

Abstract
Right ventricular failure (RVF) is the leading cause of death in pulmonary arterial hypertension (PAH). Some patients with pulmonary hypertension are adaptive remodelers and develop RV hypertrophy (RVH) but retain RV function; others are maladaptive remodelers and rapidly develop RVF. The cause of RVF is unclear and understudied and most PAH therapies focus on regressing pulmonary vascular disease. Studies in animal models and human RVH suggest that there is reduced glucose oxidation and increased glycolysis in both adaptive and maladaptive RVH. The metabolic shift from oxidative mitochondrial metabolism to the less energy efficient glycolytic metabolism may reflect myocardial ischemia. We hypothesize that in maladaptive RVH a vicious cycle of RV ischemia and transcription factor activation causes a shift from oxidative to glycolytic metabolism thereby ultimately promoting RVF. Interrupting this cycle, by reducing ischemia or enhancing glucose oxidation, might be therapeutic. Dichloroacetate, a pyruvate dehydrogenase kinase inhibitor, has beneficial effects on RV function and metabolism in experimental RVH, notably improving glucose oxidation and enhancing RV function. This suggests the mitochondrial dysfunction in RVH may be amenable to therapy. In this mini review, we describe the role of impaired mitochondrial metabolism in RVH, using rats with adaptive (pulmonary artery banding) or maladaptive (monocrotaline-induced pulmonary hypertension) RVH as models of human disease. We will discuss the possible mechanisms, relevant transcriptional factors, and the potential of mitochondrial metabolic therapeutics in RVH and RVF.
Keywords: Fatty acid oxidation inhibitors, Myc, Dichloroacetate, Pyruvate dehydrogenase kinase, Action potential duration
Introduction
Right ventricular failure (RVF) is the leading cause of death in patients with pulmonary arterial hypertension (PAH). PAH patients admitted to intensive care units with RVF have a 41% acute mortality rate [1]. Current treatment regimens for RVF are empirical and include digoxin, lasix, and intravenous inotropes. The fact these regimens vary widely amongst institutions and lack a basis in evidence from randomized clinical trials reflects an ignorance of the pathophysiology of RVF. The problem of RVF is not restricted to PAH patients. Most heart failure patients, regardless of etiology, develop a component of RVF. RV ejection fraction (EF) is “the single most important predictor of short-term prognosis in a large cohort of CHF patients who had symptoms in spite of a standardized, optimized, multipharmacologic treatment” [2]. However, the importance of preserving RV function is especially evident in PAH, where RV function trumps pulmonary vascular resistance (PVR) and other measures of pulmonary vascular obstruction as a prognostic factor [3]. Despite the significance of RVF to survival, there are no therapies that directly and selectively improve RV function and the basis for RV failure is poorly understood. Understanding the basis for RVF and developing effective therapies has huge public health relevance. In our opinion, there is confusion in the literature because of a view that the RV is a passive responder to increased afterload (PVR). Although a cure for PAH will require regression of pulmonary vascular lesions, substantial improvement in longevity and functional state can be expected with an effective treatment for RVF, much as has transpired in LV failure.
In patients with RVH due to PAH, myocardial scintigraphy suggests that the degree of RV ischemia is highly correlated with RV dysfunction [4]. There are two potential explanations for RV ischemia in RVH and they are not mutually exclusive. The simplest holds that right coronary artery perfusion pressure (Ao-RV pressure difference) is reduced. The second holds that it is the loss of small vessels (arteriolar–capillary rarefaction), rather than reduced coronary perfusion pressure, that is the main driver of RV ischemia in RVF (Fig. 1). Van Wolferen et al. found that in patients with PH, coronary artery flow (RCAF) is reduced during systole, but is preserved in diastole. However, they noted that the mean RCAF is decreased in severe RVH [5]. The reduced mean RCAF is strongly and directly correlated with RV systolic pressure (RVSP) and RV mass. While severe reduction in coronary perfusion pressure can reduce RV function, RV contractile function remains constant until perfusion pressures fall below 50 mmHg [6], which is rare, save in the worst PH. Thus, it is controversial whether reduced epicardial coronary perfusion pressure is the cause of ischemia that elicits the metabolic switch that we believe ultimately leads to RVF. It appears as or more likely that capillary rarefaction may lead to microvascular ischemia. We base this notion on published data showing capillary rarefaction is more common in maladaptive RVH [7] and on our own unpublished data, which show that epicardial perfusion pressure is reduced similarly in adaptive (pulmonary artery banding, PAB) and maladaptive (60 mg/kg monocrotaline-induced) RVH, suggesting another factor, capillary rarefaction, is a more important determinant of ischemia.
Fig. 1.
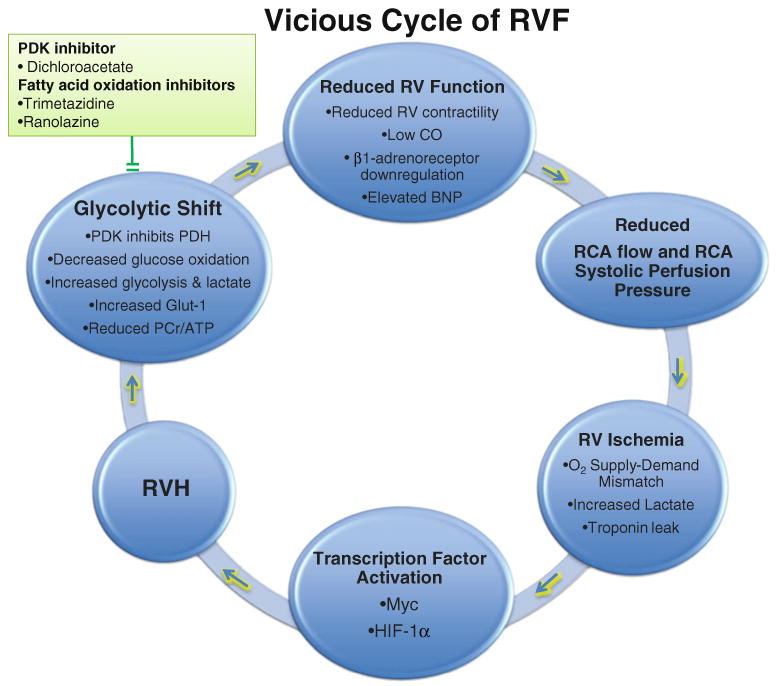
The vicious cycle of right ventricular failure (RVF). The reduced right coronary artery (RCA) flow and RCA systolic perfusion pressure in RVH ultimately cannot support the increased oxygen demand in increased RV mass. This causes RV hypoxia and ischemia. The transcriptional factors HIF-1α and Myc are activated by RV ischemia. This transcriptional reprogramming contributes to a switch from mitochondrial metabolism (glucose oxidation) to glycolysis. RV function is then further reduced by the depressed ATP production and myocardial acidosis and eventually RVF develops. Interrupting this cycle, by enhancing blood supply, inhibiting transcription factors, or restoring glucose oxidation (by inhibiting PDKs or FAO) is postulated to be therapeutic
Positron emission tomography (PET) studies suggest that there is increased RV glucose uptake in patients and animals with RVH [8]. This increased uptake of fluorodeoxyglucose (FDG) is thought to reflect enhanced glycolysis. The less efficient production of ATP by glycolysis in RVH (which leads to the formation of lactate, rather than pyruvate and thus does not fuel Krebs' cycle) is incompletely compensated for by increased expression/activity of glucose transporters, such as Glut1. The concordant development of excessive glycolysis and ventricular dysfunction in both rat and human RV during the development of severe RVH suggests dysfunction of mitochondrial metabolism is a pathologically relevant feature of RVF [8–10]. We speculate that increased glycolysis is insufficient to compensate for suppression of glucose oxidation in severe RVH, resulting in a state of RV hibernation. The term hibernating myocardium refers to hearts in which impaired ventricular metabolism and contractility are the result of a chronic reduction in coronary flow that is insufficient to cause infarction but which impairs function. Our observation in monocrotaline-induced RVH is consistent with hibernation of the RV (i.e., increased RV FDG uptake, reduced O2 consumption and enhanced glycolysis with reduced RV EF—all improved by restoring glucose oxidation) [9]. Studies in monocrotaline and PAB rats with RVH show that that mitochondrial dysfunction and decreased glucose oxidation reflect (in part) the activation of pyruvate dehydrogenase kinases (PDKs) [9]. PDKs phosphorylate and inhibit pyruvate dehydrogenase (PDH; Fig. 2). Inhibition of PDH prevents pyruvate from entering the Kreb cycle and obligates the myocyte to derive energy from glycolysis. This glycolytic shift seems to be common to many forms of RVH and can be identified noninvasively by increased FDG uptake on PET scans. The mitochondrial metabolic switch from glucose oxidation to glycolysis is associated with the impairment of RV function (reduced RV contractility and cardiac function) [9]. Understanding the basis and consequences of this metabolic switch in RVH may help suggest therapeutic strategies to improve RV function.
Fig. 2.

The interaction between glucose oxidation and fatty acid oxidation. Glycolysis: The glucose transporters (Glut1 and Glut4) transport glucose into the cytosol. Hexokinase (HK) generates glucose-6-phosphate, thus lowering glucose concentrations and maintaining the driving gradient for ongoing glucose uptake via the glucose transporters. After several steps of reactions, Glucose-6-P is converted to pyruvate. Lactate is produced from pyruvate by lactate dehydrogenase A (LDHA). Glucose oxidation: The pyruvate dehydrogenase (PDH) complex converts pyruvate (derived from glycolysis) to acetyl-CoA in the mitochondrion, thus allowing it to enter the Krebs' Cycle. PDKs activation inhibits PDH and blocks glucose oxidation. Fatty acid oxidation: Free fatty acid is transported into the cytosol by fatty acid transporters FATP1 and FATP6 and is converted to fatty acyl-CoA. Fatty acyl-CoA is transported by carnitine palmitoyltransferase 1 (CPT1) and carnitine palmitoyltransferase 2 (CPT2) into mitochondria and undergoes β-oxidation. Acetyl-CoA derived from β-oxidation then enters the Krebs' cycle. The increase in acetyl-CoA increases citrate levels, which inhibits glycolysis and activates PDKs (thereby inhibiting glucose oxidation). Either the inhibition of PDKs by dichloroacetate or the inhibition of β oxidation by trimetazidine and ranolazine could improve glucose oxidation
Substrate and oxygen utilization in normal RV
Glycolysis and glucose oxidation are the major sources of ATP production in the fetal heart which is exposed to low circulating fatty acid levels [11]. Fatty acid oxidation (FAO) becomes the major source of energy production (60–90%) after birth. Nonetheless, glucose metabolism remains important, accounting for 10–40% of ATP production [12]. In various cardiac diseases, including RVH, the metabolic fate of glucose is altered because mitochondrial metabolism is actively (and reversibly) suppressed. Glucose metabolism begins with glycolysis, a cytosolic process that converts glucose to pyruvate in the cytosol. In the normal adult RV, glycolysis provides pyruvate to PDH. If the PDH complex in mitochondria is active (i.e., expressed at normal levels and not inhibited by PDKs), this pyruvate enters the mitochondria and is converted to acetyl-COA, fueling the Krebs cycle [13]. However, if pyruvate cannot be used by mitochondrial PDH to fuel oxidative phosphorylation, only two ATP molecules/glucose are obtained (versus 36 or 38 ATP during glucose oxidation) and lactate is the end product of glycolysis, resulting in acidosis.
There is a competitive relationship between the oxidation of fatty acid and glucose [13] (see Fig. 2). Increased FAO inhibits glucose oxidation, a phenomenon referred to as “Randle Cycle.” The mechanism for this feedback inhibition is multifactorial and includes at least three factors: inhibition of phosphofructokinase by FAO-mediated citrate production; hexokinase inhibition, which reflects accumulation of glucose-6-phosphate; and inhibition of PDH activity caused by FAO-mediated acetyl-CoA-accumulation, which activates PDKs. We hypothesize that the Randle cycle can be exploited in RVH. In left ventricular ischemia, partial inhibition of FAO reduces lactate production, increases glucose oxidation, and enhances left ventricular (LV) contractility by augmenting regional contractile power and efficiency, implying the therapeutic potential of the Randle cycle [14]. This benefit is consistent with the observation that FAO costs about 12% more oxygen per unit of ATP generated, suggesting a switch to glucose oxidation may more efficiently use the limited oxygen that is available [15]. Whether inhibiting FAO can enhance glucose oxidation and improve RV function has not been tested in RVH. However, it is plausible that FAO inhibitors and PDKs inhibitors may, through unique mechanisms, both enhance mitochondrial function in RVH.
Mitochondrial metabolic adaptation in RVH
The RV has a lower oxygen requirement at rest and concordantly has lower resting coronary blood flow compared to the LV. The elevation in RV mass is the predominant adaptation to increased afterload in PH. With this comes a greater oxygen requirement. Hypertrophied RV has lower oxygen extraction reserve than normal RV and a higher dependence on coronary flow [16], suggesting an inefficient oxygen utilization despite a greater net oxygen demand. Isolated RV tissue, both in monocrotaline and PAB-induced RVH, shows lower glucose-based O2 consumption/g of myocardium in RVH [9]. This appears to reflect the consequence of impaired mitochondrial glucose oxidation, since O2 consumption is restored by inhibiting PDKs and elevating PDH activity [9].
Recognition that there is a switch in RV metabolism from oxidation of glucose to glycolysis dates back to the early 1970s. Bishop et al. discovered increased glycolysis (measured as increased lactate production in RV homogenate) and increased lactate dehydrogenase (LDH2 and LDH3) in RVH induced in dogs by PA banding [17]. Recent studies in humans and in animal models document the elevation of glycolysis in RVH using FDG PET scans. 18FDG, a tracker of glucose uptake, accumulates in RV myocardium in humans with PAH [8]. In experimental RVH, FDG accumulation occurs primarily once there is severe RVH. However, FDG uptake occurs both in adaptive and maladaptive RVH models [9]. Consistent with this, there is upregulation of the Glut1 transporter in both monocrotaline and PAB-induced RVH [9]. The notion of a glycolytic shift in the RV is directly proven by increases in the rate of RV glycolysis in RVH, as measured in the RV working heart using the dual isotope metabolic technique (Fig. 3) [9]. Abnormal mitochondrial function in RVH is further evidenced by the observation that the mitochondrial membrane potential in RV myocytes is hyperpolarized [18].
Fig. 3.
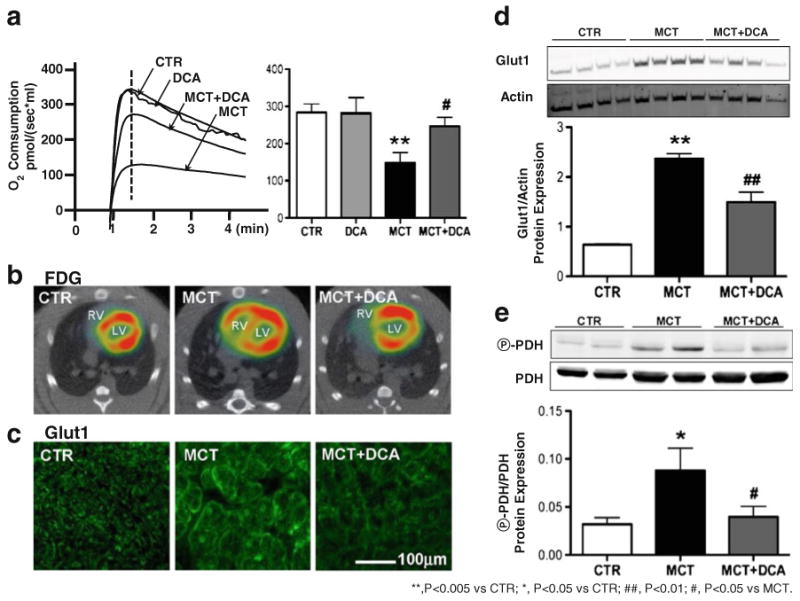
Increased glycolysis and decreased glucose oxidation in monocrotaline-induced RVH are reversed by the inhibition of pyruvate dehydrogenase kinase (PDK) by dichloroacetate (DCA; adapted from [9]). a RV O2 consumption measured by high-resolution respirometry is reduced in RVH and DCA increases it. b, c Glucose uptake measured by FDG tracking and glucose transport evaluated by Glut1 immunostaining are increased in RVH and both are reduced by DCA. d Increased protein expression of Glut1 in RVH is decreased by DCA. e The increased phosphorylation of pyruvate dehydrogenase by PDK in RVH is reduced by DCA treatment
While glucose oxidation is abnormal in RVH, changes in fatty acid metabolism are less clear. In RVH induced by chronic hypoxia, Sharma et al. examined the hypothesis that increased afterload would reactivate the fetal gene transcriptional profile. They observed substrate switching from fatty acids to glucose during early hypoxia; however the substrate preference reverted back to fatty acids by 2 weeks [19]. They observed dynamic changes of mRNA and protein for peroxisome proliferator-activated receptor (PPAR), a nuclear receptor that is considered to be a key regulator of substrate switching in the hypertrophied heart and medium-chain acyl-CoA dehydrogenase (MCAD), a PPAR-regulated gene. They interpreted this as a potential basis for the observed switching in substrate utilization but did not directly measure the rate of FAO [19]. In a small group of World Health Organization category I and III PH patients (versus normal controls), single photon emission computed tomography, using the fatty acid analog 123I β-iodophenyl pentadecanoic acid and 201Tl, demonstrated that RV FAO was impaired in severe (but not in mild) RVH [20]. FAO impairment was correlated with the reduced RV EF. Buermans et al. also reported the downregulation of carnitine palmitoytrasferase-1 β, a transporter subunit for long-chain fatty acid entering into mitochondria, in both adaptive and maladaptive RVH induced by low (20 mg/kg) and high (80 mg/kg) monocrotaline. This suggests that a reduction of FAO and switch to glucose metabolism occur with the development of RVH [21]. The above studies of FAO do not paint a consistent picture of the changes in FAO in RVH, indicating a need for further research, both in animal RVH models and in patients with RVH. As with the study of glucose oxidation, employing models of adaptive versus maladaptive RVH and being mindful of the temporality of metabolic changes will be important.
Energetic changes in RVH
Due to the mitochondrial metabolic switch from energy rich oxidative metabolism of glucose and FAO to glycolysis, the energy supply in RVH ultimately becomes unsupportable, leading to RVF. However, the combination of reduced O2 consumption and glycolysis appears adequate to compensate at least for a time. Evidence for this idea is supported by observations of high energy phosphates in ferret hearts, made 4–6 weeks after PAB. In this study in Langendorff hearts, Do et al. showed that the hypertrophied RV had lower levels of creatine phosphate (−46% versus control) but preserved ATP levels [22], likely related to the enhanced glycolysis (as evidenced by increased activities of hexokinase (+26%), aldolase (+212%), pyruvate kinase (+14%), and glucose 6-phosphate dehydrogenase (+107%)) [22]. Global ischemia was better tolerated (as measured by preservation of ATP concentration), in RVH versus control hearts, perhaps related to the lower reliance on oxidative metabolism. Nonetheless, the authors calculated that there is still a 53% decrease in energy reserve, suggesting an insufficient energy supply to fully compensate for the increased RV mass and afterload. Moreover, PAB is an adaptive (well-tolerated) form of RVH. In a maladaptive RVH model (rats studied 6 weeks after injection of monocrotaline), both ATP production and phosphocreatine levels decline [23]. Thus, in some models, the observed impairment of mitochondrial metabolism leads to insufficient ATP production, which together with acidosis caused by glycolysis, impairs RV contractility.
Electrical consequences of metabolic changes in RVH
The prolongation of action potential duration (APD) and QT interval in the LV can lead to ventricular tachycardia, cardiac arrest, or sudden death [24]. Several studies have shown that there is electrical remodeling in RVH; however, the consequences of this electrical change are unknown. In monocrotaline-induced RVH, prolongation of APD and the QT interval corrected for heart rate (QTc) have been noted [25]. Consistent with this, prolonged QTc intervals have been reported in the patients with PAH [26]. The down-regulation of voltage-gated K+ channels relevant to cardiac repolarization, including Kv1.2, Kv1.5, Kv2.1, Kv4.2, and Kv4.3, has been reported in RVH animal models [27, 28]. Our work shows that prolongation of APD and QTc only occurs once a critical level of RVH was achieved and is the consequence of reduced expression of specific repolarizing potassium channels, notably Kv1.2, Kv1.5, and Kv4.1 [9]. This study also provided evidence that the electrical changes were metabolically regulated. We showed that dichloroacetate could normalize both APD and QTc while partially restoring Kv channel expression (Fig. 4) [9]. Nonetheless, the relevance of APD prolongation to the RV function and arrhythmogenesis in RVH remains unclear.
Fig. 4.
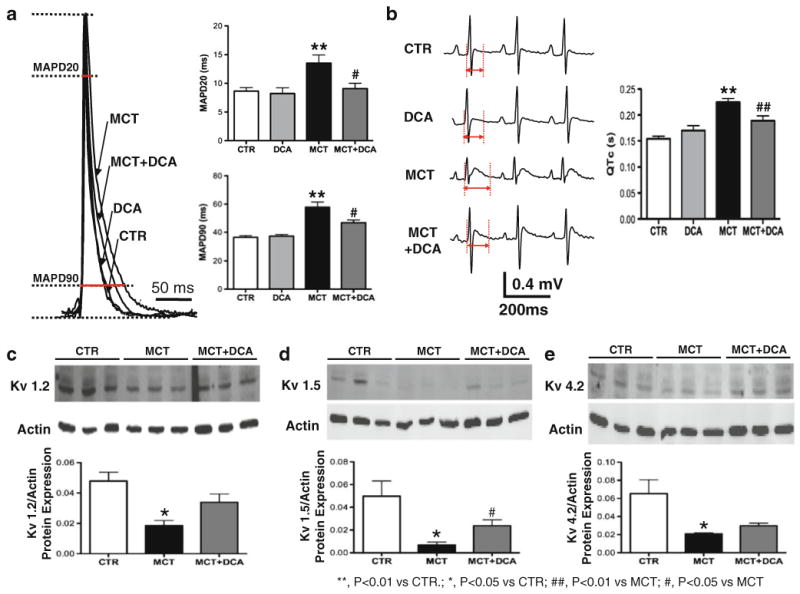
Electrical remodeling in monocrotaline-induced RVH is restored by dichloroacetate (DCA; adapted from [9]). a The representative traces of monophasic action potential recording show the prolongation of action potential durations which are calculated by measuring the durations at 20% and 90% repolarization (APD20 and APD90) in RVH. DCA shortens the prolongation. b The prolonged QT interval in RVH measured by surface EKG recording is shortened by DCA. c–e Western blot results show that the protein expressions of Kv1.2, Kv1.5, and Kv4.2 are downregulated in RVH and restored by DCA
Mechanisms of initiating and sustaining maladaptive mitochondrial metabolic adaptation in RVH
Capillary rarefaction and myocardial ischemia
The potential contribution of ischemia due to reduced coronary perfusion pressure to RV dysfunction has been demonstrated in acute PAB models (Fig. 1) [29]. Patients with PAH and RVF often present with evidence of myocardial ischemia. They experience angina-like chest pain, have elevated brain natriuretic peptide levels, and once they progress to having RV failure, frequently have small leaks of troponin, which indicate a poor prognosis [30]. The right coronary artery (RCA), which perfuses the RV, is particularly sensitive to high RVSP, since its flow occurs primarily in systole. Whether reduced RCA perfusion pressure (due to elevated RVSP) is a major cause of ischemia in PH is uncertain (although it likely contributes). RV ischemia can also be explained by RV capillary rarefaction. Decreased capillary density and vascular endothelial growth factor (VEGF) expression have been noted in two maladaptive RVH models (one induced by monocrotaline, the other by chronic hypoxia and the VEGF receptor inhibitor, SU4416) [31]. Bogaard et al. also found the activation of Akt1, a protein kinase that is involved in cellular survival pathways and hypoxia-induced factor-1α (HIF-1α) are associated with reduced VEGF expression and capillary density in maladaptive RVH [7]. We propose that reduced capillary density in RVH, perhaps exacerbated by reduced coronary perfusion pressure, fails to supply sufficient oxygen to support the increases in net RV oxygen demand in RVH in maladaptive RVH, stimulating the glycolytic shift.
Transcriptional upregulation of glycolysis and suppression of glucose oxidation (Figs. 1 and 2)
When supply of oxygen from the coronary artery fails to meet demand, myocytes adapt to low oxygen conditions by activating HIF-1α [32]. HIF-1α activation favors glycolysis by increasing transcription of Glut1, hexokinase, and lactate dehydrogenase kinase (as observed in RVH). This may account for the observed increase in glucose uptake and lactate production in RVH. At the same time, by activating transcription of PDKs genes, HIF-1α inhibits glucose oxidation [33]. Activation of HIF-1α has been reported in RVH induced by monocrotaline [34] and chronic hypoxia [7]. In chronic hypoxic RVH activation of HIF occurs in parallel with increased VEGF expression [7]. In our unpublished work in maladaptive RVH, there is a suppression of VEGF, consistent with the notion of capillary rarefaction. In monocrotaline-induced RVH [34], the activation of HIF-1α is highly correlated with complex II-associated production of reactive oxygen species in RVH, suggesting mitochondrial dysfunction may be caused by the activation of HIF-1α [34].
Another transcriptional mechanism for the metabolic switch in RVH, and one that can act cooperatively with HIF-1α, is the activation of the oncogene Myc. Myc activation has been detected 2 h after PA banding [35]. Although there is no direct evidence of the involvement of Myc in the metabolic switch in RVH, it has been reported that Myc mediates the hypertrophic growth in cardiac myocytes [36]. The role of Myc and HIF-1α as transcriptional mediators of the glycolytic shift in RVH merits further study.
By the administration of different doses of monocrotaline, Buermans et al. demonstrated maladaptive RVH model is accompanied by the activation of p38-MAP kinase (MAPK), a stress-responsive protein kinase. Conversely, in adaptive RVH (rats exposed to low-dose monocrotaline which caused somewhat milder hypertrophy and no mortality within 12 weeks), there was no p38-MAPK activation [21]. p38-AMPK activation elevates glycolysis and limits cardioprotection in ischemic rat hearts and in acute studies, the p38-MAPK inhibitor, SB-202190, decreases glycolysis, and restores cardiac function [37]. The function of p38-MAPK on mitochondrial metabolism in RVH needs to be further addressed.
Targeting the mitochondria to reverse RVH and improve RV function
The recognition that the mitochondrial metabolic shift in RVH is reversible has therapeutic consequences [9]. Our prior work suggests that enhancing glucose oxidation improves RV function in experimental RVF. Here we propose two mitochondrial targeted therapies for RVH and RV failure: PDKs inhibition and partial inhibition of FAO.
Dichloroacetate-PDKs inhibition
Activated PDKs phosphorylate and inhibit PDH. This inhibits formation of acetyl-CoA and therefore slows Krebs' cycle, accounting for the reduced O2 consumption/g tissue in RVH and reduced energy production (Fig. 2) [38]. PDH phosphorylation in MCT RVH, and its consequences, can be reversed with dichloroacetate. Both in monocrotaline and PAB-induced RVH, dichloroacetate reduces PDH phosphorylation and improves glucose oxidation [9]. Moreover, dichloroacetate reduces the upregulated expression of Glut1, partially restores Kv1.5 channel expression, and shortens the prolonged APD and QTc (Figs. 3 and 4). As a consequence of these effects, dichloroacetate increases cardiac output and improves cardiac function in RVH [9]. Intriguingly, as described in this issue by Michelakis et al., these beneficial effects also occur in the pulmonary vasculature, where PDKs inhibition is well established to reduce PVR by inhibiting proliferation of pulmonary artery smooth muscle cells and by promoting apoptosis [39]. Thus PDKs inhibition is a strategy that should be well suited for a clinical trial in patients with PH and RVF.
Trimetazidine and ranolazine—partial inhibition of fatty acid oxidation
One may be able to exploit the Randle cycle to enhance glucose oxidation. Although no study on the beneficial effects of trimetazidine and ranolazine has been published in RVH, both inhibitors improve cardiac function in LVH, ischemic heart, and heart failure. Moreover, preliminary data indicate that trimetazidine may be beneficial in PAB-induced RVH. Trimetazidine, a long-chain 3-ketoacyl coenzyme A thiolase, is used in Europe to treat refractory ischemia (Fig. 2) [40–43]. In 19 nondiabetic patients with idiopathic dilated cardiomyopathy, randomized to single-blind trimetazidine (n=12) or placebo (n=7) for 3 months, PET studies (using palmitate and acetate tracers) showed that trimetazidine caused a modest (∼10%) decrease in FAO without altering myocardial oxidative rate, implying a compensatory increase in the oxidation of glucose [44].
Another partial inhibition of fatty acid oxidation, ranolazine, is approved for refractory ischemia in America [45–47] (although there is some controversy whether it works through inhibition of FAO and activation of PDH [48–50]). Ranolazine inhibits FAO leading to decreased acetyl-CoA and activation of PDH [48]. McCormack et al. has confirmed the activation of PDH and the stimulation of glucose oxidation by ranolazine in ischemia and/or reperfusion models [49]. It has been reported that in canine hearts with experimentally induced heart failure, ranolazine improves abnormal depolarization and contractility [51]. In rats with chronic heart failure induced by myocardial infarction, ranolazine did not improve exercise capacity, although it reduced lactate production [52].
Preclinical study of trimetazidine and ranolazine as a means of promoting glucose oxidation and improving RV function in experimental RVH are underway in our laboratory. Clinical trials of PDKs and FAO inhibitors in PH and RVH are desirable in light of the current limitations in the therapy for RVF.
There are other ways of intervening on mitochondria to improve RV function, in addition to metabolic modulation. For example, EUK-1340, a superoxide dismutase and catalase mimetic, has shown beneficial effect in decreasing RV oxidative stress and preventing the development of cardio-myocyte hypertrophy and fibrosis by scavenging ROS [53].
Summary
In RVH, there is a mitochondrial metabolic switch from glucose oxidation to glycolysis due to myocardial ischemia. Transcriptionally mediated increases in glycolysis may temporarily preserve the energetic balance, but ultimately this becomes maladaptive. The mitochondrial dysfunction in RVH is reversible and both PDKs and FAO inhibitors may offer selective strategies for improving RV function.
Acknowledgments
This work is supported by NIH-RO1-HL071115 and 1RC1HL099462-01, the American Heart Association (AHA), and the Roche Foundation for Anemia Research.
Footnotes
Disclosure of potential conflict of interests The authors declare no conflict of interests related to this study.
Contributor Information
Lin Piao, Section of Cardiology, Department of Medicine, University of Chicago, Chicago, IL, USA.
Glenn Marsboom, Section of Cardiology, Department of Medicine, University of Chicago, Chicago, IL, USA.
Stephen L. Archer, Section of Cardiology, Department of Medicine, University of Chicago, Chicago, IL, USA, Harold Hines Jr. Professor of Medicine, University of Chicago, 5841 South Maryland Avenue (MC6080), Chicago, IL 60637, USA, sarcher@medicine.bsd.uchicago.edu
References
- 1.Sztrymf B, Souza R, Bertoletti L, Jais X, Sitbon O, Price LC, Simonneau G, Humbert M. Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur Respir J. 2010;35:1286–1293. doi: 10.1183/09031936.00070209. [DOI] [PubMed] [Google Scholar]
- 2.Gavazzi A, Berzuini C, Campana C, Inserra C, Ponzetta M, Sebastiani R, Ghio S, Recusani F. Value of right ventricular ejection fraction in predicting short-term prognosis of patients with severe chronic heart failure. J Heart Lung Transplant. 1997;16:774–785. [PubMed] [Google Scholar]
- 3.van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG, Spreeuwenberg MD, Postmus PE, Vonk-Noordegraaf A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28:1250–1257. doi: 10.1093/eurheartj/ehl477. [DOI] [PubMed] [Google Scholar]
- 4.Gomez A, Bialostozky D, Zajarias A, Santos E, Palomar A, Martinez ML, Sandoval J. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol. 2001;38:1137–1142. doi: 10.1016/s0735-1097(01)01496-6. [DOI] [PubMed] [Google Scholar]
- 5.van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, Henkens IR, Gan CT, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J. 2008;29:120–127. doi: 10.1093/eurheartj/ehm567. [DOI] [PubMed] [Google Scholar]
- 6.Bian X, Williams AG, Jr, Gwirtz PA, Downey HF. Right coronary autoregulation in conscious, chronically instrumented dogs. Am J Physiol. 1998;275:H169–H175. doi: 10.1152/ajpheart.1998.275.1.H169. [DOI] [PubMed] [Google Scholar]
- 7.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 8.Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, Takahashi T, Nawata J, Ido T, Watanabe J, Shirato K. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45:1849–1855. doi: 10.1016/j.jacc.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 9.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD, Archer SL. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med. 2010;88:47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handa N, Magata Y, Mukai T, Nishina T, Konishi J, Komeda M. Quantitative FDG-uptake by positron emission tomography in progressive hypertrophy of rat hearts in vivo. Ann Nucl Med. 2007;21:569–576. doi: 10.1007/s12149-007-0067-2. [DOI] [PubMed] [Google Scholar]
- 11.Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007;12:331–343. doi: 10.1007/s10741-007-9034-1. [DOI] [PubMed] [Google Scholar]
- 12.Neely JR, Morgan HE. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu Rev Physiol. 1974;36:413–459. doi: 10.1146/annurev.ph.36.030174.002213. [DOI] [PubMed] [Google Scholar]
- 13.Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. 1997;33:243–257. doi: 10.1016/s0008-6363(96)00245-3. [DOI] [PubMed] [Google Scholar]
- 14.Chandler MP, Chavez PN, McElfresh TA, Huang H, Harmon CS, Stanley WC. Partial inhibition of fatty acid oxidation increases regional contractile power and efficiency during demand-induced ischemia. Cardiovasc Res. 2003;59:143–151. doi: 10.1016/s0008-6363(03)00327-4. [DOI] [PubMed] [Google Scholar]
- 15.Abozguia K, Clarke K, Lee L, Frenneaux M. Modification of myocardial substrate use as a therapy for heart failure. Nat Clin Pract Cardiovasc Med. 2006;3:490–498. doi: 10.1038/ncpcardio0583. [DOI] [PubMed] [Google Scholar]
- 16.Saito D, Tani H, Kusachi S, Uchida S, Ohbayashi N, Marutani M, Maekawa K, Tsuji T, Haraoka S. Oxygen metabolism of the hypertrophic right ventricle in open chest dogs. Cardiovasc Res. 1991;25:731–739. doi: 10.1093/cvr/25.9.731. [DOI] [PubMed] [Google Scholar]
- 17.Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol. 1970;218:153–159. doi: 10.1152/ajplegacy.1970.218.1.153. [DOI] [PubMed] [Google Scholar]
- 18.Nagendran J, Gurtu V, Fu DZ, Dyck JR, Haromy A, Ross DB, Rebeyka IM, Michelakis ED. A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J Thorac Cardiovasc Surg. 2008;136:168–178. 178 e161–163. doi: 10.1016/j.jtcvs.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 19.Sharma S, Taegtmeyer H, Adrogue J, Razeghi P, Sen S, Ngumbela K, Essop MF. Dynamic changes of gene expression in hypoxia-induced right ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2004;286:H1185–H1192. doi: 10.1152/ajpheart.00916.2003. [DOI] [PubMed] [Google Scholar]
- 20.Kim Y, Goto H, Kobayashi K, Sawada Y, Miyake Y, Fujiwara G, Chiba H, Okada T, Nishimura T. Detection of impaired fatty acid metabolism in right ventricular hypertrophy: assessment by I-123 beta-methyl iodophenyl pentadecanoic acid (BMIPP) myocardial single-photon emission computed tomography. Ann Nucl Med. 1997;11:207–212. doi: 10.1007/BF03164765. [DOI] [PubMed] [Google Scholar]
- 21.Buermans HP, Redout EM, Schiel AE, Musters RJ, Zuidwijk M, Eijk PP, van Hardeveld C, Kasanmoentalib S, Visser FC, Ylstra B, Simonides WS. Microarray analysis reveals pivotal divergent mRNA expression profiles early in the development of either compensated ventricular hypertrophy or heart failure. Physiol Genomics. 2005;21:314–323. doi: 10.1152/physiolgenomics.00185.2004. [DOI] [PubMed] [Google Scholar]
- 22.Do E, Baudet S, Verdys M, Touzeau C, Bailly F, Lucas-Heron B, Sagniez M, Rossi A, Noireaud J. Energy metabolism in normal and hypertrophied right ventricle of the ferret heart. J Mol Cell Cardiol. 1997;29:1903–1913. doi: 10.1006/jmcc.1997.0429. [DOI] [PubMed] [Google Scholar]
- 23.Daicho T, Yagi T, Abe Y, Ohara M, Marunouchi T, Takeo S, Tanonaka K. Possible involvement of mitochondrial energy-producing ability in the development of right ventricular failure in monocrotaline-induced pulmonary hypertensive rats. J Pharmacol Sci. 2009;111:33–43. doi: 10.1254/jphs.08322fp. [DOI] [PubMed] [Google Scholar]
- 24.Antzelevitch C. Ionic, molecular, and cellular bases of QT-interval prolongation and torsade de pointes. Europace. 2007;9(Suppl 4):iv4–iv15. doi: 10.1093/europace/eum166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JK, Kodama I, Honjo H, Anno T, Kamiya K, Toyama J. Stage-dependent changes in membrane currents in rats with monocrotaline-induced right ventricular hypertrophy. Am J Physiol. 1997;272:H2833–H2842. doi: 10.1152/ajpheart.1997.272.6.H2833. [DOI] [PubMed] [Google Scholar]
- 26.Hlaing T, Guo D, Zhao X, DiMino T, Greenspon L, Kowey PR, Yan GX. The QT and Tp-e intervals in left and right chest leads: comparison between patients with systemic and pulmonary hypertension. J Electrocardiol. 2005;38:154–158. doi: 10.1016/j.jelectrocard.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 27.Lee JK, Nishiyama A, Kambe F, Seo H, Takeuchi S, Kamiya K, Kodama I, Toyama J. Downregulation of voltage-gated K (+) channels in rat heart with right ventricular hypertrophy. Am J Physiol. 1999;277:H1725–H1731. doi: 10.1152/ajpheart.1999.277.5.H1725. [DOI] [PubMed] [Google Scholar]
- 28.Zhang TT, Cui B, Dai DZ. Downregulation of Kv4.2 and Kv4.3 channel gene expression in right ventricular hypertrophy induced by monocrotaline in rat. Acta Pharmacol Sin. 2004;25:226–230. [PubMed] [Google Scholar]
- 29.Vlahakes GJ, Turley K, Hoffman JI. The pathophysiology of failure in acute right ventricular hypertension: hemodynamic and biochemical correlations. Circulation. 1981;63:87–95. doi: 10.1161/01.cir.63.1.87. [DOI] [PubMed] [Google Scholar]
- 30.Kurzyna M, Zylkowska J, Fijalkowska A, Florczyk M, Wieteska M, Kacprzak A, Burakowski J, Szturmowicz M, Wawrzynska L, Torbicki A. Characteristics and prognosis of patients with decompensated right ventricular failure during the course of pulmonary hypertension. Kardiol Pol. 2008;66:1033–1039. discussion 1040–1031. [PubMed] [Google Scholar]
- 31.Partovian C, Adnot S, Eddahibi S, Teiger E, Levame M, Dreyfus P, Raffestin B, Frelin C. Heart and lung VEGF mRNA expression in rats with monocrotaline- or hypoxia-induced pulmonary hypertension. Am J Physiol. 1998;275:H1948–H1956. doi: 10.1152/ajpheart.1998.275.6.H1948. [DOI] [PubMed] [Google Scholar]
- 32.Graham RM, Frazier DP, Thompson JW, Haliko S, Li H, Wasserlauf BJ, Spiga MG, Bishopric NH, Webster KA. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J Exp Biol. 2004;207:3189–3200. doi: 10.1242/jeb.01109. [DOI] [PubMed] [Google Scholar]
- 33.Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 34.Redout EM, Wagner MJ, Zuidwijk MJ, Boer C, Musters RJ, van Hardeveld C, Paulus WJ, Simonides WS. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc Res. 2007;75:770–781. doi: 10.1016/j.cardiores.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 35.Pollack PS, Houser SR, Budjak R, Goldman B. c-myc gene expression is localized to the myocyte following hemodynamic overload in vivo. J Cell Biochem. 1994;54:78–84. doi: 10.1002/jcb.240540109. [DOI] [PubMed] [Google Scholar]
- 36.Zhong W, Mao S, Tobis S, Angelis E, Jordan MC, Roos KP, Fishbein MC, de Alboran IM, MacLellan WR. Hypertrophic growth in cardiac myocytes is mediated by Myc through a cyclin D2-dependent pathway. EMBO J. 2006;25:3869–3879. doi: 10.1038/sj.emboj.7601252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaswal JS, Gandhi M, Finegan BA, Dyck JR, Clanachan AS. Inhibition of p38 MAPK and AMPK restores adenosine-induced cardioprotection in hearts stressed by antecedent ischemia by altering glucose utilization. Am J Physiol Heart Circ Physiol. 2007;293:H1107–H1114. doi: 10.1152/ajpheart.00455.2007. [DOI] [PubMed] [Google Scholar]
- 38.Sugden MC, Langdown ML, Harris RA, Holness MJ. Expression and regulation of pyruvate dehydrogenase kinase isoforms in the developing rat heart and in adulthood: role of thyroid hormone status and lipid supply. Biochem J. 2000;352(Pt 3):731–738. [PMC free article] [PubMed] [Google Scholar]
- 39.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–250. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- 40.Grabczewska Z, Bialoszynski T, Szymanski P, Sukiennik A, Swiatkiewicz I, Kozinski M, Kochman W, Grzesk G, Kubica J. The effect of trimetazidine added to maximal anti-ischemic therapy in patients with advanced coronary artery disease. Cardiol J. 2008;15:344–350. [PubMed] [Google Scholar]
- 41.Gunes Y, Guntekin U, Tuncer M, Sahin M. Improved left and right ventricular functions with trimetazidine in patients with heart failure: a tissue Doppler study. Heart Vessels. 2009;24:277–282. doi: 10.1007/s00380-008-1118-x. [DOI] [PubMed] [Google Scholar]
- 42.Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86:580–588. doi: 10.1161/01.res.86.5.580. [DOI] [PubMed] [Google Scholar]
- 43.Rosano GM, Vitale C, Sposato B, Mercuro G, Fini M. Trimetazidine improves left ventricular function in diabetic patients with coronary artery disease: a double-blind placebo-controlled study. Cardiovasc Diabetol. 2003;2:16. doi: 10.1186/1475-2840-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tuunanen H, Engblom E, Naum A, Nagren K, Scheinin M, Hesse B, Juhani Airaksinen KE, Nuutila P, Iozzo P, Ukkonen H, Opie LH, Knuuti J. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118:1250–1258. doi: 10.1161/CIRCULATIONAHA.108.778019. [DOI] [PubMed] [Google Scholar]
- 45.Fragasso G, Spoladore R, Cuko A, Palloshi A. Modulation of fatty acids oxidation in heart failure by selective pharmacological inhibition of 3-ketoacyl coenzyme-A thiolase. Curr Clin Pharmacol. 2007;2:190–196. doi: 10.2174/157488407781668776. [DOI] [PubMed] [Google Scholar]
- 46.Stanley WC. Partial fatty acid oxidation inhibitors for stable angina. Expert Opin Investig Drugs. 2002;11:615–629. doi: 10.1517/13543784.11.5.615. [DOI] [PubMed] [Google Scholar]
- 47.Wang P, Fraser H, Lloyd SG, McVeigh JJ, Belardinelli L, Chatham JC. A comparison between ranolazine and CVT-4325, a novel inhibitor of fatty acid oxidation, on cardiac metabolism and left ventricular function in rat isolated perfused heart during ischemia and reperfusion. J Pharmacol Exp Ther. 2007;321:213–220. doi: 10.1124/jpet.106.115519. [DOI] [PubMed] [Google Scholar]
- 48.Clarke B, Wyatt KM, McCormack JG. Ranolazine increases active pyruvate dehydrogenase in perfused normoxic rat hearts: evidence for an indirect mechanism. J Mol Cell Cardiol. 1996;28:341–350. doi: 10.1006/jmcc.1996.0032. [DOI] [PubMed] [Google Scholar]
- 49.McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation. 1996;93:135–142. doi: 10.1161/01.cir.93.1.135. [DOI] [PubMed] [Google Scholar]
- 50.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aaker A, McCormack JG, Hirai T, Musch TI. Effects of ranolazine on the exercise capacity of rats with chronic heart failure induced by myocardial infarction. J Cardiovasc Pharmacol. 1996;28:353–362. doi: 10.1097/00005344-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Redout EM, van der Toorn A, Zuidwijk MJ, van de Kolk CW, van Echteld CJ, Musters RJ, van Hardeveld C, Paulus WJ, Simonides WS. Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H1038–H1047. doi: 10.1152/ajpheart.00097.2009. [DOI] [PubMed] [Google Scholar]