

Abstract
BACKGROUND AND PURPOSE
Sphingosine kinases (SK) catalyse the formation of sphingosine 1-phosphate, which is a key lipid mediator regulating cell responses such as proliferation, survival and migration. Here we have investigated the effect of targeted inhibition of SK-1 on cell damage and elucidated the mechanisms involved.
EXPERIMENTAL APPROACH
Three human carcinoma cell lines (colon HCT-116, breast MDA-MB-231, lung NCI-H358) were used, which were either transduced with shRNA constructs to deplete SK-1, or treated with a SK-1 inhibitor. Cell growth and viability were assayed by [3H]thymidine incorporation and colony formation. Reactive oxygen species (ROS) were measured by fluorescence and apoptosis by annexin V with flow cytometry. Proteins were analysed by Western blotting. DNA damage was induced by doxorubicin.
KEY RESULTS
Knock-down of SK-1 by shRNA strongly inhibited DNA synthesis and colony formation of carcinoma cells. SK-1 knock-down (SK-1kd) cells revealed dysfunctional extracellular signal-regulated protein kinase and PKB/Akt cascades, and contained increased levels of ROS. After SK-1kd, treatment with doxorubicin increased DNA damage, measured by histone-2AX phosphorylation. Similar effects were found in cells with a SK-1 inhibitor and doxorubicin. The increased damage response in SK-1kd cells was accompanied by greater reduction of DNA synthesis and colony formation, and by more pronounced apoptosis. Addition of a NADPH oxidase inhibitor reduced the increased apoptosis in doxorubicin-treated SK-1kd cells.
CONCLUSIONS AND IMPLICATIONS
SK-1kd in carcinoma cells triggered oxidative stress by increasing intracellular Ros production. Targeted inhibition of SK-1 represents a promising approach to sensitize cells to DNA damage and facilitate apoptosis upon doxorubicin treatment.
Keywords: cancer drug resistance, DNA damage, doxorubicin, oxidative stress, sphingosine 1-phosphate, sphingosine kinase-1
Introduction
Resistance to chemotherapy is a hallmark of many solid tumours and a major obstacle to clinical success. DNA damage induced by routinely used chemotherapeutic agents such as doxorubicin, etoposide and cisplatin triggers a well-described damage response, leading to cell cycle arrest followed by DNA repair or apoptosis. Mechanistically, it is thought that DNA lesions provide the initial signal detected by ‘sensors’ that regulate the ultimate response. Over the last few years, efforts have been made to identify sensor proteins, which include the ataxia telangiectasia mutated (ATM) protein kinases, the ATM- and Rad3-related (ATR) protein kinases, and the DNA-dependent protein kinase (DNA-PK) (Yang et al., 2003; 2004;). These kinases belong to the family of phosphatidylinositol 3-kinase-like enzymes and are rapidly activated by DNA double strand breaks (DSB) occurring after exposure of cells to genotoxic stress. These damage sentinel kinases can phosphorylate and hence activate histone-2AX (H2AX) at the site of damage and over a distance of approximately one megabase of chromatin surrounding the DSB (Rogakou et al., 1999). As a consequence of activation, H2AX specifically stimulates the recruitment of DNA repair proteins to the sites of damage (Fillingham et al., 2006). In addition to H2AX, a number of other downstream targets are activated by the phosphatidylinositol 3-kinase-like enzymes (Hiom, 2005), which regulate cell proliferation and survival, including the checkpoint kinases Chk1 and Chk2 (Chen and Poon, 2008).
Sphingolipids are important constituents of biological membranes, but have recently been recognized to act also as signalling molecules (Olivera and Spiegel, 1993; Huwiler et al., 2000; Huwiler and Zangemeister-Wittke, 2007). In particular, ceramide and sphingosine 1-phosphate (S1P) play opposing roles in the regulation of cell proliferation and death (Obeid et al., 1993; Olivera and Spiegel, 1993; for review, see: Huwiler et al., 2000; Huwiler and Zangemeister-Wittke, 2007). Whereas ceramide is considered a pro-apoptotic and anti-proliferative molecule, S1P counteracts ceramide with anti-apoptotic and pro-proliferative effects. S1P is generated from ceramide by the action of two classes of enzyme, the ceramidases, which catalyse the deacylation of ceramide to form sphingosine, and the sphingosine kinases (SK), which phosphorylate sphingosine to form S1P (Huwiler et al., 2000; Alemany et al., 2007; Huwiler and Zangemeister-Wittke, 2007).
Two subtypes of SK have been identified, SK-1 and SK-2 (Alemany et al., 2007), but so far most investigations have been centred around the SK-1 subtype, which is activated by many growth factors and proinflammatory cytokines by transcriptional and posttranslational mechanisms (Alemany et al., 2007). In contrast, the molecular mechanisms of SK-2 activation are still largely unknown. Recently, overexpression of SK-1 has been associated with drug resistance in various cancer cell types (Huwiler et al., 2000; Pchejetski et al., 2005); however, its role within the DNA damage sensor, repair and cell death signalling network remains to be clarified.
In the present study, we have investigated the role of SK-1 in cancer drug resistance and shown that, in carcinoma cells of various histotypes, doxorubicin-induced DNA damage was significantly enhanced upon genetic down-regulation or pharmacologic inhibition of SK-1. As doxorubicin induces oxidative stress that may assist in DNA damage, we also measured changes in reactive oxygen species (ROS) upon SK-1 depletion. We demonstrate for the first time that carcinoma cells of various histotypes with down-regulated SK-1 also contain elevated levels of ROS, which primes them for additional DNA damage and facilitates apoptosis induced by cytotoxic chemotherapy.
Methods
Carcinoma cell lines
The human breast carcinoma cell line MDA-MB-231 and the colon carcinoma cell line HCT-116 were obtained from the European Collection of Cell Cultures through Sigma Aldrich, Buchs, Switzerland; the non-small cell lung carcinoma cell line NCI-H358 was obtained from the American Tissue Culture Collection. MDA-MB-231 and NCI-H358 cells were cultured in RPMI medium supplemented with 10% fetal bovine serum, 10 mM HEPES pH 7.4, penicillin and streptomycin. HCT-116 cells were cultured in McCoy's medium supplemented with 10% fetal bovine serum, 10 mM HEPES pH 7.4, penicillin and streptomycin.
SK-1 down-regulation by lentiviral short hairpin (sh) RNA transduction
For stable gene silencing of SK-1, five different lentiviral SK-1-specific shRNA-containing constructs were tested and transduced into cells as recommended by the manufacturer. The following shRNA constructs were used: TRC number: 36964 (#1), 36965 (#2), 36966 (#3), 36967 (#4), 36968 (#5). All MISSIONR shRNA constructs were obtained as bacterial glycerol stocks from Sigma Aldrich Fine Chemicals, St. Louis, US. Transduced cells were selected in medium containing 1 µg·mL−1 puromycin. The silencing efficiency was confirmed by Western blotting. For all subsequent experiments, cells transduced with shRNA#4, are referred to as SK-1 knock-down (SK-1kd).
Western blot analysis
Stimulated cells were homogenized in lysis buffer and processed as previously described (Döll et al., 2005). Thirty microgram of protein was separated by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membrane and Western blotting was performed as previously described (Döll et al., 2005) using antibodies as indicated in the figure legends.
Sphingolipid quantification by LC/MS/MS
Equal numbers of cells in 6-well plates were scraped into methanol containing internal C17-S1P and C17-sphingosine standards and subjected to lipid extraction as previously described (Hofmann et al., 2008). Lipids were redissolved in dimethyl sulfoxide containing 2% HCl and taken for LC/MS/MS analysis exactly as previously described (Hofmann et al., 2008).
[3H]thymidine incorporation
4 × 104 cells were seeded into 24-well plates and after 8 h when cells had become adherent, they were incubated for the indicated time periods in growth medium including 0.2 µCi·mL−1 of [3H]methyl-thymidine. Thereafter, cells were washed with phosphate-buffered saline and incubated for 30 min with ice cold 5% (w/v) trichloroacetic acid, then washed twice with ice cold 5% trichloroacetic acid and solubilized in 0.5 M sodium hydroxide. Solubilized radioactivity representing [3H]thymidine incorporated into newly synthesized DNA was counted in a liquid scintillation analyser (Packard BioScience Company, Meriden, CT, USA).
Colony formation assay
Cells were subcultured in 60 mm diameter dishes at a density of 700 cells per dish. After 24 h, cells were treated with the indicated concentrations of doxorubicin in growth medium and incubated for a further 10 days (HCT-116) or 14 days (MDA-MB-231 and NCI-H358 cells) to allow colony formation. Cells were then washed with phosphate-buffered saline, air dried and stained for 30 min with 2% (w/v) crystal violet, washed with water and air dried again. The number of colonies was quantified using a ColCount (Mammalian Cell Colony Counter, Oxford Optronix, Oxford, UK). Only colonies containing more than 50 cells were evaluated.
Measurement of ROS formation
Equal numbers of cells (104 for HCT-116 and MDA-MB-231, 3 × 104 for NCI H358) were subcultured into black bottom-clear 96-well plates (Greiner, Bio-One Schweiz GmbH, St. Gallen, Switzerland) and cultured for 24 h to reach approximately 80% confluency. In a parallel experiment, cells were seeded and cell numbers were counted after 24 h to ensure that the different cell lines did not vary in cell number before labelling. Then, cells were labelled for 30 min with 10 µM 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) in Hanks buffered saline solution (HBSS). Cells were then washed with HBSS to remove excess dye and further incubated in HBSS for 2 h. Fluorescence was monitored at 480 nm excitation and 540 nm emission wavelengths using a Spectramax M2 plate reader (Molecular Devices, Sunnyvale, CA, USA).
Comet assay
DNA damage was quantified using a Comet assay kit (Trevigen Inc. Gaithersburg, MD, USA), which is based on the alkaline lysis of labile DNA at sites of damage. The unwound, relaxed DNA is able to migrate out of the cell during electrophoresis and can be visualized using SYBR Green nucleic acid gel stain. Cells with DNA damage appear as fluorescent comets with tails of DNA fragmentation, whereas normal undamaged DNA does not migrate far from the origin. Comet quantification was performed by automatic scoring using the Comet assay IV scoring software (Perspective Instruments, Haverhill, UK).
Apoptosis assay
The exposure of phosphatidylserine (PS) on the cell surface as an early marker of apoptosis was measured by quantification of annexin V binding to PS using flow cytometry. In brief, cells were harvested by trypsinization and cell pellets were resuspended at a concentration of 6 × 105 cells·mL−1 in binding buffer. PS-exposing cells were stained with GFP-coupled annexin V at a dilution of 1:1000 to stain apoptotic cells. The flow cytometry analyses were performed using a FACSCalibur (Becton Dickinson AG, Allschwil, Switzerland). Analyses were performed using CELLQuest as part of the FACSCalibur-operating system.
Statistical analysis
Data are presented as means ± SD Statistical analysis was performed by one way analysis of variance (anova). For multiple comparisons with the same control group, the limit of significance was divided by the number of comparisons according to Bonferroni.
Materials
[3H]methyl-thymidine (specific activity, 6.7 Ci·mmol−1), secondary horseradish peroxidase-coupled IgGs, Hyperfilm MP, and enhanced chemiluminescence reagents were from GE Health Care Systems GmbH (Freiburg, Germany); five different lentiviral shRNA vectors of human SK-1 (MISSIONR), DPI, doxorubicin and puromycin were from Sigma Aldrich Fine Chemicals (Deisenhofen, Germany); CM-H2DCFDA was from Molecular Probes (Göttingen, Germany); the SK inhibitor SKI II [2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole] was from Merck Biosciences (Schwalbach, Germany); phospho-Ser139-H2AX, total DNA-PK, phospho-ERK1/2, phospho-Thr308-PKB/Akt and cleaved caspase-3 antibodies were from Cell Signaling Technology (Frankfurt am Main, Germany); phospho-Thr2609-DNA-PK (sc-101664), and GAPDH (V-18; sc-20357) antibodies were from Santa Cruz Biotechnology (Heidelberg, Germany); an affinity-purified antibody against human SK-1 was generated and characterized as previously described (Döll et al., 2005). Recombinant GFP-labelled annexin V was kindly provided by Prof Thomas Kaufmann, Bern. Lipofectamine and all cell culture nutrients were from Invitrogen AG, Basel, Switzerland. Drug and receptor nomenclature used here follows Alexander et al. (2009).
Results
Loss of SK-1 inhibits cell growth and colony formation of carcinoma cells
Because the sphingolipid S1P is a well-known mitogen for many cell types and the S1P-generating enzyme SK-1 was previously shown to be up-regulated in various tumour tissues (French et al., 2003; 2006;) and in tumour cells exposed to growth factors (Döll et al., 2005; 2007; Sarkar et al., 2005), we investigated the consequence of SK-1 knock-down for the growth of various carcinoma cell types.
We first tested five different lentiviral shRNA constructs on the breast carcinoma cell line MDA-MB-231. As shown in Figure 1A, shRNA#4 and shRNA#1 resulted in an almost complete down-regulation of SK-1 protein expression, whereas shRNA#3 and shRNA#5 were non-functional. For further studies, the shRNA#4 transduced cells were used. In addition to the MDA-MB-231 cells, we also transduced the colon carcinoma cell line HCT-116 and the non-small cell lung carcinoma cell line NCI-H358, which all express SK-1 protein though at various levels (Figure 1B). The cell lines with stably down-regulated SK-1 showed a reduction of SK-1 protein by 82% in HCT-116, 96% in MDA-MB231 and 98% in NCI-H358 (Figure 1B). SK-2 protein expression was also detected in the three Wt and SK-1kd cell lines. SK-2 was highest expressed in the HCT-116 cells, but hardly detectable in MDA-MB-231 cells and NCI-H358 cells (data not shown). There was no compensatory up-regulation of SK-2 in any of the SK-1kd cells (data not shown). Additionally, we have quantified the sphingolipid levels in the three Wt and SK-1kd cell lines. S1P levels were very low and changes hardly detectable (Figure 1C). However, sphingosine levels significantly increased in all SK-1kd cell lines (Figure 1C).
Figure 1.
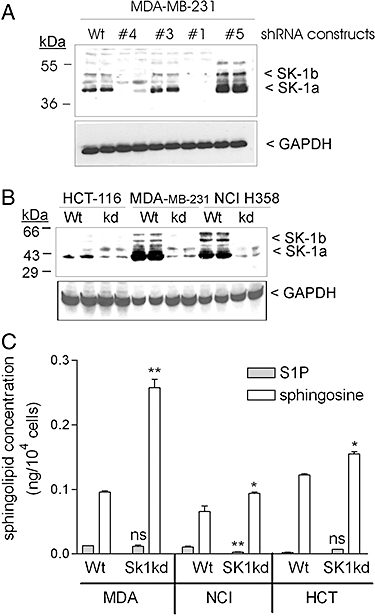
Characterization of various carcinoma cell lines stably depleted of SK-1 by lentiviral transduction of a shRNA construct. A. MDA-MB-231 cells were either taken non-transduced (Wt), or transduced with shRNA constructs #1, #4, #3 and #5 from MISSIONR-Sigma. B. HCT-116, MDA-MB-231 and NCI-H358 were transduced with a SK-1 shRNA#4 construct for stable knock-down of SK-1 (SK-1kd) or taken without transduction (Wt) as described in the Methods section. Lysates containing equal amounts of protein were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using an antibody against SK-1 (dilution 1:2000, upper panel), or GAPDH (dilution 1:3000, lower panel). The two splice variants of SK-1, that is, SK-1a and SK-1b run at 43 and 51 kDa, respectively. C. HCT-116, MDA-MB-231 and NCI-H358 of Wt origin or Sk-1kd were taken for lipid extraction and quantification by mass spectrometry. Results are indicated as ng/104 cells and are means SD (n = 3). *P < 0.05, **P < 0.01, significantly different from the Wt control values.
All SK-1kd cell lines showed a strongly diminished rate of [3H]thymidine incorporation compared with their Wt counterparts (Figure 2A). To further assess the potential anti-tumour effect of SK-1kd and whether this included also proliferating cells, we measured colony formation in clonogenic assays. HCT-116 cells showed the highest colony forming capacity (25%) followed by MDA-MB-231 cells (21%) and NCI-H358 cells (19%). After knock-down of SK-1, colony formation was suppressed, in all cell lines, by 50–90% (Figure 2B).
Figure 2.
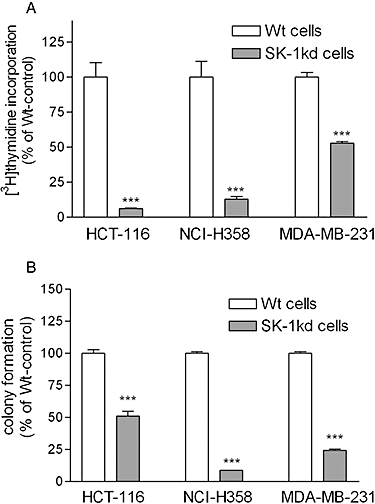
Effect of SK-1 knock-down on [3H]thymidine incorporation and colony formation in HCT-116, MDA-MB-231 and NCI H358 cells. A. 4 × 104 cells of either wild-type (Wt) or SK-1 depleted (SK-1kd) HCT-116, NCI-H358 and MDA-MB-231 were seeded and incubated for 24 h in growth medium containing [3H]thymidine. Cells were harvested and [3H]thymidine incorporated into DNA was determined. Data are expressed as % of Wt-controls and are means ± SD (n = 4); ***P < 0.001 significantly different from the respective Wt-control values. B. 700 cells of either wild-type (Wt) or SK-1 knock-down (SK-1kd) HCT-116, NCI-H358 and MDA-MB-231 were seeded and incubated for 10 days in growth medium. Thereafter, colonies were stained and counted. Data are expressed as % of Wt-controls and are means ± SD (n = 3); ***P < 0.001 significantly different from the Wt-control values.
To investigate whether this effect of SK-1kd on carcinoma cell growth could be reproduced by inhibition of SK-1 catalytic activity, we treated HCT-116 cells with a recently developed SK inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole (SKI II) (French et al., 2003). As shown in Figure 3A, [3H]thymidine incorporation into HCT-116 cells was dose-dependently reduced by SKI II with an IC50 value of about 100 nM. Similarly, suppression of colony formation was also detectable, although higher SKI II concentrations were required for this effect (Figure 3B).
Figure 3.
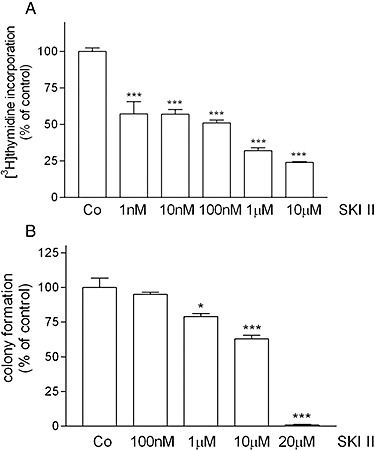
Effect of an inhibitor of SK activity, SKI II, on [3H]thymidine incorporation and colony formation of HCT-116 cells. A. Wild-type HCT-116 cells were treated for 24 h with either vehicle (Co) or the indicated concentrations of the SK inhibitor SKI II in the presence of [3H]thymidine. Cells were harvested and [3H]thymidine incorporated into DNA was determined. Data are expressed as % of controls and are means ± SD (n = 4). B. Wild-type HCT-116 cells were treated for 10 days with either vehicle (Co) or the indicated concentrations of the SK inhibitor SKI II. Thereafter, colonies were stained and counted. Data are expressed as % of controls and are means ± SD (n = 3); *P < 0.05, ***P < 0.001 significantly different from the control values.
Loss of SK-1 impairs PKB/Akt and MAPK signalling, and increases intracellular ROS formation in carcinoma cells
Because loss of SK-1 inhibits cell growth and colony formation, we investigated the activation status of two crucial survival pathways, the protein kinase B (PKB)/Akt pathway (Bonni et al., 1999) and the classical extracellular signal-regulated protein kinase (ERK) pathway (Nicholson and Anderson, 2002) in Wt and SK-1kd cell lines. Interestingly, as shown by Western blot analysis both pathways were defective in all three SK-1kd cell lines. Consequently, phospho-PKB and phospho-ERK levels, which are considered to represent the active enzymes, were strongly reduced in SK-1kd cells compared with Wt cells (Figure 4). Total protein levels of the kinases were not affected. On the contrary, no changes in the phosphorylation status of the stress-activated protein kinase cascades, that is, the N-terminal c-Jun kinases and the p38-mitogen-activated protein kinase were detected (data not shown).
Figure 4.
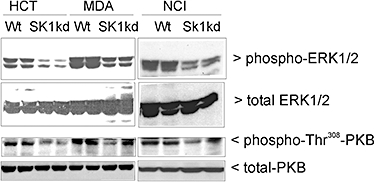
Effect of SK-1 knock-down on the expression and activation profile of ERK1/2 and PKB/Akt in HCT-116, MDA-MB-231 and NCI-H358 cells. Protein lysates of wild-type cells (Wt) or SK-1 knock-down (SK-1kd) HCT-116 (HCT), MDA-MB-231 (MDA) and NCI-H358 (NCI) cells were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using specific antibodies against phospho-ERK1/2, total ERK1/2, phospho-Thr308-PKB/Akt and total PKB/Akt (all at dilutions of 1:1000). Bands were visualized by the ECL method. Data are representative of three independent experiments.
The finding that SK-1kd cells had dysfunctional survival pathways prompted us to measure oxidative stress by determining intracellular ROS levels in Wt and SK-1kd cells using the fluorophore CM-H2DCFDA. As shown in Figure 5A for MDA-MB-231 cells, all cell lines in which SK-1 was down-regulated, that is, shRNA#1 and shRNA#4, had significantly increased ROS levels when compared with Wt cells, whereas the cell line containing the non-functional shRNA#3 had even slightly reduced ROS levels. Similarly, HCT-116 cells and NCI-H358 cells with SK-1 knock-down showed significantly higher levels of ROS compared with Wt cells (Figure 5B). Furthermore, the presence of the NAPDH oxidase inhibitor diphenyleneiodonium (DPI) significantly reduced ROS level in Wt and SK-1kd MDA-MB-231 cells. Similar data were also obtained in HCT-116 and NCI-H358 cells (not shown).
Figure 5.
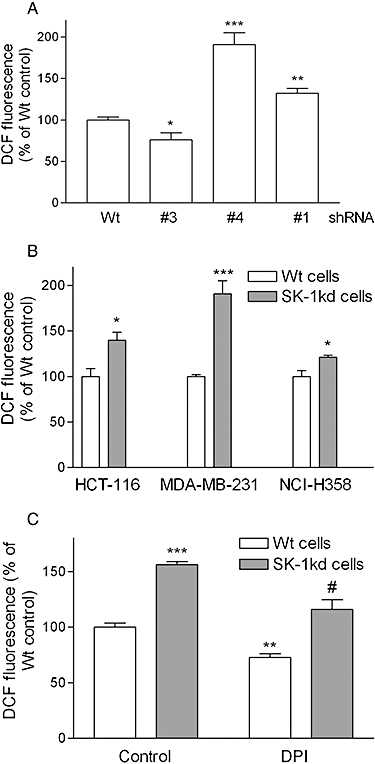
Effect of SK-1 knock-down on ROS formation in MDA-MB-231, HCT-116 and NCI-H358 cells. A. MDA-MB-231 cells of wild-type (Wt) origin or cells stably transduced with shRNA#3, shRNA#4 or shRNA#1 specific for human SK-1 were seeded into 96-well plates and incubated for 20 h before ROS determination. B. HCT-116, MDA-MB-231 and NCI-H358 cells of Wt origin or SK-1kd were seeded into 96-well plates and incubated for 20 h before ROS determination. C. MDA-MD-231 cells of Wt or SK-1kd were pre-incubated for 1 h with either vehicle (Control) or diphenyleneiodonium (DPI, 10 µM) before ROS determination. Data are expressed as % of Wt control values and are means ± SD (n = 3–4); *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from the respective Wt control values. #P < 0.05 significantly different from the SK-1kd control values.
Loss of SK-1 in carcinoma cells increases DNA damage by doxorubicin
Doxorubicin is a potent anticancer agent that induces DSB by increasing lipid peroxidation and generation of ROS, and by inhibition of topoisomerase II (Swift et al., 2006). One early marker of double-strand DNA breaks is the phosphorylation of H2AX at Ser139 at the free DNA ends (Banáth and Olive, 2003). To investigate whether SK-1kd and Wt cells differed in the extent of DNA damage, as a result of protective mechanisms acting before drug-target interaction or in the mode of handling and responding to damaged DNA, phosphorylation analyses of H2AX and damage-sensitive kinases were performed. As shown in Figure 6A, doxorubicin, as expected, induced phosphorylation of H2AX in Wt HCT-116 cells. Interestingly, in SK-1kd cells, the doxorubicin-induced phosphorylation of H2AX was much higher suggesting that more DNA damage occurred in these cells. The protein kinases responsible for H2AX phosphorylation include the DNA damage ‘sensors’ DNA-PK, ATM and ATR (Yang et al., 2003; 2004;). Accordingly, we found that DNA-PK was also phosphorylated and more strongly activated upon doxorubicin treatment in SK-1kd cells (Figure 6A, middle panel). In contrast to DNA-PK, we could not detect SK-1-dependent differences in ATM and ATR phosphorylation (data not shown).
Figure 6.

Effect of SK-1 knock-down and of ROS on doxorubicin-induced phosphorylation of histone-2AX and DNA-PKc in HCT-116, MDA-MB-231 and NCI-H358 cells. A. HCT-116, MDA-MB-231 and NCI-H358 cells of wild-type origin (Wt) or SK-1 knock-down (SK-1kd) were treated for 24 h with either vehicle (Co) or 3 µM doxorubicin (doxo). B. Wt MDA-MB-231 cells were treated for 6 h with either vehicle (Co), or the redox cycler 2,3-dimethoxy-1-naphthoquinone (DMNQ, 50 µM) in the absence (−) or presence of vitamin C (0.1 mM). C. SK-1kd MDA-MB-231 cells were treated for 16 h with either vehicle (−), diphenyleneiodonium (DPI, 10 µM), N-acetylcysteine (NAC, 10 mM) or vitamin C (0.3 mM). Thereafter, cell lysates containing 50 µg of protein were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using specific antibodies against phospho-Ser139-H2AX, phospho-Thr2609-DNA-PKc and GAPDH (all diluted 1:1000). Bands were visualized by the ECL method according to the manufacturer's recommendation. Data shown are duplicates and representative of four independent experiments, giving similar results.
Interestingly, in all SK-1kd cell lines, even in the absence of doxorubicin treatment, there was a basal level of DNA damage, compared with Wt cells, which may at least partly explain their higher sensitivity to doxorubicin, when used as an additional damage inducer. We further tested whether the redox cycler 2,3-dimethoxy-1-naphthoquinone, which generates superoxide radicals, had an effect on H2AX phosphorylation in Wt cells. As seen in Figure 6B, H2AX phosphorylation was increased by 2,3-dimethoxy-1-naphthoquinone and this effect was reversed in the presence of the antioxidant vitamin C. Additionally, the direct NAPDH oxidase inhibitor DPI and the antioxidant vitamin C, but not N-acetyl-cysteine, reduced the increased H2AX phosphorylation in SK-1kd cells down to levels in Wt cells (Figure 6C). These data strongly suggested that the effect of SK-1kd on H2AX phosphorylation was mediated by ROS.
To confirm that the increased phosphorylation of H2AX was a consequence of DNA damage and not due to functional dysregulation of a DNA damage sensor protein, we further performed Comet assays to quantify the amount of DNA breaks. In this assay, the extent of DNA damage induced by doxorubicin was again higher in Sk-1kd than in Wt cells (data not shown). A dose-dependent enhancement of the doxorubicin-induced phosphorylation of H2AX and of DNA-PK was also seen in Wt HCT-116 cells treated with the SK inhibitor SKI II (Figure 7).
Figure 7.
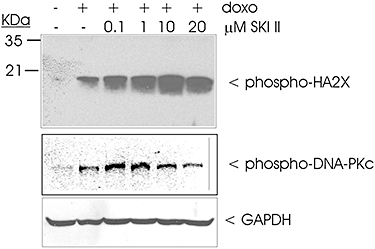
Effect of the SK inhibitor SKI II on doxorubicin-induced phosphorylation of H2AX and DNA-PKc in HCT-116 cells. Wild-type HCT-116 cells were treated for 24 h with either vehicle (−), or doxorubicin (3 µM) in the presence of increasing concentrations of SKI II. Thereafter, cell lysates containing 50 µg of protein, were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using specific antibodies against phospho-Ser139-H2AX (upper panel), phospho-Thr2609-DNA-PKc (middle panel) or GAPDH (lower panel). Bands were visualized by the ECL method according to the manufacturer's recommendation. Data are representative of three independent experiments giving similar results.
Loss of SK-1 sensitizes carcinoma cells to the cytotoxic effect of doxorubicin
To determine whether the increased DNA damage induced by doxorubicin in SK-1kd cells increased cytotoxicity, cell growth was measured by [3H]thymidine incorporation. Treatment of Wt HCT-116 cells for 24 h with doxorubicin led to a dose-dependent reduction of [3H]thymidine uptake into DNA (Figure 8A, open columns) with an IC50 value below 1 nM. After SK-1 knock-down, doxorubicin-induced growth inhibition was further increased. Similarly, also in MDA-MB-231 cells, doxorubicin inhibited DNA synthesis, although they were less sensitive with an IC50 of approximately 3 µM (Figure 8B, open columns). NCI-H358 cells showed an intermediate sensitivity and doxorubicin inhibited DNA synthesis with an IC50 of 300 nM (Figure 8C, open columns). Again, in all three SK-1kd cell lines the sensitivity to doxorubicin was increased. Notably, the sensitivity of the Wt cells to doxorubicin correlated with the amount of SK-1 expression as shown in Figure 1B.
Figure 8.
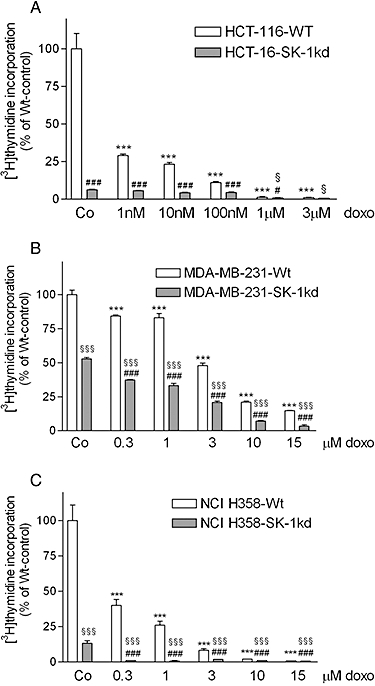
Dose-dependent effect of doxorubicin on [3H]thymidine incorporation in HCT-116, MDA-MB-231 and NCI H358 cells. 4 × 104 HCT-116 (A), MDA-MB-231 (B) and NCI-H358 cells (C), which were either wild-type (Wt) or SK-1 knock-down (SK-1kd) were seeded and incubated for 24 h in growth medium containing vehicle (Co) or the indicated concentrations of doxorubicin (doxo) in the presence of [3H]thymidine. Cells were harvested and [3H]thymidine incorporated into DNA was determined. Data are expressed as % of Wt-controls and are means ± SD (n = 4); ***P < 0.001 significantly different from the Wt-vehicle control values, #P < 0.05, ###P < 0.001 significantly different from the corresponding doxorubicin concentrations in Wt cells; §P < 0.05, §§§P < 0.001 significantly different from the SK-1kd-vehicle control values.
Furthermore, we examined whether loss of SK-1 also sensitized carcinoma cells to drug-induced inhibition of clonogenicity. In all three cell lines, colony formation was dose-dependently reduced by doxorubicin (Figure 9A–C, open columns). After SK-1 knock-down, the inhibition of colony formation by doxorubicin was more pronounced (Figure 9A–C, open columns vs. closed columns). To investigate this phenomenon further, in a control experiment with HCT-116 cells we also tested the combination of doxorubicin with the pharmacological SK inhibitor SKI II. As shown in Figure 10, SKI II indeed significantly increased the effects of all concentrations of doxorubicin on DNA synthesis.
Figure 9.
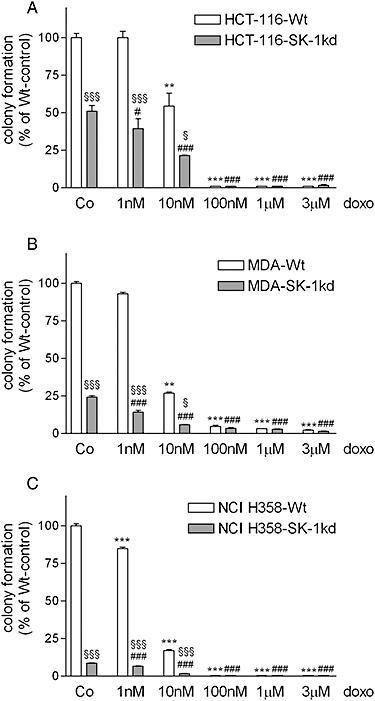
Effect of SK-1 knock-down and doxorubicin on carcinoma cell colony formation. 700 wild-type cells (Wt) and SK-1 knock-down cells (SK-1kd) of HCT-116 (A), MDA-MB-231 (B) and NCI-H358 (C) were incubated for 10 days in growth medium in the absence or presence of the indicated concentrations of doxorubicin. Thereafter, colonies were stained and counted. Data are expressed as % of Wt-controls and are means ± SD (n = 3); ***P < 0.001 significantly different from the Wt-vehicle control values, #P < 0.05, ###P < 0.001 significantly different from the corresponding doxorubicin concentrations in Wt cells; §P < 0.05, §§§P < 0.001 significantly different from the SK-1kd-vehicle control values.
Figure 10.
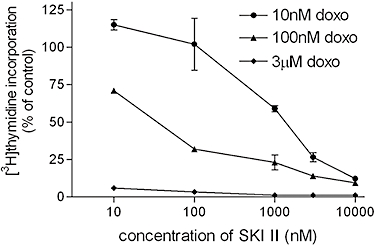
Effect of a combination of SKI II with doxorubicin on DNA synthesis of HCT-116 cells. Wild-type HCT-116 cells were treated for 24 h with the indicated concentrations of the SK inhibitor SKI II together with either 10 nM (circles), 100 nM (triangles) or 3 µM (squares) doxorubicin in the presence of [3H]thymidine. Cells were harvested and [3H]thymidine incorporated into DNA was determined. Data are expressed as % of controls and are means ± SD (n = 4).
Finally, we investigated whether, in the presence of doxorubicin, the increased DNA damage and the defects in survival and cell cycle signalling detected in SK-1kd cells would facilitate apoptosis. As a marker of increased apoptosis following SK-1 knock-down, processing of caspase-3 to the active 17 and 19 kDa fragments was enhanced in all SK-1kd cell lines (Figure 11A). Moreover, we analysed apoptotic cells by flow cytometry measuring annexin V binding to PS exposed on the cell surface. As shown in Figure 11B and C, the binding of annexin V after doxorubicin treatment was clearly more pronounced in SK-1kd HCT-116 and MDA-MB231 cells, respectively. In NCI-H358 cells, annexin V staining showed a strong signal in untreated cells (data not shown), probably as a consequence of membrane disruption by trypsinization. Therefore, for this cell line, we used Trypan blue staining to quantify non-viable cells. As shown in Figure 11D, doxorubicin decreased cell viability and this cytotoxic effect was significantly more pronounced in SK-1kd cells. Finally, to see whether the potentiation by SK-1kd of doxorubicin-induced apoptosis was mediated by ROS, we treated the cells in the presence of DPI, an inhibitor of NADPH oxidase, a major generator of ROS. This regimen led to a partial but significant reduction of apoptosis (Figure 12).
Figure 11.
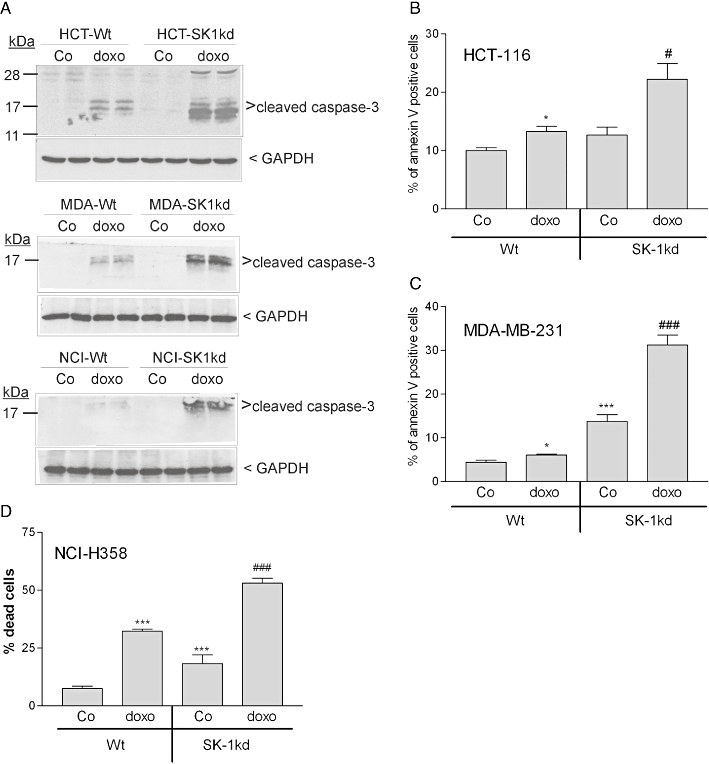
Effect of SK-1 knock-down on doxorubicin-induced caspase-3 processing, annexin V binding or Trypan blue exclusion in HCT-116, MDA-MB-231 and NCI-H358 cells. A. To detect caspase-3 activation, MDA-MB-231 (MDA), HCT-116 (HCT) and NCI-H358 (NCI) cells were treated for 6 h with either vehicle (Co) or 3 µM doxorubicin (doxo). Thereafter, cell lysates were taken for SDS-PAGE, transferred to nitrocellulose and subjected to Western blotting using a cleaved caspase-3-specific antibody (dilution 1:1000) or a GAPDH antibody (1:1000). Data are representative of two independent experiments performed in duplicates that gave similar results. B HCT-116 and C MDA-MB-231 cells were treated for 6 h with either vehicle (Co) or 3 µM doxorubicin (doxo). Thereafter, cells were taken for annexin V surface staining of apoptotic cells. Data show the % of annexin V positive cells and are means ± SD (n = 3). D. NCI-H358 cells were treated for 24 h with either vehicle (Co) or 3 µM doxorubicin (doxo), and subsequently analysed for Trypan blue exclusion to quantify non-viable cells. Data show the % of dead cells and are means ± SD (n = 3), *P < 0.05, ***P < 0.001 significantly different from the vehicle treated samples; #P < 0.05, ###P < 0.001 significantly different from the doxo-treated Wt samples.
Figure 12.
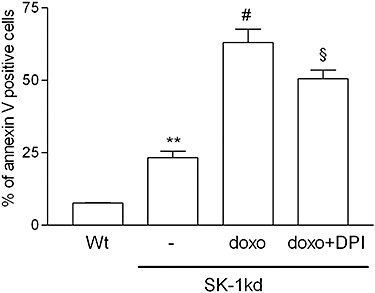
Effect of the NADPH oxidase inhibitor DPI on doxorubicin-induced apoptosis in MDA-MB-231 cells. MDA-MB-231 cells of wild-type origin (Wt) or SK-1kd cells were treated for 6 h with either vehicle (−), doxorubicin (doxo, 3 µM) or doxo plus diphenyleneiodonium (DPI, 10 µM). Thereafter, cells were taken for annexin V surface staining of apoptotic cells. Data show the % of annexin V positive cells and are means ± SD (n = 3). **P < 0.01 significantly different from the vehicle-treated Wt values; #P < 0.05 significantly different from the vehicle-treated SK-1kd values; §P < 0.05 significantly different from the doxo-treated SK-1kd values.
Discussion
Despite the use of new anticancer agents and more effective combination treatments, drug resistance remains a major obstacle to the successful treatment of advanced solid tumours. Therefore, new therapeutic targets are desperately needed, which are relevant for the malignant phenotype and are selectively expressed in tumours. SK-1 is overexpressed in many human tumours and has been identified as a negative prognostic marker in various carcinomas and brain tumours (French et al., 2003; Li et al., 2008; 2009; Ruckhäberle et al., 2008). Further evidence from preclinical studies also suggests that SK-1 is an attractive target for cancer therapy, as its overexpression is associated with tumour angiogenesis and resistance to radiation and chemotherapy (Shida et al., 2008).
In this study we have demonstrated that targeted inhibition of SK-1 expression using shRNA suppressed the growth of carcinoma cells of various histotypes from colon, breast and lung as measured in 3H-thymidine incorporation assays. Further evidence for the potential therapeutic implication of SK-1 targeting came from the finding that loss of SK-1 also affected actively proliferating cells as measured by suppression of colony formation. Because SK-1 is responsible for the generation of S1P, which is a mitogenic factor for many cell types (Olivera and Spiegel, 1993; Huwiler et al., 2000; Huwiler and Zangemeister-Wittke, 2007), it is very likely that the loss of DNA synthesis and cell cycle progression is due to the interruption of SK-1/S1P signalling. The mechanism seems to strictly involve intracellular S1P action and not extracellular S1P acting via surface S1P receptors, because the strong reduction of DNA synthesis in SK-1kd cells occurred in the presence of serum, which already contains a high concentration of S1P, in the range of 500–900 nM (Okajima, 2002). Thus, if S1P receptors were to be involved, serum S1P would be expected to compensate for the lack of intracellular S1P generation and export in SK-1kd cells. The molecular mechanism by which intracellularly generated S1P triggers proliferation is still unclear. Recently, an export of intracellularly generated S1P was proposed, which then acts via S1P receptors to activate the cell cycle machinery (Ancellin et al., 2002; Venkataraman et al., 2006; Kim et al., 2009). In this context it was shown that the multi-drug resistance transporters P-glycoprotein (MDR1) and MRP1 (ABCC1) can translocate sphingolipids, including S1P, across the plasma membrane (Raggers et al., 1999; Mitra et al., 2006). Alternatively, an intracellular action of S1P may also be possible. Van Brocklyn et al. (1998) showed that microinjection of S1P into mouse fibroblast cells stimulated DNA synthesis without involving S1P receptors. This result, however, seems to be cell type-specific as microinjection into neuronal cells was ineffective (Postma et al., 1996) and microinjection into keratinocytes reduced cell proliferation (Vogler et al., 2003).
Furthermore, our data demonstrate that in all carcinoma cell lines with SK-1kd, activation of the classical ERKs and PKB/Akt was strongly diminished. These protein kinases are crucial for cell growth and survival (Bonni et al., 1999; Nicholson and Anderson, 2002), and their deregulated expression or hyperactivation confers resistance to apoptosis in many cell types. In this context it was shown that ERK mediates Bcl-2 phosphorylation (Deng et al., 2001) thereby activating this anti-apoptotic survival protein, whereas PKB/Akt phosphorylates pro-apoptotic Bad to promote its dissociation from anti-apoptotic Bcl-2 and Bcl-XL to maintain their anti-apoptotic function (Zha et al., 1996). S1P is a strong activator of both ERK and PKB/Akt activation (Baudhuin et al., 2004; Xin et al., 2004), and it is thus conceivable that S1P is cytoprotective by triggering these pathways.
In all tested cell lines we found that loss of SK-1 expression increased oxidative stress as measured by increased ROS generation, and sensitized cells to DNA damage by the anticancer agent doxorubicin. This observation was confirmed in the presence of the recently developed SK inhibitor SKI II, which was also able to sensitize HCT-116 cells to doxorubicin. However, dependent on the cell line the use of higher doses of inhibitor alone not only inhibited cell proliferation but showed signs of non-specific toxicity (data not shown), and thus the use of this inhibitor on its own, does not constitute a reliable test system to investigate the effect of SK-1 inactivation in the presence of an additional cytotoxic agent. The reason for the constitutively increased ROS production in the absence of SK-1 remains to be investigated. ROS production in cells is usually the result of oxidative stress when antioxidants are depleted. Treatment with S1P was shown to protect cultured cardiac myocytes and isolated hearts from apoptosis (Karliner et al., 2001). Conversely, there is also evidence that the sphingolipid rheostat plays a role in the cellular response to oxidative stress and ceramide has been implicated in ROS-induced cell death, in a variety of normal cells and tissues (Huwiler et al., 2001; Maceyka et al., 2007). This is consistent with our earlier finding in glomerular endothelial cells that the enzymatic activity of neutral sphingomyelinase is activated by superoxide generation, but is inhibited by increasing the level of reduced glutathione (Huwiler et al., 2001). Enhanced ROS can lead to DNA damage as shown here by increased H2AX phosphorylation, but it may also contribute to malignant progression by activating survival signalling pathways. As, in the present study, all tested SK-1kd carcinoma cell lines showed reduced proliferation and colony formation, we can rule out a survival advantage at least under standard cell culture conditions. Interestingly, exogenous S1P also stimulated ROS generation in isolated hamster resistance arteries and triggered vessel contraction (Keller et al., 2006). This is reminiscent of recent findings that extracellular and intracellularly S1P can have opposing effects on cell responses as exemplified by the expression and secretion of the profibrotic connective tissue growth factor in renal podocytes (Ren et al., 2009). Clearly, the S1P-triggered mechanisms, either via S1P receptors or via intracellular targets, may be completely different.
Loss of SK-1 indeed resulted in slightly increased amounts of DSB as demonstrated by increased phospho-H2AX and phospho-DNA-PK. Doxorubicin is frequently used for the treatment of solid tumours and induces cell cycle arrest and apoptosis by induction of DNA damage. Anthracyclines are known for their complex cytotoxic mechanism involving inhibition of topoisomerase II, RNA polymerase and cytochrome c oxidase, intercalation into DNA, chelation of iron and generation of ROS (Müller et al., 1998). More recently, it was also suggested that doxorubicin increases the intracellular levels of the lipid messengers ceramide and diacylglycerol, and decreases intracellular glutathione (Martínez et al., 2009). Based on this observation, the enhanced cytotoxic effect of doxorubicin on SK-1kd cells with their higher pre-existing levels of DNA damage is not surprising, as it is reasonable that such damage primes the cells for additional damage, thereby increasing drug sensitivity and facilitating apoptosis. Recently, SK-1 was shown to modulate also the function of the multi-drug transporter protein P-glycoprotein (Pilorget et al., 2007) for which doxorubicin is a substrate. However, we could not measure changes in P-glycoprotein expression in SK-1kd cells (data not shown) and therefore consider that that the increased DNA damage in these cells did not result from enhanced intracellular drug accumulation. Furthermore, as measured by rhodamine 123 uptake, drug efflux was not affected in SK-1kd cells compared with Wt cells (data not shown).
In all tested carcinoma cell lines, doxorubicin-induced death occurred by classical apoptosis involving caspase-3 processing. SK-1kd cells also showed, in general, higher levels of spontaneous apoptosis compared with their Wt counterparts, with the biggest difference being observed in HCT-116 cells. Thus, SK-1 loss not only diminished the proliferative capacity of carcinoma cells but also facilitated apoptosis induced by genotoxic therapy by increasing ROS as an intracellular stress factor. There was no correlation between the cell's spontaneous apoptosis or sensitivity to doxorubicin and their p53 status.
In summary, we described, for the first time, that targeted inhibition of SK-1 in various carcinoma cell types increased oxidative stress, thereby inhibiting cell proliferation and facilitating drug-induced apoptosis by sensitizing the cells to DNA damage. Our finding may have considerable implication for the design of more effective cancer therapy based on the use of synergistic combinations of SK-1 targeting and the anticancer agent doxorubicin.
Acknowledgments
We thank Mrs Marianne Maillard-van Laer, Susanne Probst, Svetlana Bubnova and Isolde Römer for excellent technical assistance. This work was supported by the Wilhelm Sander Foundation, the Swiss National Foundation (310000-119859), the German Research Foundation (FOG748, HU 842/5-1, PF367/6-1, SFB 815) and the Krebsliga of the Kanton Bern.
Glossary
Abbreviations
- ATM
ataxia telangiectasia mutated
- ATR
ATM- and Rad3-related
- CM-H2DCFDA
5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester
- DNA-PK
DNA-dependent protein kinase
- DPI
diphenyleneiodonium
- DSB
DNA double strand breaks
- ERK
extracellular signal-regulated protein kinase
- H2AX
histone-2AX
- PKB
protein kinase B/Akt
- ROS
reactive oxygen species
- S1P
sphingosine 1-phosphate
- SDS-PAGE
sodium dodecylsulfate-polyacrylamide gel electrophoresis
- shRNA
small hairpin RNA
- SK
sphingosine kinase
- SK-1kd
SK-1 knock-down
- SKI
II, 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–12 as PowerPoint slide.
References
- Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer zu Heringdorf D. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:413–428. doi: 10.1007/s00210-007-0132-3. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancellin N, Colmont C, Su J, Li Q, Mittereder N, Chae SS, et al. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J Biol Chem. 2002;277:6667–6675. doi: 10.1074/jbc.M102841200. [DOI] [PubMed] [Google Scholar]
- Banáth JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003;63:4347–4350. [PubMed] [Google Scholar]
- Baudhuin LM, Jiang Y, Zaslavsky A, Ishii I, Chun J, Xu Y. S1P3-mediated Akt activation and cross-talk with platelet-derived growth factor receptor (PDGFR) FASEB J. 2004;18:341–343. doi: 10.1096/fj.03-0302fje. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Chen Y, Poon RY. The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Front Biosci. 2008;13:5016–5029. doi: 10.2741/3060. [DOI] [PubMed] [Google Scholar]
- Deng X, Kornblau SM, Ruvolo PP, May WS., Jr Regulation of Bcl2 phosphorylation and potential significance for leukemic cell chemoresistance. J Natl Cancer Inst Monogr. 2001;28:30–37. doi: 10.1093/oxfordjournals.jncimonographs.a024254. [DOI] [PubMed] [Google Scholar]
- Döll F, Pfeilschifter J, Huwiler A. The epidermal growth factor stimulates sphingosine kinase-1 expression and activity in the human mammary carcinoma cell line MCF7. Biochim Biophys Acta. 2005;1738:72–81. doi: 10.1016/j.bbalip.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Döll F, Pfeilschifter J, Huwiler A. Prolactin upregulates sphingosine kinase-1 expression and activity in the human breast cancer cell line MCF7 and triggers enhanced proliferation and migration. Endocr Relat Cancer. 2007;14:325–335. doi: 10.1677/ERC-06-0050. [DOI] [PubMed] [Google Scholar]
- Fillingham J, Keogh MC, Krogan NJ. GammaH2AX and its role in DNA double-strand break repair. Biochem Cell Biol. 2006;84:568–577. doi: 10.1139/o06-072. [DOI] [PubMed] [Google Scholar]
- French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor activity of sphingosine kinase inhibitors. J Pharmacol Exp Ther. 2006;318:596–603. doi: 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- Hiom K. DNA repair: how to PIKK a partner. Curr Biol. 2005;15:R473–R475. doi: 10.1016/j.cub.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Hofmann LP, Ren S, Schwalm S, Pfeilschifter J, Huwiler A. Sphingosine kinase 1 and 2 regulate the capacity of mesangial cells to resist apoptotic stimuli in an opposing manner. Biol Chem. 2008;389:1399–1407. doi: 10.1515/BC.2008.160. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Zangemeister-Wittke U. Targeting the conversion of ceramide to sphingosine 1-phosphate as a novel strategy for cancer therapy. Crit Rev Oncol Hematol. 2007;63:150–159. doi: 10.1016/j.critrevonc.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Kolter T, Pfeilschifter J, Sandhoff K. Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim Biophys Acta. 2000;1485:63–99. doi: 10.1016/s1388-1981(00)00042-1. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Böddinghaus B, Pautz A, Dorsch S, Briner VA, Brade V, et al. Superoxide potently induces ceramide formation in glomerular endothelial cells. Biochem Biophys Res Commun. 2001;284:404–410. doi: 10.1006/bbrc.2001.4941. [DOI] [PubMed] [Google Scholar]
- Karliner JS, Honbo N, Summers K, Gray MO, Goetzl EJ. The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J Mol Cell Cardiol. 2001;33:1713–1717. doi: 10.1006/jmcc.2001.1429. [DOI] [PubMed] [Google Scholar]
- Keller M, Lidington D, Vogel L, Peter BF, Sohn HY, Pagano PJ, et al. Sphingosine kinase functionally links elevated transmural pressure and increased reactive oxygen species formation in resistance arteries. FASEB J. 2006;20:702–704. doi: 10.1096/fj.05-4075fje. [DOI] [PubMed] [Google Scholar]
- Kim RH, Takabe K, Milstien S, Spiegel S. Export and functions of sphingosine-1-phosphate. Biochim Biophys Acta. 2009;1791:692–696. doi: 10.1016/j.bbalip.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Guan HY, Gong LY, Song LB, Zhang N, Wu J, et al. Clinical significance of sphingosine kinase-1 expression in human astrocytomas progression and overall patient survival. Clin Cancer Res. 2008;14:6996–7003. doi: 10.1158/1078-0432.CCR-08-0754. [DOI] [PubMed] [Google Scholar]
- Li W, Yu CP, Xia JT, Zhang L, Weng GX, Zheng HQ, et al. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2009;15:1393–1399. doi: 10.1158/1078-0432.CCR-08-1158. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Milstien S, Spiegel S. Shooting the messenger: oxidative stress regulates sphingosine-1-phosphate. Circ Res. 2007;100:7–9. doi: 10.1161/01.RES.0000255895.19868.a3. [DOI] [PubMed] [Google Scholar]
- Martínez R, Navarro R, Lacort M, Ruiz-Sanz JI, Ruiz-Larrea MB. Doxorubicin induces ceramide and diacylglycerol accumulation in rat hepatocytes through independent routes. Toxicol Lett. 2009;190:86–90. doi: 10.1016/j.toxlet.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci USA. 2006;103:16394–16399. doi: 10.1073/pnas.0603734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller I, Niethammer D, Bruchelt G. Anthracycline-derived chemotherapeutics in apoptosis and free radical cytotoxicity. Int J Mol Med. 1998;1:491–494. doi: 10.3892/ijmm.1.2.491. [DOI] [PubMed] [Google Scholar]
- Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- Okajima F. Plasma lipoproteins behave as carriers of extracellular sphingosine 1-phosphate: is this an atherogenic mediator or an anti-atherogenic mediator? Biochim Biophys Acta. 2002;1582:132–137. doi: 10.1016/s1388-1981(02)00147-6. [DOI] [PubMed] [Google Scholar]
- Olivera A, Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365:557–560. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V, et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005;65:11667–11675. doi: 10.1158/0008-5472.CAN-05-2702. [DOI] [PubMed] [Google Scholar]
- Pilorget A, Demeule M, Barakat S, Marvaldi J, Luis J, Béliveau R. Modulation of P-glycoprotein function by sphingosine kinase-1 in brain endothelial cells. J Neurochem. 2007;100:1203–1210. doi: 10.1111/j.1471-4159.2006.04295.x. [DOI] [PubMed] [Google Scholar]
- Postma FR, Jalink K, Hengeveld T, Moolenaar WH. Sphingosine-1-phosphate rapidly induces Rho-dependent neurite retraction: action through a specific cell surface receptor. EMBO J. 1996;15:2388–2392. [PMC free article] [PubMed] [Google Scholar]
- Raggers RJ, van Helvoort A, Evers R, van Meer G. The human multidrug resistance protein MRP1 translocates sphingolipid analogs across the plasma membrane. J Cell Sci. 1999;112:415–422. doi: 10.1242/jcs.112.3.415. [DOI] [PubMed] [Google Scholar]
- Ren S, Babelova A, Moreth K, Xin C, Eberhardt W, Doller A, et al. Transforming growth factor-beta2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int. 2009;76:857–867. doi: 10.1038/ki.2009.297. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckhäberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat. 2008;112:41–52. doi: 10.1007/s10549-007-9836-9. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Maceyka M, Hait NC, Paugh SW, Sankala H, Milstien S, et al. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005;579:5313–5317. doi: 10.1016/j.febslet.2005.08.055. [DOI] [PubMed] [Google Scholar]
- Shida D, Fang X, Kordula T, Takabe K, Lépine S, Alvarez SE, et al. Cross-talk between LPA1 and epidermal growth factor receptors mediates up-regulation of sphingosine kinase 1 to promote gastric cancer cell motility and invasion. Cancer Res. 2008;68:6569–6577. doi: 10.1158/0008-5472.CAN-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift LP, Rephaeli A, Nudelman A, Phillips DR, Cutts SM. Doxorubicin-DNA adducts induce a non-topoisomerase II-mediated form of cell death. Cancer Res. 2006;66:4863–4871. doi: 10.1158/0008-5472.CAN-05-3410. [DOI] [PubMed] [Google Scholar]
- Van Brocklyn JR, Lee MJ, Menzeleev R, Olivera A, Edsall L, Cuvillier O, et al. Dual actions of sphingosine-1-phosphate: extracellular through the Gi-coupled receptor Edg-1 and intracellular to regulate proliferation and survival. J Cell Biol. 1998;142:229–240. doi: 10.1083/jcb.142.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman K, Thangada S, Michaud J, Oo ML, Ai Y, Lee YM, et al. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem J. 2006;397:461–471. doi: 10.1042/BJ20060251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler R, Sauer B, Kim DS, Schäfer-Korting M, Kleuser B. Sphingosine-1-phosphate and its potentially paradoxical effects on critical parameters of cutaneous wound healing. J Invest Dermatol. 2003;120:693–700. doi: 10.1046/j.1523-1747.2003.12096.x. [DOI] [PubMed] [Google Scholar]
- Xin C, Ren S, Kleuser B, Shabahang S, Eberhardt W, Radeke H, et al. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J Biol Chem. 2004;279:35255–35262. doi: 10.1074/jbc.M312091200. [DOI] [PubMed] [Google Scholar]
- Yang J, Yu Y, Hamrick HE, Duerksen-Hughes PJ. ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis. 2003;24:1571–1580. doi: 10.1093/carcin/bgg137. [DOI] [PubMed] [Google Scholar]
- Yang J, Xu ZP, Huang Y, Hamrick HE, Duerksen-Hughes PJ, Yu YN. ATM and ATR: sensing DNA damage. World J Gastroenterol. 2004;10:155–160. doi: 10.3748/wjg.v10.i2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.