

Abstract
Any protein synthesized in the secretory pathway has the potential to misfold and would need to be recognized and ubiquitylated for degradation. This is astounding since only a few ERAD-specific E3 ligases have been identified. To begin to understand substrate recognition, we wished to map the ubiquitylation sites on the NS-1 non-secreted immunoglobulin light chain, which is an ERAD substrate. Ubiquitin is usually attached to lysine residues and less frequently to the N-terminus of proteins. In addition, several viral E3s have been identified that attach ubiquitin to cysteine or serine/threonine residues. Mutation of lysines, serines, and threonines in the NS-1 variable region was necessary to significantly reduce ubiquitylation and stabilize the protein. The Hrd1 E3 ligase was required to modify all three amino acids. Our studies argue that ubiquitylation of ER proteins relies on very different mechanisms of recognition and modification than those used to regulate biological processes.
INTRODUCTION
The folding and assembly of nascent proteins in the mammalian ER is carefully monitored by a process referred to as “ER quality control” (Ellgaard and Helenius, 2003). Proteins that pass this inspection can exit the ER for residence in other organelles of the secretory pathway, secretion, or expression at the cell surface. However, proteins that fail to mature properly in the ER are identified, retrotranslocated to the cytosol, and targeted for degradation by the 26S proteasome via an incompletely understood process termed ER-associated degradation (ERAD) (Werner et al., 1996; Vembar and Brodsky, 2008). Like the degradation of cytosolic proteins by the 26S proteasome, this pathway is dependent on ubiquitylation of the unfolded substrates. The ERAD pathway was first described in yeast (Werner et al., 1996) and is conserved in mammalian cells (Wiertz et al., 1996; Plemper et al., 1997). The role of ER chaperones (Brodsky et al., 1999; Taxis et al., 2003) and ubiquitylation (Hiller et al., 1996; Meusser et al., 2005) in disposing of the misfolded proteins is well documented. Whereas, the proteins that play a role in the extraction and ubiquitylation of the ERAD substrates have more recently been identified.
A number of proteins have been identified that assist in the dislocation and degradation of misfolded ER proteins. In S. cerevisiae, these include the multi-pass transmembrane protein Der1p (Knop et al., 1996), which may form part of the channel. Two cytosolically-oriented, integral membrane E3 ubiquitin ligases, Hrd1p (Bays et al., 2001) and Doa10p (Swanson et al., 2001), are essential for ubiquitylation of many different ERAD substrates. Lumenally oriented Hrd3, forms a complex with Hrd1 (Wilhovsky et al., 2000), and Usa1 links the Hrd1p/Hrd3p complex to Der1p (Carvalho et al., 2006). Finally, the Cdc48p AAA ATPase complex supplies the energy to extract the substrates from the ER (Rabinovich et al., 2002). Different compliments of these proteins are required depending on the substrate.
Mammalian equivalents of these yeast proteins have been identified, including three Der1p homologues, Derlin-1-3 (Ye et al., 2004; Lilley and Ploegh, 2004; Oda et al., 2006). Sel1L is an orthologue of Hrd3p (Mueller et al., 2006) and plays a role in targeting glycoproteins that are associated with EDEM to the retrotranslocon (Cormier et al., 2009). Herp is an orthologue of Usa1 and contains a ubiquitin-like domain (Kokame et al., 2000), which might account for its ability to bind both to the 26S proteasome and ubiquitylated substrates (Okuda-Shimizu et al., 2007). Herp also associates with the E3 ligase Hrd1 and the p97 AAA ATPase (Schulze et al., 2005). At least two E3 mammalian ubiquitin ligases have been identified that show broad substrate specificity; gp78 (Fang et al., 2001) and Hrd1 (Kikkert et al., 2004), with several additional E3 ligases, Rma1 (Matsuda et al., 2001; Wang et al., 2008; Delaunay et al., 2008), TEB4 (Hassink et al., 2005 ; Zavacki et al., 2009), and parkin (Hassink et al., 2005; Wang et al., 2008; Kitada et al., 1998), showing much more limited substrate specificity.
A particularly perplexing, unresolved issue is how so few E3 ligases can potentially be responsible for the disposal of any protein synthesized in the ER that fails to mature properly. To begin to understand how ERAD substrates might be recognized, we wished to identify the site(s) of ubiquitylation on a soluble ERAD substrate. For this study, we chose the non-secreted NS-1 immunoglobulin κ LC (Skowronek et al., 1998; Okuda-Shimizu et al., 2007). Ubiquitylation of proteins occurs in a multi-step transfer between a series of ubiquitin ligases (Pickart and Eddins, 2004; Scheffner et al., 1995). The first step is the covalent attachment of the C-terminal glycine of ubiquitin to a cysteine on the E1 enzyme via a thioester bond. The next step is the transfer of ubiquitin from the E1 to a cysteine residue on one of many different E2s. The association of an E2 with an E3 provides the specificity of the reaction, as there are even more E3s, which interact specifically with a very limited group or in some cases even a single substrate (Pickart, 2001). The E2/E3 pair most commonly transfers ubiquitin to the ε-amino group of a lysine residue on the substrate via an isopeptide bond (Pickart, 2001). However, there are a number of instances where ubiquitin is placed on the N-terminal amino group of the substrate protein (Ciechanover and Ben-Saadon, 2004). Notably, several viral E3s induce ubiquitylation of non-amino groups including cysteine (Cadwell and Coscoy, 2005) or serine/threonines (Wang et al., 2007) on their target proteins. Once the ubiquitin chain is elongated to about 4 or more molecules, the substrate can be recognized by the proteasome and degraded.
To begin to understand how so many ERAD substrates are recognized by so few E3 ligases, we set out to map the site(s) of ubiquitylation on the NS-1 LC. We found that it was apparently ubiquitylated predominantly on serine/threonine residues, but if these amino acids were mutated it was then modified on lysines. Mutation of all three types of amino acids was required to significantly reduce ubiquitylation and stabilize the NS-1 LC. Modification of all three amino acids was dependent on the Hrd1 E3 ligase.
RESULTS
NS-1 ubiquitylation and turnover is not dependent on lysine residues
The non-secreted NS-1 κ LC was previously shown to be an ERAD substrate that is degraded by the 26S proteasome (Knittler and Haas, 1992; Skowronek et al., 1998). Our previous data demonstrated that this LC is ubiquitylated, which occurs on a folding intermediate in which the VL domain is reduced but the CL domain remains oxidized (Okuda-Shimizu et al., 2007). We first confirmed the ubiquitylation of this substrate by treating the LC with SDS. The LC was still ubiquitylated, demonstrating that the attachment was covalent (Figure S1A). We also used a rabbit antiserum to detect ubiquitin (Figure S1B), so that the secondary antibody would not interact directly with the NS-1 LC. Again, ubiquitin was detected on the immunoprecipitated LC, ruling out the possibility that the signal at the top of the blot represented aggregated LC instead of ubiquitylated LC. Finally, we used two linkage-specific antiserum to probe the LC and found that the anti-K48 linkage antibody reacted with precipitated material but not the anti-K63 linkage antibody (Figure S1C). These three pieces of data, coupled with our previous 2D data showing that the ubiqutin signal emanates only from the partially oxidized (ox1) form of LC (Okuda-Shimizu et al., 2007), assured us that the NS-1 LC was indeed ubiquitylated. Thus, we began our studies by making a number of mutants in which the 6 lysine residues present in the VL domain were changed to arginine either singly or in various combinations, including one in which all 6 lysines were mutated (Figure 1 & S2A). Mutant proteins lacking any (data not shown) or all of the lysines in the VL domain were still ubiquitylated (Figure 1) and were degraded by the 26S proteasome with kinetics similar to that of the wild-type protein; demonstrating that ubiquitylation of the lysines in the VL domain was not required for the degradation of this ERAD substrate. Although the CL domain remained oxidized and was unlikely to be ubiquitylated, we proceeded to mutate the seven lysines in this domain alone (K7-13R) or together with the lysines in the VL domain (K1-13R). Examination of these mutants revealed that even the lysine-less LC was still ubiquitylated and readily degraded by the proteasome (Figure 1). The K1-13R mutant was consistently degraded somewhat faster than the wild-type protein, which might reflect additional destabilization of the LC by these mutations. Thus, either lysine ubiquitylation does not occur on this ERAD substrate or it makes only a minor contribution to the ubiquitin signal and does not affect its degradation.
Figure 1. NS-1 κ LC is ubiquitylated at non-lysine residues.

(A) The indicated LC constructs were expressed in COS-1 cells. Lysates were prepared from cells treated with or without lactacystin and isolated with anti-κ LC antiserum. Samples were subjected to immunoblot analyses with a mouse monoclonal anti-Ub. (B) WT or a lysine-less mutant of NS-1 κ LC (K1-13R) was expressed in 293T cells. Cells were preincubated with or without lactacystin, pulse-labeled for 15 min, and chased for the indicated times. Immunoprecipitated samples were analyzed by SDS-PAGE under reducing conditions and visualized by autoradiography. The percent of signal remaining as detected by phosphoimager are indicated under the lanes and reflect the average of three experiments.
The N-terminus does not represent a major site of ubiquitylation
As a number of cellular proteins are ubiquitylated at the N-terminus, we examined this possibility using two approaches. First, we engineered a Factor Xa cleavage site four residues after the N-terminus of the mature sequence of the NS-1 LC (Xa4) (Figure S2B). Ubiquitylated wild-type and Xa4 LC were isolated and incubated with or without Factor Xa (Figure 2A). Factor Xa recognizes the sequence Ile-Glu-Gly-Arg and cleaves immediately after the arginine (Nagai and Thogersen, 1987). As expected, Factor Xa did not remove ubiquitin from wild-type LC that does not have a Xa cleavage site. When the Xa4 LC was similarly treated, there was no significant decrease in the ubiquitin signal, arguing that the N-terminus either was not ubiquitylated or did not contribute significantly to the signal. As an alternative method to determine if N-terminal ubiquitylation contributed to the degradation of the NS-1 LC, we mutated the first two amino acids of the mature N-terminus from Asn-Ile to either Ser-Asp or Ala-Asp. Cytosolic proteins possessing these residues are resistant to N-terminal ubiquitylation due to acetylation of these amino acids (Ciechanover and Ben-Saadon, 2004; Polevoda and Sherman, 2003), although it is important to note that this has not been tested for a protein synthesized in the ER. In both cases, the protein was still ubiquitylated (Figure 2B) and turned over with kinetics very similar to that observed for the wild-type protein (Figure 2C). Together these two approaches argue that the NS-1 LC is not ubiquitylated at the N-terminus either.
Figure 2. NS-1 κ LC is not ubiquitylated at the N-terminus.

(A) COS-1 cells expressing either WT NS-1 LC or the Xa4 mutant were treated with MG132 for 5 hr and isolated LC were cleaved with Factor Xa followed by a second immunoprecipitation with an anti-κ LC antibody that recognizes the CL domain. Ubiquitylation of the LC were analyzed by immunoblotting. The uncleaved form is indicated with an asterisk and the cleaved form with two asterisks. (B) The indicated untagged LC constructs were expressed in COS-1 cells and lysates were prepared from cells treated with or without lactacystin. LC were isolated and subjected to immunoblot analyses with the mouse anti-Ub antibody. (C) 293T cells expressing the indicated mutants were pulse labeled and chased for the indicated times. Isolated LC were separated by reducing SDS-PAGE, and detected by autoradiography. Percent LC remaining at each time point is indicated under each lane.
NS-1 ubiquitylation is not occurring on cysteine residues
Previous reports demonstrated that the MIR1 E3 ligase of the Kaposi’s sarcoma-associated herpes virus ubiquitylates the cytosolic tail of the MHC I heavy chain on a single cysteine residue, which is sufficient to target this protein to the proteasome for degradation (Cadwell and Coscoy, 2005). While this activity was performed by a viral E3 not a cellular E3, it does demonstrate that this linkage can occur and be recognized on an ERAD substrate, in addition to the well characterized ubiquitylation of E1s and E2s on cysteine residues (Pickart and Eddins, 2004). There are two cysteines available in the reduced VL domain of the ox1 form of LC that would be potential sites for ubiquitylation. These were mutated either alone (C23,88A) or in combination with the six lysines in the VL domain (K1-6R, C23,88A) (Figure S2C). Because the thioester bond can be broken with the β-mercaptoethanol present in reducing buffers, we analyzed the mutants under both reducing and non-reducing conditions. We found that the wild-type and mutant LC were similarly ubiquitylated under both conditions (Figures 3A and B). The cysteine mutants were degraded even more rapidly than the wild-type protein (Figure 3C), which may reflect the fact that these mutants are only present in the partially oxidized form that is the substrate for degradation (Okuda-Shimizu et al., 2007), as opposed to the wild-type NS-1 LC in which the fully oxidized (ox2) form must be converted to the partially oxidized (ox1) form for degradation.
Figure 3. Mutation of cysteines and lysines in the V-region of NS-1 κ LC does not affect ubiquitylation or inhibit degradation.

(A) The indicated untagged LC constructs were expressed in COS cells. Lysates were prepared from cells treated with or without MG132, and isolated LC were subjected to reducing (A) or non-reducing (B) SDS PAGE followed by immunoblot analyses with an anti-Ub antibody. (C) The indicated LC constructs were expressed in 293T cells. Cells were pulse labeled for 15 min and chased for the indicated times. Immunoprecipitated samples from cell extracts were subjected to SDS-PAGE under reducing conditions, and the signal was detected by autoradiography. Percent LC signal remaining is indicated under the lanes.
NS-1 ubiquitylation is largely restricted to the unfolded VL domain
Given the unexpected findings obtained thus far, we wished to verify that ubiquitylation was restricted to the reduced VL domain before proceeding. To do so, we inserted a Factor Xa site at position 50 in the mature sequence (Xa50) of the VL domain and a second one just at the end of this domain at position 109 (Xa109) (Figure S2D). LC were isolated from untreated and MG132 treated cells, and ubiquitylation was monitored before and after incubating with Factor Xa (Figure 4A). In the case of the Xa50 mutant, Factor Xa treatment produced a band at ~18 kDa. This was the expected size of the remaining LC, which could be recognized by the anti-κ antibody that is specific for the CL domain (Figure 4B). We found that removing the first 50 amino acids reduced the amount of ubiquitylation (Figure 4A) associated with the LC, arguing that at least one modified residue exists between amino acids 4 and 50. Cleavage of the Xa109 mutant produced an even smaller LC fragment of the expected size (Figure 4B) and resulted in almost complete removal of the ubiquitin signal from the remaining LC fragment (Figure 4A). In fact the signal is nearly the same as that obtained with lysing buffer and immunoprecipitating antibody alone (Figure 4A, buffer). These data demonstrate that the NS-1 LC is ubiquitylated on at least two residues in the VL domain and that this domain possesses the majority, if not all, of the ubiquitylation sites.
Figure 4. NS-1 κ LC is ubiquitylated at multiple sites in the VL region.

COS-1 cells expressing the Xa50 and Xa109 mutants were incubated with or without MG132 prior to lysing, and LC were isolated and analyzed as in Figure 2. Membranes were blotted for Ub (A) or κ LC (B). The LC fragments remaining after cleavage from the Xa50 (single asterisk) and Xa109 (double asterisks) mutants are indicated.
NS-1 ubiquitylation is sensitive to high pH
The mK3 E3 from the γ-herpes virus modifies serines and threonines on the cytosolic tail of the MHC I heavy chain and targets it for proteasomal degradation (Wang et al., 2007). Again, these data at least provide proof of principal for this type of modification on an ERAD substrate. However, unlike the cytosolic tail of the MHC I heavy chain, which contained only 1 threonine and 4 serines, the VL domain of the NS-1 LC has a total of 24 serine/threonine residues, making it an unwieldy target for PCR-induced mutagenesis. Therefore, because the ester bond found in this type of linkage is sensitive to high pH (Wang et al., 2007), we began by isolating the ubiquitylated LC and incubating it with and without NaOH (Figure 5A). This treatment readily released ubiquitin from the NS-1 LC but only slightly reduced the background signal associated with the vector control (Figure 5A, lanes 2 and 3). Free ubiquitin cannot be detected under these conditions or with this antibody. Thus, we repeated the experiment on NS-1 LC isolated from the P3U.1 cells, but in this case the membrane was autoclaved before probing with an antibody that recognizes both free and bound ubiquitin. We detected a ladder of bands that reacted with the anti-ubiquitin antibody starting as small as 3-ubiquitin chains but not with anti-κ LC (Figure S3A). No free ubiquitin was detected arguing that this substrate was not monoubiquitylated and that the elongation of ubiquitin chains is likely to occur primarily through more conventional isopeptide bonds in keeping with our data showing that K48 linkages could be detected (Figure S1C). The NaOH-eluted material was reprecipitated with either anti-ubiquitin or anti- κ LC and blotted for ubiquitin (Figure S3B). The strong signal obtained when the sample was both immunoprecipitated and blotted with anti-ubiquitin made it difficult to prove that ubiquitin was released by this treatment, although the ubiquitin signal was clearly lost from LC. Thus, COS cells were transfected with His-tagged ubiquitin and the κ LC. In this case, nickel agarose was clearly able to re-precipitate the released His-tagged ubiquitin (Figure S3C). In toto, these data strongly suggest that this ERAD substrate is modified on a serine/threonine residue(s) in the VL domain.
Figure 5. Ubiquitylation of NS-1κ LC occurs on serine/threonine and lysine residues.

(A) COS-1 cells were transfected with an empty vector or NS-1 κ LC, and lysates were prepared from cells treated with or without MG132. Isolated LC were treated with or without NaOH and analyzed by immunoblotting with the anti-Ub antibody. (B) COS-1 cells were transfected with the indicated constructs, and LC were isolated and immunoblotted with monoclonal anti-Ub. The ST− mutant migrated slightly faster than the WT and K− mutant which co-migrated with the precipitating antibody. (C) LC were isolated from COS-1 cells expressing either the K− or ST− mutant, incubated with NaOH, and Ub was detected by western blotting. Membranes were stripped and reblotted with an HRP-conjugated anti-κ antiserum. (D) 293T cells were transfected with the indicated mutants and subjected to pulse-chase analyses. LC remaining at each time point is indicated.
Mutation of serine, threonine, and lysine residues is required to significantly inhibit ubiquitylation and stabilize the NS-1 LC
Given the largely unprecedented nature of these results, we synthesized a VL domain in which all 24 serines and threonines alone were mutated to alanine (ST−) and one in combination with the lysines in this domain being mutated to arginine (STK−). Cells were transfected with these two constructs, the wild-type NS-1 or the lysine-less VL domain mutant, and the effects on ubiquitylation were determined. Even though NaOH had removed a very significant amount of the ubiquitin signal from the wild-type LC, we found that changing all of the serines and threonines to alanine had little or no affect on the overall ubiquitylation signal (Figure 5B), which is similar to what was seen when LC were examined in which all of the lysines had been mutated to arginine (Figures 1 and 5B). However, when lysine and serine/threonine residues were mutated together (STK−), the signal was significantly reduced to a point similar to that seen when the entire VL region was cleaved from the protein with Factor Xa (Figure 4A). The most straightforward interpretation of this data is that in the absence of serines and threonines the LC is now modified on lysines, which is similar to what the mK3 E3 is able to do to the MHC heavy chain (Wang et al., 2007). However, lysine does not seem to be a preferred amino acid on the wild-type NS-1 protein, since NaOH treatment removes most of the ubiquitin signal (Figure 5A). To examine this possibility, the VL K− and ST− mutants were treated with NaOH. Indeed high pH removed ubiquitin from the lysine-less mutant but not from the serine/threonine-less mutant arguing that lysines were now modified via isopeptide bonds (Figures 5C and S3D), which should be resistant to NaOH treatment.
In keeping with the ubiquitylation data, we found that the lysine-less and the serine/threonine-less VL domain mutants were turned over at a similar rate as the wild-type LC (Figure 5D). Only when all the lysines, serines, and threonines in this domain were mutated was the LC significantly stabilized. The VL K− and ST− mutants entered the ER as evidenced by their binding to BiP and were still degraded by the proteasome, as only MG132 but not NH4Cl, an inhibitor of lysosomal degradation, blocked their turnover (Figure S4), thus arguing they are still recognized by the ERAD machinery. Of note, a small amount of ubiquitylation remains even after mutating all of the lysines, serines, and threonines in the VL domain. It was possible that a serine or threonine at the N-terminus of the CL domain was modified, as there are several in the first 10 amino acids of this domain. To examine this possibility, we synthesized a NS-1 mutant in which the 26 serine/threonine residues in the CL domain were additionally mutated to alanine, as well as one in which all lysines and serine/threonines in the protein were substituted with arginines and alanines respectively. These mutants behaved very similar to the corresponding VL domain mutants in terms of both ubiquitylation and turnover (data not shown). This argues that the VL domain possesses the major sites of ubiquitylation and controls the turnover of this substrate.
Hrd1 regulates the ubiquitylation of serine, threonine, and lysine residues and controls the turnover of the NS-1 κ LC
We next determined if the same cellular E3 was modifying both types of amino acids. Our previous data demonstrated that expression of a dominant negative Hrd1 mutant interfered with the turnover of wild-type NS-1 (Okuda-Shimizu et al., 2007). We first examined the effects of the dominant negative Hrd1 mutant on the turnover of wild-type LC and the two VL domain mutants. As expected, the dominant negative Hrd1 inhibited the turnover of the wild-type NS-1 LC, but it also affected the degradation of both the K− and ST− VL domain mutants (Figure 6A). In addition, 293T cells were transfected with these NS-1 constructs and a vector expressing siRNA specific for either firefly luciferase or human Hrd1. Pulse chase experiments revealed that siRNA to Hrd1 significantly inhibited the turnover of not only wild-type NS-1, but also of both VL mutants (Figure 6C). In all cases, interfering with Hrd1 activity reduced the initial signal of these ERAD substrates, although we do not know the cause. To determine if the stabilization of the various LC constructs was due to a reduction in their ubiquitylation, we co-expressed them with an empty vector (C) or the dominant negative Hrd1 (D). Indeed, ubiquitylation of all LC constructs was reduced when they were co-expressed with the dominant negative Hrd1 mutant (Figure 6B). As the lysine-less mutant still has serines and threonines and the serine/threonine-less mutant has lysines, these data imply that Hrd1 is required for the ubiquitylation all three amino acids.
Figure 6. Hrd1 activity is required for ubiquitylation and degradation of wild-type LC as well as the lysine-less and serine/threonine-less VL mutants.

(A) The indicated LC constructs were co-expressed in 293T cells with an empty vector or a dominant negative Hrd1 mutant. Cells were subjected to pulse-chase analyses as described above. (B) The indicated LC constructs were co-expressed in COS cells with an empty control vector (C) or a dominant negative Hrd1 mutant (D). After MG132 treatment, cell lysates were prepared for western blotting with anti-Ub (top panel) or an HRP-conjugated anti-κ LC (bottom panel). The top panel represents two different exposures of the anti-Ub blot. (C) 293T cells were transfected with the indicated LC vectors along with siRNA to luciferase (siLuc) or to Hrd1 (siHrd1). Pulse-chase experiments were conducted and LC were isolated and analyzed by reducing SDS-PAGE. Signals were quantitated from two experiments and expressed as a percent of that present immediately after the pulse. (D) Lysates were prepared from cells expressing the indicated siRNAs and blotted for Hrd1 or actin as a control. A separate dish was transfected with a GFP vector, which revealed that the transfection efficiency was ~40% in this experimental group.
Other ERAD substrates are modified by NaOH sensitive ubiquitylation
To determine if modification of serines and threonines was unique to the NS-1 LC or could be detected on other ERAD substrates, we expressed the mini-HC (γ VH-CH1) (Lee et al., 1999) or the NHK α1-anti-trypsin mutant (Hosokawa et al., 2003) in COS-1 cells. p53 was used as a control for this experiment, as it is known to be modified on lysines by its E3 ligase, MDM2 (Nakamura et al., 2000). We found that ubiquitylation of the mini-HC and the NHK mutants was diminished by treating the isolated proteins with NaOH, whereas the ubiquitylation of p53 was not obviously affected (Figure 7A). As another approach to assess how widespread this modification might be, we treated NIH3T3 cells with tunicamycin to inhibit the folding of ER proteins; followed by MG132 to allow them to accumulate. We then isolated Herp and p97, since both proteins bind to a wide variety of ubquitylated ERAD substrates (Ye et al., 2003; Okuda-Shimizu et al., 2007). In both cases, the signal for ubiquitylation was reduced after NaOH treatment (Figure 7B), arguing that ubiquitylation of serine/threonine residues is likely to occur on numerous ERAD substrates. We next asked if reducing Hrd1 levels or activity affected the ubiquitylation and turnover of the mini HC or NHK mutant. When the ubiquitin signal was normalized to the amount of NHK isolated, we found that both siRNA (Figure 7C) and expression of the dominant negative Hrd1 mutant (Figure S5A) significantly reduced ubiquitylation of the NHK mutant, and in both cases the protein was stabilized (Figure 7D and Figure S5B). Similar data were obtained for turnover of the mini-HC (Figure S5C and D).
Figure 7. Ubiquitylation of other ERAD proteins are sensitive to NaOH.
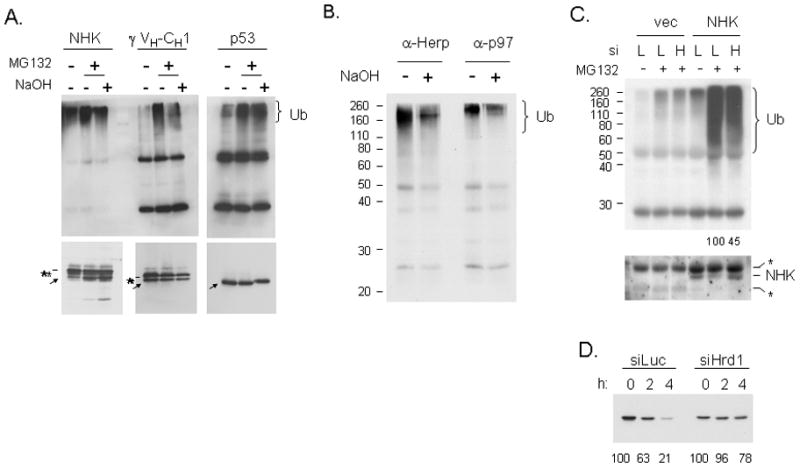
(A) Cells expressing the NHK α1-antitrypsin variant, an HA-tagged mini-HC, or p53 were isolated and treated with NaOH. The transfected proteins were isolated with the appropriate antibody and prepared for immunoblotting with mouse anti-Ub (top panel) or with substrate specific antibodies (bottom panel). Substrates are indicated with an arrow and the bands from the precipitating antibodies with an asterisk. (B) NIH3T3 cells were treated with tunicamycin for 24 hr and MG132 was included for the final 5 hrs. Cell lysates were prepared for immunoprecipitation with anti-Herp or anti-p97. Associated proteins were treated with or without NaOH and prepared for immunoblotting with the monoclonal anti-ubiquitin antibody. (C) 293T cells were co-transfected with an empty vector or NHK and a vector producing siRNA to either luciferase (L) or Hrd1 (H). Cells were treated with or without MG132 and α1-anti-trypsin was isolated and subjected to western blot analyses for ubiquitin (top panel) or NHK (bottom panel) expression. Relative amounts of ubiquitin remaining in the knock-down cells after correcting for NHK signal is indicated under the lanes. (D) 293T cells were co-transfected with NHK and the indicated siRNA vectors. Pulse-chase experiments were performed and the NHK mutant was immunoprecipitated and analyzed by reducing SDS-PAGE.
DISCUSSION
The ubiquitylation and attendant degradation of proteins is a widespread mechanism to regulate biological activity. E3s bind to both E2s and the substrate and catalyze the transfer of ubiquitin to either a very small subset of proteins or in some cases to only a single protein (Pickart and Eddins, 2004). If the identification and modification of misfolded proteins was regulated in the same way, the cell would have to produce an E3 for every different type of protein synthesized in the ER, because any of these proteins could fail to mature properly even under normal physiological conditions, but particularly in response to changes in ER homeostasis. Thus, it is plausible to assume that the rules for recognizing misfolded substrates could be quite different. This possibility is all the more likely given that only a small handful of mammalian E3 ligases have been identified that participate in the ubiquitylation of all ERAD substrates that have been studied thus far. We have found that at least three ERAD substrates have pH sensitive ubiquitin linkages, suggesting that they are modified by ubiquitin on serine/threonine residues prior to being degraded by the 26S proteasome, and all of them appear to be substrates of the Hrd1 E3. It is not clear how many other Hrd1 substrates are ubiquitylated on serine or threonine residues or if gp78 is also capable of this type of activity, but the finding that ubiquitylation of proteins associated with Herp and p97 are somewhat sensitive to high pH, argues that a number of ERAD substrates are likely to be modified on serine and threonine residues. This modification may even be more widespread, as the non-ER, anti-apoptotic protein Bid was reported to be ubiquitylated on serine/threonine residues in response to proapoptotic signals, although the responsible E3 was not identified (Tait et al., 2007).
Despite the large number of studies on ERAD, we were surprised to learn that the site of ubiquitylation by the cellular machinery had not been identified for any ERAD substrate when we initiated this study. The most concerted effort to do so was on the α chain of the T cell antigen receptor, TCRα (Yu et al., 1997), which was one of the first non-transported secretory pathway proteins to be shown to be degraded by a non-lysosomal pathway. In this case, all of the lysines were mutated to arginine, which did not affect its turnover or sensitivity to proteasome inhibitors (Yu et al., 1997). It was assumed that this protein must be ubiquitylated at the N-terminus, as this was the only other residue known to be modified by ubiquitin at that time. Recently Hrd1 was shown to modify serines on the cytosolic tail of TCR α (Ishikura et al., 2010), although the substrate was previously reported to be ubiquitylated by the gp78 E3 (Chen et al., 2006). Elongation of the ubiquitin chains is usually performed by the same E2/E3 pair that initiates the modification. Since Hrd1 seems capable of catalyzing attachment of ubiquitin to amine groups as well as to hydroxyl groups and we did not see any free ubiquitin released with NaOH, it is reasonable to argue that Hrd1 is elongating chains through more conventional lysine-48 linkages.
As a mechanism for escaping immune surveillance, several viruses produce E3 ligases that ubiquitylate the MHC Class I heavy chain and target it for degradation, which allows the virally infected cell to go undetected. In the case of the Kaposi’s sarcoma associated virus, the MIR 1 protein ubiquitylates the cytosolic tail of the MHC heavy chain on a single cysteine residue (Cadwell and Coscoy, 2005), which is sufficient to target it for proteasomal degradation. The γ herpes virus 68 encodes the mK3 E3 ligase, which again targets the MHC heavy chain tail, although in this case the E3 ubiquitylates either serine or threonine. However if these are mutated, mK3 will now ubiquitylate lysines on this substrate (Wang et al., 2007; Wang et al., 2009), demonstrating that it is capable of modifying all three amino acids. This latter finding is pertinent to the fact that gp78 was shown to ubiquitylate Herp in vitro on lysine residues (Li et al., 2007) and to serve as the E3 for the TCR α chain (Chen et al., 2006), which can be ubiquitylated on non-lysine residues (Yu et al., 1997). The data presented here argue that normally NS-1 is ubiquitylated largely on serine/threonine residues, as NaOH treatment of the wild-type protein removes the majority of the ubiquitin. However, when all of the serines/threonines in the VL domain were mutated to alanine the LC was then modified on lysines, and Hrd1 ligase activity was required for the ubiquitylation and turnover of both the ST− and the K− mutant. Together these data argue that the Hrd1 E3 ligase is required for the ubiquitylation of all three types of residues, and suggests that ERAD substrates modified by Hrd1 could show varying degrees of serine/threonine versus lysine ubiquitylation. In vitro ubiquitylation assays will be required to determine if it directly catalyzes both types of linkages or if it utilizes other cellular proteins to do so, but very recently, the mK3 ligase was demonstrated to perform both types of linkages in combination with cellular E2, Ube2j2 (Wang et al., 2009). As viruses often hijack cellular processes and adapt them to their own needs, it is plausible that mK3 evolved from Hrd1 or gp78.
It is striking that the two viral E3s and mammalian Hrd1 use non-traditional linkages for attaching ubiquitin. The reason for this is not clear but there are several interesting possibilities. First, the ability to modify multiple types of amino acids would increase the likelihood that very soon after a substrate begins to emerge into the cytosol it could be ubiquitylated. Ubiquitylation occurs prior to the interaction of the substrate with the AAA ATPase p97 complex, which provides the energy for extracting the unfolded protein from the ER. Thus, it would seem critical, at least in the case of soluble, luminal proteins, that the substrate be modified by ubiquitin as soon as possible to ensure that it does not slip back into the lumen before it can be engaged by p97. A second possibility is that the attachment of ubiquitin via non-isopeptide bonds might make them more resistant to cytosolic deubiquitin ligases. Over 100 deubiquitin ligases have been identified in the human genome and play a role in reversing ubiquitylation to regulate specific biological responses (Amerik and Hochstrasser, 2004). However, once a protein has been identified as misfolded and extracted from the ER, it is reasonable to speculate that it would be detrimental to allow this protein to be deubiquitylated and remain in the cytosol where it would perhaps be disruptive to cytosolic or nuclear functions. Of note, a recent paper demonstrates that the serine/threonine ubiquitylation of MHC heavy chain by the mK3 E3 is more resistant to deubiquitylation than that of a mutant modified on lysines (Wang et al., 2009).
In conclusion, we found that several ERAD substrates can be ubiquitylated via pH-sensitive linkages by Hrd1, allowing them to be targeted to the 26S proteasome and degraded. In the case of the LC, mutating the serine/threonine residues then allowed lysines to become targets of the E3 ligase. Unlike the regulation of biological functions, in which the E3s show great specificity, only a limited number of ERAD-associated E3s have been identified, which must potentially recognize and modify any protein in the secretory pathway that fails to fold properly. It would appear that this involves the ability of the ERAD E3s to modify multiple types of amino acids as they emerge into the cytosol, and as such, represents a paradigm shift in the ubiquitin field.
EXPERIMENTAL PROCEDURES
Cell lines and antibodies
293T, COS-1, and NIH3T3 cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, and 100 U/ml penicillin-streptomycin and cultured under 5% (293T and NIH3T3) or 3% CO2 (COS-1). The 26S proteasome was inhibited with 5 μM lactacystin (Sigma) or with 10 μM MG-132 (Calbiochem) for the indicated times. As indicated, cells were treated with 2.5 μg/ml tunicamycin (Sigma) for 24 hr. Lysosomal degradation was inhibited with 10 mM NH4Cl (Sigma).
The rabbit anti-Herp antibody was described previously (Okuda-Shimizu et al., 2007). The monoclonal anti-HA (12CA5) antibody was provided by Dr. Al Reynolds (Vanderbilt University, USA), the Hrd1 antibody by Dr. Richard Wojcikiewicz (SUNY Upstate Medical University, USA), and the anti-p97 antiserum by Dr. George DeMarino (UT Southwestern, USA). All other antibodies were purchased; anti-mouse κ LC, HRP-conjugated anti-mouse Ig, and HRP-conjugated anti-mouse κ LC (Southern Biotechnology Associates), mouse anti-poly Ub FK2 (BIOMOL), mouse anti-free and poly Ub (Imgenex), rabbit anti-Ub (Dako), anti-p53 (Santa Cruz), anti-α1 anti-trypsin (Cappel), and anti-Flag epitope (Santa Cruz).
Construction and transfection of expression vector
C-terminal epitope-tagged versions of the NS-1 κ LC were created using PCR mutagenesis. PCR-directed mutagenesis was used to mutate the 13 lysines of the NS-1 κ LC to arginine alone or in various combinations. The two cysteines present in the VL domain were mutated to alanine on the wild-type background (C23,88A) and on the VL mutant in which all six lysines were substituted with arginines (K1-6R, C23,88A). Two additional mutants were made to change the first two amino acids of the mature N-terminus from Asn-Ile to either Ser-Asp or Ala-Asp (Ciechanover and Ben-Saadon, 2004; Polevoda and Sherman, 2003).
A Factor Xa cleavage site (Ile-Glu-Gly-Arg) was introduced after amino acid 4, 50, or 109. Two cDNAs were synthesized at Geneart (Regensburg, Germany); one in which all 24 serine/threonines in the VL domain were substituted with alanine (NS-1 ST−), and one where these serine/threonine substitutions were made in combination with changing the six lysines in this domain to arginine (NS-1 STK−). The cDNAs were inserted into the NS-1 κ LC expression vector in place of the wild-type VL sequence.
The wild-type and dominant negative Hrd1 constructs were provided by Dr. Yihong Yi (NIDDK, USA), and the pSuperstar vector encoding siRNA specific for firefly luciferase or human Hrd1 were a gift from Dr. Ron Kopito (Stanford University, USA). A truncated version of the γ Ig HC (γ VH-CH1) was described previously (Lee et al., 1999), as was the NHK-AAT mutant (Le et al., 1990). A wild type p53 expression vector was supplied by Dr. Michael Kastan (St. Jude Children’s Research Hospital, USA). The recombinant plasmids were introduced into cells using calcium phosphate precipitation, the FuGENE 6 Transfection Reagent (Roche), or GeneCellin (BioCellChallenge).
Preparation of cell extracts, immunoprecipitations, and pulse-chase experiments
Pulse-chase experiments were done as described previously (Okuda-Shimizu et al., 2007). To inhibit proteasomal function during pulse-chase experiments, lactacystin or MG132 was added 3 hr prior to the starvation and included through the labeling and chase. To determine half-lives of the various proteins, the signal for each band was quantified by phosphorimager and the bands present in the chase samples were expressed as a percent of the signal present in the pulse lane (0). The numbers under the autoradiographs represent the average of at least three experiments.
To examine ubiquitylation of the various ERAD substrates, cells were lysed in a pH 6.8 RIPA buffer [20 mM Na-PO4 (pH 6.8), 0.15 M NaCl, 1 mM EDTA, 1% Nonidet P-40 (NP-40), 0.1% SDS, 0.2% deoxycholate, and 10% glycerol] supplemented with protease inhibitors, 10 mM N-ethylmaleimide (NEM), and 10 μM MG132. Where indicated the Ub signal was detected on a LAS-1000 plus Luminescent Image Analysis System and quantified using Multi Gauge software (Fuji Film). The vector control Ub signal was subtracted from the test signal, which was normalized to that of the ERAD substrate.
Cleavage of proteins with Factor Xa
COS-1 cells were transfected with vectors encoding a NS-1 κ LC with Factor Xa cleavage sites at the indicated positions. The NS-1-Flag-tagged proteins were isolated with anti-Flag agarose (Sigma), and precipitated proteins were eluted with 300 μg/ml 3x Flag-peptide in Xa buffer [20 mM Tris-HCl (pH 6.8), 100 mM NaCl, 2 mM CaCl2]. Eluted proteins were divided and treated with or without 60 μg/ml Factor Xa (New England Biolabs) for 4 hr at 32ºC. Samples were diluted with Xa buffer and reisolated with anti-κ LC antibodies and prepared for western blotting.
NaOH treatment of ubiquitylated ERAD substrates
COS-1 cells expressing ERAD substrates were treated with 10 μM MG132 for 5 hr and lysed in pH 6.8 RIPA buffer. Proteins were immunoprecipitated from the lysates and eluted with SDS elution buffer [10 mM Tris-HCl (pH 6.8) with 0.5% SDS] at 95°C for 3 min. The eluate was split and re-incubated for 1hr at 32ºC with 50 mM NaOH or PBS and analyzed by western blotting. To detect released poly-Ub chains, the membranes were autoclaved for 15 min after transferring before immunoblotting. Released Ub chains were reprecipitated with anti-Ub and anti-κ LC, or in cells that were tranfected with His-tagged Ub with Ni+2 agarose beads.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant GM54068 (LMH), the Cancer Center CORE Grant CA21765, and the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital. We are extremely grateful to Drs. Ryan Tyler and Steve Kaiser of Dr. Ron Kopito’s lab for their valiant efforts to detect the sites of NS-1 ubiquitylation by mass spectrometry and thank Dr. Jonathan Weissman for suggesting we synthesize the VL gene to obtain a construct without serines and threonines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB, McCracken AA. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem. 1999;274:3453–3460. doi: 10.1074/jbc.274.6.3453. [DOI] [PubMed] [Google Scholar]
- Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309:127–130. doi: 10.1126/science.1110340. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Chen B, Mariano J, Tsai YC, Chan AH, Cohen M, Weissman AM. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci U S A. 2006;103:341–346. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 2004;14:103–106. doi: 10.1016/j.tcb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Cormier JH, Tamura T, Sunryd JC, Hebert DN. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell. 2009;34:627–633. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay A, Bromberg KD, Hayashi Y, Mirabella M, Burch D, Kirkwood B, Serra C, Malicdan MC, Mizisin AP, Morosetti R, Broccolini A, Guo LT, Jones SN, Lira SA, Puri PL, Shelton GD, Ronai Z. The ER-bound RING finger protein 5 (RNF5/RMA1) causes degenerative myopathy in transgenic mice and is deregulated in inclusion body myositis. PLoS ONE. 2008;3:e1609. doi: 10.1371/journal.pone.0001609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassink G, Kikkert M, van VS, Lee SJ, Spaapen R, van LT, Coleman CS, Bartee E, Fruh K, Chau V, Wiertz E. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem J. 2005;388:647–655. doi: 10.1042/BJ20041241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- Ishikura S, Weissman AM, Bonifacino JS. Serine residues in the cytosolic tail of the T-cell antigen receptor α-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J Biol Chem. 2010;285:23916–23924. doi: 10.1074/jbc.M110.127936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van VS, Thanedar S, Roitelman J, Chau V, Wiertz E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- Knittler MR, Haas IG. Interaction of BiP with newly synthesized immunoglobulin light chain molecules: cycles of sequential binding and release. EMBO J. 1992;11:1573–1581. doi: 10.1002/j.1460-2075.1992.tb05202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Kokame K, Agarwala KL, Kato H, Miyata T. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J Biol Chem. 2000;275:32846–32853. doi: 10.1074/jbc.M002063200. [DOI] [PubMed] [Google Scholar]
- Le A, Graham KS, Sifers RN. Intracellular degradation of the transport-impaired human PiZ alpha 1-antitrypsin variant. Biochemical mapping of the degradative event among compartments of the secretory pathway. J Biol Chem. 1990;265:14001–14007. [PubMed] [Google Scholar]
- Lee YK, Brewer JW, Hellman R, Hendershot LM. BiP and Ig light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Mol Biol Cell. 1999;10:2209–2219. doi: 10.1091/mbc.10.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Tu D, Brunger AT, Ye Y. A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature. 2007;446:333–337. doi: 10.1038/nature05542. [DOI] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Mueller B, Lilley BN, Ploegh HL. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J Cell Biol. 2006;175:261–270. doi: 10.1083/jcb.200605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai K, Thogersen HC. Synthesis and sequence-specific proteolysis of hybrid proteins produced in Escherchia coli. Methods Enzymol. 1987;153:461–481. doi: 10.1016/0076-6879(87)53072-5. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Roth JA, Mukhopadhyay T. Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Mol Cell Biol. 2000;20:9391–9398. doi: 10.1128/mcb.20.24.9391-9398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J Cell Biol. 2006;172:383–393. doi: 10.1083/jcb.200507057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which requires Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- Polevoda B, Sherman F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J Mol Biol. 2003;325:595–622. doi: 10.1016/s0022-2836(02)01269-x. [DOI] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83. doi: 10.1038/373081a0. [DOI] [PubMed] [Google Scholar]
- Schulze A, Standera S, Buerger E, Kikkert M, van VS, Wiertz E, Koning F, Kloetzel PM, Seeger M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol. 2005;354:1021–1027. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Skowronek MH, Hendershot LM, Haas IG. The variable domain of non-assembled Ig light chains determines both their half-life and binding to BiP. Proc Natl Acad Sci U S A. 1998;95:1574–1578. doi: 10.1073/pnas.95.4.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001;15:2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, de VE, Maas C, Keller AM, D'Santos CS, Borst J. Apoptosis induction by Bid requires unconventional ubiquitination and degradation of its N-terminal fragment. J Cell Biol. 2007;179:1453–1466. doi: 10.1083/jcb.200707063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C, Hitt R, Park SH, Deak PM, Kostova Z, Wolf DH. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem. 2003;278:35903–35913. doi: 10.1074/jbc.M301080200. [DOI] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Dong H, Soroka CJ, Wei N, Boyer JL, Hochstrasser M. Degradation of the bile salt export pump at endoplasmic reticulum in progressive familial intrahepatic cholestasis type II. Hepatology. 2008;48:1558–1569. doi: 10.1002/hep.22499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Herr RA, Chua WJ, Lybarger L, Wiertz EJ, Hansen TH. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol. 2007;177:613–624. doi: 10.1083/jcb.200611063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Herr RA, Rabelink M, Hoeben RC, Wiertz EJ, Hansen TH. Ube2j2 ubiquitinates hydroxylated amino acids on ER-associated degradation substrates. J Cell Biol. 2009;187:655–668. doi: 10.1083/jcb.200908036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci U S A. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- Wilhovsky S, Gardner R, Hampton R. HRD gene dependence of endoplasmic reticulum-associated degradation. Mol Biol Cell. 2000;11:1697–1708. doi: 10.1091/mbc.11.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- Yu H, Kaung G, Kobayashi S, Kopito RR. Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J Biol Chem. 1997;272:20800–20804. doi: 10.1074/jbc.272.33.20800. [DOI] [PubMed] [Google Scholar]
- Zavacki AM, Arrojo E Drigo, Freitas BC, Chung M, Harney JW, Egri P, Wittmann G, Fekete C, Gereben B, Bianco AC. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol Cell Biol. 2009;29:5339–5347. doi: 10.1128/MCB.01498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.