]
- Bordey A, Sontheimer H, Trouslard J. Muscarinic activation of BK channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. J Membr Biol. 2000;176:31–40. doi: 10.1007/s00232001073. [DOI] [PubMed] [Google Scholar]
- Boulenc X, Roques C, Joyeux H, Berger Y, Fabre G. Bisphosphonates increase tight junction permeability in the human intestinal epithelial (Caco-2) model. Int J Pharm. 1995;123:13–24. [Google Scholar]
- Brainard AM, Miller AJ, Martens JR, England SK. Maxi-K channels localize to caveolae in human myometrium: a role for an actin-channel-caveolin complex in the regulation of myometrial smooth muscle K+ current. Am J Physiol Cell Physiol. 2005;289:C49–C57. doi: 10.1152/ajpcell.00399.2004. [DOI] [PubMed] [Google Scholar]
- Burg ED, Remillard CV, Yuan JX. K+ channels in apoptosis. J Membr Biol. 2006;209:3–20. doi: 10.1007/s00232-005-0838-4. [DOI] [PubMed] [Google Scholar]
- Busto JV, Sot J, Goni FM, Mollinedo F, Alonso A. Surface-active properties of the antitumour ether lipid 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine (edelfosine) Biochim Biophys Acta. 2007;1768:1855–1860. doi: 10.1016/j.bbamem.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Candal FJ, Bosse DC, Vogler WR, Ades EW. Inhibition of induced angiogenesis in a human microvascular endothelial cell line by ET-18-OCH3. Cancer Chemother Pharmacol. 1994;34:175–178. doi: 10.1007/BF00685937. [DOI] [PubMed] [Google Scholar]
- Chajes V, Cambot M, Moreau K, Lenoir GM, Joulin V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res. 2006;66:5287–5294. doi: 10.1158/0008-5472.CAN-05-1489. [DOI] [PubMed] [Google Scholar]
- Chantome A, Girault A, Potier M, Collin C, Vaudin P, Pages JC, et al. KCa2.3 channel-dependent hyperpolarization increases melanoma cell motility. Exp Cell Res. 2009;315:3620–3630. doi: 10.1016/j.yexcr.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Engebraaten O, Bjerkvig R, Berens ME. Effect of alkyl-lysophospholipid on glioblastoma cell invasion into fetal rat brain tissue in vitro. Cancer Res. 1991;51:1713–1719. [PubMed] [Google Scholar]
- Gajate C, Mollinedo F. Biological activities, mechanisms of action and biomedical prospect of the antitumor ether phospholipid ET-18-OCH(3) (edelfosine), a proapoptotic agent in tumor cells. Curr Drug Metab. 2002;3:491–525. doi: 10.2174/1389200023337225. [DOI] [PubMed] [Google Scholar]
- Gajate C, Mollinedo F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood. 2007;109:711–719. doi: 10.1182/blood-2006-04-016824. [DOI] [PubMed] [Google Scholar]
- Gajate C, Del Canto-Janez E, Acuna AU, Amat-Guerri F, Geijo E, Santos-Beneit AM, et al. Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis. J Exp Med. 2004;200:353–365. doi: 10.1084/jem.20040213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajate C, Gonzalez-Camacho F, Mollinedo F. Involvement of raft aggregates enriched in Fas/CD95 death-inducing signaling complex in the antileukemic action of edelfosine in Jurkat cells. PLoS ONE. 2009;4:e5044. doi: 10.1371/journal.pone.0005044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannier F, White E, Lacampagne A, Garnier D, Le Guennec JY. Streptomycin reverses a large stretch induced increases in [Ca2+]i in isolated guinea pig ventricular myocytes. Cardiovasc Res. 1994;28:1193–1198. doi: 10.1093/cvr/28.8.1193. [DOI] [PubMed] [Google Scholar]
- Gessner G, Schonherr K, Soom M, Hansel A, Asim M, Baniahmad A, et al. BKCa channels activating at resting potential without calcium in LNCaP prostate cancer cells. J Membr Biol. 2005;208:229–240. doi: 10.1007/s00232-005-0830-z. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Starrett JE, Jr, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, et al. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med. 2001;7:471–477. doi: 10.1038/86546. [DOI] [PubMed] [Google Scholar]
- Hac-Wydro K, Dynarowicz-Latka P. Effect of edelfosine on tumor and normal cells model membranes – a comparative study. Colloids Surf B Biointerfaces. 2010;76:366–369. doi: 10.1016/j.colsurfb.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Haugland HK, Nygaard SJ, Tysnes OB. Combined effect of alkyl-lysophospholipid and vincristine on proliferation, migration and invasion in human glioma cell lines in vitro. Anticancer Res. 1999;19:149–156. [PubMed] [Google Scholar]
- Holland M, Langton PD, Standen NB, Boyle JP. Effects of the BKCa channel activator, NS1619, on rat cerebral artery smooth muscle. Br J Pharmacol. 1996;117:119–129. doi: 10.1111/j.1476-5381.1996.tb15163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R, Krause P, Jung S, Basrai D, Liebmann L, Bolz J, et al. BK channel openers inhibit migration of human glioma cells. Pflugers Arch. 2003;446:248–255. doi: 10.1007/s00424-003-1012-4. [DOI] [PubMed] [Google Scholar]
- Lamson M, Fox J, Higuchi W. Calcium and 1-hydroxyethylidene-1,1-bisphosphonic acid: polynuclear complex formation in the physiological range of pH. Int J Pharm. 1984;21:143–154. [Google Scholar]
- Lohmeyer M, Workman P. Growth arrest vs direct cytotoxicity and the importance of molecular structure for the in vitro anti-tumour activity of ether lipids. Br J Cancer. 1995;72:277–286. doi: 10.1038/bjc.1995.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honore E. Lysophospholipids open the two-pore domain mechano-gated K(+) channels TREK-1 and TRAAK. J Biol Chem. 2000;275:10128–10133. doi: 10.1074/jbc.275.14.10128. [DOI] [PubMed] [Google Scholar]
- Mollinedo F, Gajate C, Martin-Santamaria S, Gago F. ET-18-OCH3 (edelfosine): a selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor. Curr Med Chem. 2004;11:3163–3184. doi: 10.2174/0929867043363703. [DOI] [PubMed] [Google Scholar]
- Nieto-Miguel T, Gajate C, Mollinedo F. Differential targets and subcellular localization of antitumor alkyl-lysophospholipid in leukemic versus solid tumor cells. J Biol Chem. 2006;281:14833–14840. doi: 10.1074/jbc.M511251200. [DOI] [PubMed] [Google Scholar]
- Nieto-Miguel T, Fonteriz RI, Vay L, Gajate C, Lopez-Hernandez S, Mollinedo F. Endoplasmic reticulum stress in the proapoptotic action of edelfosine in solid tumor cells. Cancer Res. 2007;67:10368–10378. doi: 10.1158/0008-5472.CAN-07-0278. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Lazdunski M, Honore E. Lipid and mechano-gated 2P domain K(+) channels. Curr Opin Cell Biol. 2001;13:422–428. doi: 10.1016/s0955-0674(00)00231-3. [DOI] [PubMed] [Google Scholar]
- Potier M, Joulin V, Roger S, Besson P, Jourdan ML, Leguennec JY, et al. Identification of SK3 channel as a new mediator of breast cancer cell migration. Mol Cancer Ther. 2006;5:2946–2953. doi: 10.1158/1535-7163.MCT-06-0194. [DOI] [PubMed] [Google Scholar]
- Principe P, Braquet P. Advances in either phospholipids treatment of cancer. Crit Rev Oncol Hematol. 1995;18:155–178. doi: 10.1016/1040-8428(94)00118-d. [DOI] [PubMed] [Google Scholar]
- Pushkareva MY, Janoff AS, Mayhew E. Inhibition of cell division but not nuclear division by 1-O- octadecyl-2-O-methyl-Sn-glycero-3-phosphocholine. Cell Biol Int. 1999;23:817–828. doi: 10.1006/cbir.1999.0478. [DOI] [PubMed] [Google Scholar]
- Roger S, Potier M, Vandier C, Le Guennec JY, Besson P. Description and role in proliferation of iberiotoxin-sensitive currents in different human mammary epithelial normal and cancerous cells. Biochim Biophys Acta. 2004;1667:190–199. doi: 10.1016/j.bbamem.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Roos G, Berdel WE. Sensitivity of human hematopoietic cell lines to an alkyl-lysophospholipid-derivative. Leuk Res. 1986;10:195–202. doi: 10.1016/0145-2126(86)90042-1. [DOI] [PubMed] [Google Scholar]
- Schwab A, Reinhardt J, Schneider SW, Gassner B, Schuricht B. K(+) channel-dependent migration of fibroblasts and human melanoma cells. Cell Physiol Biochem. 1999;9:126–132. doi: 10.1159/000016309. [DOI] [PubMed] [Google Scholar]
- Schwab A, Nechyporuk-Zloy V, Fabian A, Stock C. Cells move when ions and water flow. Pflugers Arch. 2007;453:421–432. doi: 10.1007/s00424-006-0138-6. [DOI] [PubMed] [Google Scholar]
- Slaton JW, Hampton JA, Selman SH. Exposure to alkyllysophospholipids inhibits in vitro invasion of transitional cell carcinoma. J Urol. 1994;152:1594–1598. doi: 10.1016/s0022-5347(17)32485-0. [DOI] [PubMed] [Google Scholar]
- Steelant WF, Goeman JL, Philippe J, Oomen LC, Hilkens J, Krzewinski-Recchi MA, et al. Alkyl-lysophospholipid 1-O-octadecyl-2-O-methyl- glycerophosphocholine induces invasion through episialin-mediated neutralization of E-cadherin in human mammary MCF-7 cells in vitro. Int J Cancer. 2001;92:527–536. doi: 10.1002/ijc.1216. [DOI] [PubMed] [Google Scholar]
- Storme GA, Berdel WE, Van Blitterswijk WJ, Bruyneel EA, De Bruyne GK, Mareel MM. Antiinvasive effect of racemic 1-O-octadecyl-2-O-methylglycero-3-phosphocholine on MO4 mouse fibrosarcoma cells in vitro. Cancer Res. 1985;45:351–357. [PubMed] [Google Scholar]
- Syme CA, Hamilton KL, Jones HM, Gerlach AC, Giltinan L, Papworth GD, et al. Trafficking of the Ca2+-activated K+ channel, hIK1, is dependent upon a C-terminal leucine zipper. J Biol Chem. 2003;278:8476–8486. doi: 10.1074/jbc.M210072200. [DOI] [PubMed] [Google Scholar]
- Van Blitterswijk WJ, Hilkmann H, Storme GA. Accumulation of an alkyl lysophospholipid in tumor cell membranes affects membrane fluidity and tumor cell invasion. Lipids. 1987;22:820–823. doi: 10.1007/BF02535537. [DOI] [PubMed] [Google Scholar]
- Villalonga N, Ferreres JC, Argiles JM, Condom E, Felipe A. Potassium channels are a new target field in anticancer drug design. Recent Patents Anticancer Drug Discov. 2007;2:212–223. doi: 10.2174/157489207782497181. [DOI] [PubMed] [Google Scholar]
- Vogler WR, Liu J, Volpert O, Ades EW, Bouck N. The anticancer drug edelfosine is a potent inhibitor of neovascularization in vivo. Cancer Invest. 1998;16:549–553. doi: 10.3109/07357909809032884. [DOI] [PubMed] [Google Scholar]
- Wang Z. Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch. 2004;448:274–286. doi: 10.1007/s00424-004-1258-5. [DOI] [PubMed] [Google Scholar]
- Wiese A, Wieder T, Mickeleit M, Reinohl S, Geilen CC, Seydel U, et al. Structure-dependent effects of glucose-containing analogs of platelet activating factor (PAF) on membrane integrity. Biol Chem. 2000;381:135–144. doi: 10.1515/BC.2000.019. [DOI] [PubMed] [Google Scholar]
- Zaremberg V, Gajate C, Cacharro LM, Mollinedo F, Mcmaster CR. Cytotoxicity of an anti-cancer lysophospholipid through selective modification of lipid raft composition. J Biol Chem. 2005;280:38047–38058. doi: 10.1074/jbc.M502849200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.