

Abstract
In the mammalian forebrain, most glutamatergic excitatory synapses occur on small dendritic protrusions called dendritic spines. Dendritic spines are highly plastic and can rapidly change morphology in response to numerous stimuli. This dynamic remodeling of dendritic spines is thought to be critical for information processing, memory and cognition. Conversely, multiple studies have revealed that neuropathologies such as autism spectrum disorders (ASDs) are linked with alterations in dendritic spine morphologies and miswiring of neural circuitry. One compelling hypothesis is that abnormal dendritic spine remodeling is a key contributing factor for this miswiring. Ongoing research has identified a number of mechanisms that are critical for the control of dendritic spine remodeling. Among these mechanisms, regulation of small GTPase signaling by guanine-nucleotide exchange factors (GEFs) is emerging as a critical mechanism for integrating physiological signals in the control of dendritic spine remodeling. Furthermore, multiple proteins associated with regulation of dendritic spine remodeling have also been implicated with multiple neuropathologies, including ASDs. Epac2, a GEF for the small GTPase Rap, has recently been described as a novel cAMP(yet PKA-independent) target localized to dendritic spines. Signaling via this protein in response to pharmacological stimulation or cAMP accumulation, via the dopamine D1/5 receptor, results in Rap activation, promotes structural destabilization, in the form of dendritic spine shrinkage, and functional depression due to removal of GluR2/3-containing AMPA receptors. In addition, Epac2 forms macromolecular complexes with ASD-associated proteins, which are sufficient to regulate Epac2 localization and function. Furthermore, rare nonsynonymous variants of the EPAC2 gene associated with the ASD phenotype alter protein function, synaptic protein distribution, and spine morphology. We review here the role of Epac2 in the remodeling of dendritic spines under normal conditions, the mechanisms that underlie these effects, and the implications these disease-associated variants have on our understanding of the pathophysiology of ASD.
Keywords: dendritic spine, autism, neuroligin, GEF, Rap, GTPase, GluR2, cAMP, dopamine
Introduction
The mechanisms that neurons employ to encode patterns of activity remain a mystery despite decades of intensive research. Neurons must translate fleeting ionic fluctuations into long-lasting changes that will influence cellular responses to future stimuli. A promising theory for the cellular mechanism of information storage is that neurons alter the strength and number of their synaptic connections by coordinated changes in synaptic structure and protein content. Given the diversity of synaptic proteins and the complexity of synaptic signaling, dissecting the signaling pathways responsible for controlling synaptic strength is a daunting task. Nevertheless, a growing body of work has capitalized on new imaging, biochemical and genetic techniques to yield unprecedented insights into the molecular mechanisms of learning and memory. While great advances have been made in understanding the molecular mechanisms underlying the strengthening of synapses, there is currently a paucity of information regarding mechanisms that actively promote destabilization of central synapses (Tada and Sheng, 2006; Woolfrey et al., 2009). In this review we will give an overview of our current understanding of dendritic spine remodeling and its implication for synaptic plasticity, memory and cognition. We will also describe some of the molecular components that are thought to be important for dendritic spine remodeling, focusing on the characterization of a novel role for the cAMP effector, Epac2, in actively promoting synapse destabilization. Further, we will describe the effects of rare nonsynonymous variants of the EPAC2 gene, which segregate with the autistic phenotype, on dendritic spine structure and synaptic signaling, and discuss the implication these findings have on our understanding of normal and abnormal brain function.
Dendritic spines and structural plasticity
In the mammalian forebrain, synapses are the predominant site of neuronal communication. These highly specialized sites readily undergo structural and functional changes resulting in altered communication between neurons. These changes are considered to be a major contributor to the processing and storage of information within the forebrain. At the cellular level, the majority of excitatory synapses are located on dendritic spines, mushroom-shaped protrusions of dendrites (Bourne and Harris, 2008) (Fig. 1). These structures are highly plastic: they can be rapidly formed, eliminated, or change shape/size (collectively referred to as morphology) during synaptogenesis, plasticity, and maintenance, or in response to a number of stimuli.
Figure 1.

Dendritic spines are small protrusions along dendrites that contain postsynaptic densities. (A) Example of a cortical neuron expressing green fluorescent protein (GFP). The main dendrite is branched and has dendritic spines along its length. The axon of the neuron is much thinner than the dendrite and has no spines. The inset shows a high magnification image of dendritic spines. (B) Dendrite of a cortical neuron immunofluorescence staining for phalloidin, a marker of endogenous β-actin. Note the enrichment of β-actin in the dendritic spines. (C) Schematic of a mature dendritic spine making contact with an axon.
Early in development, following neurite extension, dendritic spines are absent. Instead, dynamic, frequently transient cytoplasmic extensions known as filopodia line the dendritic arbor of pyramidal neurons (Bonhoeffer and Yuste, 2002). These protrusions are thought to sample the surrounding neuropil for presynaptic contacts. As the neuron matures, filopodia are lost as spines emerge. Evidence currently supports a model of spinogenesis whereby filopodia make presynaptic contacts, stabilize and transform into mature dendritic spines (Bhatt et al., 2009; Grutzendler et al., 2002; Yoshihara et al., 2009; Zuo et al., 2005a).
The mature brain differs substantially from developing brain in that filopodia are rare and dendritic spines with mature phenotypes, namely with discernible heads and necks, predominate (Fig. 1C). Though less labile than filopodia, spines are formed and pruned throughout an animal s life span. Unprecedented insight into the dynamism of spines in vivo has been afforded by recent advances in 2-photon laser scanning microscopy that has allowed for long-term imaging of dendritic spines in living animals (Grutzendler and Gan, 2006). Ascertaining the basal rate of spine turnover has not been trivial, as this parameter is age- (Zuo et al., 2005a), cell type- (Trachtenberg et al., 2002) and brain region- (Majewska et al., 2006) dependent. Spine formation and retraction are common events in developing brain but are more rare occurrences in mature brain (Bhatt et al., 2009; Holtmaat and Svoboda, 2009). Thus the emerging trend is that spines gradually become more stable over the lifespan while a small minority of spines continues to turn over in adulthood (this topic is extensively reviewed in Bhatt et al., 2009 and Holtmaat and Svoboda, 2009). It has been proposed that this pattern of general stability with sparse plasticity is an ideal mechanism for memory storage (Yang et al., 2009).
Regulation of dendritic spine morphology modulates synaptic properties and the ability of synapses to undergo plasticity (Bourne and Harris, 2008). Thus it is thought that alterations in dendritic spine morphology is a key mechanism in the remodeling of neural circuits, memory and cognition (Holtmaat and Svoboda, 2009; Kasai et al., 2010). Conversely, aberrant dendritic spine number/morphology has been extensively associated with neuropsychiatric disorders, including schizophrenia, autism spectrum disorders (ASD) and mental retardation (Dierssen and Ramakers, 2006; Fiala et al., 2002; Glantz and Lewis, 2000; Hutsler and Zhang, 2010; Irwin et al., 2000). Conversely, increased dendritic spine plasticity has been suggested to promote functional recovery from neurological disorders (Lewis and Sweet, 2009). Thus, a major focus of current cellular neuroscience is to uncover the mechanisms underlying the control of dendritic spine morphology and remodeling.
Dendritic spine dynamics: stabilization vs. destabilization
Remodeling of central neural circuits depends on the bidirectional control of synapse stability, structure, and strength (Bhatt et al., 2009; Kasai et al., 2010). Synapse stabilization, enlargement, and potentiation contribute to the establishment of long-lasting connections, and has been extensively reviewed previously (Bhatt et al., 2009; Kasai et al., 2010). Conversely, synapse destabilization entails increased spine motility, turnover, spine shrinkage, and depressed glutamatergic transmission. This process is thought to contribute not only to neural circuit remodeling during development, but also the experience-dependent refinement and plasticity of mature circuits (Bonhoeffer and Yuste, 2002; Kasai et al., 2010; Zuo et al., 2005b). It is of note that synapse destabilization can result in the shrinkage of dendritic spines, but not elimination (Woolfrey et al., 2009). Though spine shrinkage and spine elimination are related concepts, the former does not necessarily result in the later. Rather, it is thought that dendritic spine shrinkage could be a mechanism to destabilize existing spines, which can then either be further potentiated, or actively eliminated (Kasai et al., 2010; Segal, 2005; Srivastava et al., 2008; Yoshihara et al., 2009).
Dendritic spine motility, caused by the rapid rearrangement of the spine actin cytoskeleton, is another form of dendritic spine remodeling which has recently received a great deal of research attention. Using two-photon microscopy, spines have been shown to be motile in adult mouse cortex (Bhatt et al., 2009; Holtmaat and Svoboda, 2009; Zuo et al., 2005a). In dissociated culture systems where temporal and spatial resolution is greatly enhanced, spine motility has been carefully described and a variety of spine “behaviors” have been identified (Bonhoeffer and Yuste, 2002; Jones et al., 2009; Srivastava et al., 2008; Woolfrey et al., 2009). Although the physiological relevance of spine dynamism is still under debate, it has been suggested that spine motility may be a mechanism that controls the amount of overlap between the pre- and postsynapses, thus regulating synaptic strength. Alternatively, spine motility may allow for the creation and elimination of new connections; together these two mechanisms could have implications for learning and memory.
Functional correlates of dendritic spine structure
Recent studies have started to show that dendritic spine structure is correlated with synaptic function. Seminal work a decade ago used a glutamate uncaging approach to demonstrate that large spines contain greater amounts of AMPA receptors than thin spines (Kasai et al., 2010; Matsuzaki et al., 2001). This finding led to the prominent model that dendritic structure and stability are tightly correlated with function (Kasai et al., 2010). Specifically, large spines on average feature larger PSDs (Bourne and Harris, 2008), persist for longer periods of time (Holtmaat and Svoboda, 2009; Kasai et al., 2010; Trachtenberg et al., 2002) and are resistant to plasticity-inducing stimuli. These large, stable spines have thus been labeled “memory spines” (Kasai et al., 2003). Conversely, smaller spines are often short-lived and can be readily potentiated to become stable spines, leading to the moniker “learning spines” (Kasai et al., 2003). Therefore, according to this model, spine morphology modulates synaptic properties and the ability to undergo plasticity (Bourne and Harris, 2008; Segal, 2005). However, it is of note that this is not always the case; recent studies have demonstrated that in a few circumstances changes in spine size do not always correlate with synaptic strength (Segal, 2010).
By what mechanisms can spines become stable? Long-term potentiation (LTP), a process involving the activity-dependent strengthening of synapses, is thought to be the cellular equivalent of learning and memory. In classical LTP, NMDA receptor-dependent postsynaptic calcium influx elicits the phosphorylation and insertion of AMPA receptors into the PSD of already active synapses (Shepherd and Huganir, 2007) or may alter functional activity by the activation of previously “silent” (AMPA receptor-lacking) synapses (Isaac et al., 1997; Shepherd and Huganir, 2007). LTP induction is associated with persistent increases in dendritic spine size (Kasai et al., 2010; Park et al., 2006) that are actin-dependent (Okamoto et al., 2009). The initial actin-mediated increase in spine size is strongly correlated with but ultimately dissociable from long-term changes in synapse potentiation (Yang et al., 2008). Indeed, long-term consolidation of potentiation (late phase LTP) requires additional elements such as protein synthesis (Sutton and Schuman, 2006).
Interestingly, dendritic spine destabilization and shrinkage is also associated with forebrain long-term depression (LTD) (Nagerl et al., 2004; Zhou et al., 2004). Though functionally opposite to LTP, NMDA receptor-dependent LTD shares a similar mechanism. In this case, calcium influx (albeit with different concentration and kinetics from LTP-inducing circumstances) induces elevated phosphatase activity, dephosphorylation and synaptic removal of AMPA receptors (Malenka and Bear, 2004). LTP and LTD are just two of the many mechanisms employed by neurons of the forebrain to bidirectionally modulate dendritic spine morphology, synaptic strength and functional connectivity (Feldman, 2009).
Dendritic spines play key roles in normal brain function
Neural circuits need to exhibit functional plasticity to encode information about the environment. A variety of stimuli, both physiological and pathological can influence spine dynamics (Alvarez and Sabatini, 2007; Tau and Peterson, 2010). Sensory experience, as modeled by mouse whisker stimulation, resulted in a transient increase in dendritic spine density in the corresponding primary cortical sensory region (Wilbrecht et al., 2010). On the other hand, selective sensory deprivation elicited through whisker trimming increased the fraction of transient spines (Trachtenberg et al., 2002). Rearing animals in enriched environments also results in elevated spine density in forebrain (Bose et al., 2009). Very recent evidence has shown rapid spine formation and stabilization in association with motor learning, followed by homeostatic pruning of synapses in layer V motor cortex (Yang et al., 2009). Thus structural reorganization of forebrain spiny synapses appears to be a powerful mechanism for information storage.
Dendritic spine dysfunction in disease
Deficits in cognitive function, notably in working, spatial and reference memory, as well as social interactions, are core features of a great number of neurological disorders (DSM-IV, 2000). As dendritic spine morphology has been intimately linked to cognitive function (Holtmaat and Svoboda, 2009; Kasai et al., 2010; Ramakers, 2002), it is not surprising that multiple neuropathologies are strongly associated with disruptions of neural circuits (van Spronsen and Hoogenraad, 2010). Indeed, numerous neuropatholgical postmortem studies have strongly linked abnormal spine morphology with the pathogenesis of a number of neuropsychiatric disorders and neurodevelopmental disorders (Fiala et al., 2002); such as mental retardation (MR) (Dierssen and Ramakers, 2006), fragile-X (Irwin et al., 2000), Down’s syndrome (Takashima et al., 1989), autism spectrum disorders (ASDs) (Hutsler and Zhang, 2010; Pickett and London, 2005; Zoghbi, 2003), schizophrenia (Glantz and Lewis, 2000; Lewis and Sweet, 2009), and depression (Gorman and Docherty, 2010). It is currently posited that dendritic spine dysmorphogenesis can lead to defective or excessive synapse function and connectivity, resulting in disruptions in neural circuitry (Tau and Peterson, 2010; van Spronsen and Hoogenraad, 2010). Dysregulation of the complex mechanisms that control dendritic spine structure and function may contribute to these synaptic irregularities. Understanding the mechanisms by which dendritic spine morphogenesis occurs will therefore, not only expand our knowledge of normal brain function, but that of abnormal brain function as well.
Molecular control of dendritic spine morphology
Recently major advances have been made into our understanding of the mechanisms that underlie changes in synapse structure. The postsynaptic density contains hundreds of distinct proteins, and the organizational complexity of this structure is becoming increasingly apparent. A significant challenge is to identify the proteins that are responsible for determining postsynaptic ultrastructure. Dendritic spines are actin rich structures (Fig 1B); actin is the primary cytoskeletal component in spines, and actin modulation is essential for changes in spine morphology (Frost et al., 2010; Hotulainen and Hoogenraad, 2010). Multiple signaling pathways are know to converge on the actin cytoskeleton, and stimuli that induce filamentous actin rearrangements are associated with the formation, elimination and changes in morphology of dendritic spines (Jones et al., 2009; Lise and El-Husseini, 2006; Penzes et al., 2008; Srivastava et al., 2008; Tada and Sheng, 2006; Woolfrey et al., 2009; Xie et al., 2008; Xie et al., 2007; Yoshihara et al., 2009). One family of proteins that have been shown to have potent effects on the actin cytoskeleton are the small GTPases.
Small GTPases are G proteins that comprise a superfamily of more than 100 proteins with diverse cellular functions. As their name implies, these proteins are of low molecular weight (20–40kD) and are related to heterotrimeric proteins (e.g. Gs and Gi). These proteins are highly evolutionarily conserved and expressed in many tissues including brain (Takai et al., 2001). There are five families (Ras, Rho, Rab, Sar1/Arf, and Ran) within the small GTPase superfamily which are categorized by structure and function. All small GTPases feature similar gross morphology which permits binding to GDP and GTP (Takai et al., 2001). Diversity in small GTPases arises from unique effector-interacting domains and C-terminal regions which can be modified by post-translational modification. The ability of Rho- and Ras-family GTPases to regulate cytoskeleton dynamics and gene transcription places them as idea candidates to control fundamentally important neuronal functions.
The small GTPases exist in discrete inactive or active states dependent on whether GDP (inactivating) or GTP (activating) is bound, and thus are often referred to as molecular switches. Activation of these proteins permits interaction with a vast array of effector proteins, some of which converge on the actin cytoskeleton. GTPases also exhibit weak enzymatic activity, slowly hydrolyzing GTP to GDP resulting in self-inactivation over time. Tight regulation of small GTPase activity is achieved by upstream regulators from two classes of proteins GEFs and GAPs. Guanine-nucleotide exchange factors (GEFs) activate GTPases by facilitating the exchange of GDP for GTP while GTPase activating proteins (GAPs) catalyze the hydrolysis of GTP to GDP and thus inactivate small GTPases. GEFs and GAPs respond to a wide range of upstream signals, and GTPases generally are regulated by multiple GEFs and GAPs. Importantly, a single GEF or GAP is specific for only one small GTPase. Thus GEFs and GAPs provide specificity in GTPase signaling.
Small GTPases control dendritic spine morphology and function
The roles of GTPases themselves have been thoroughly explored through overexpression and knockout studies, but the pathways that govern GTPase activity in vivo remain obscure. In efforts to characterize these pathways, the immediate upstream regulators of GEFs and GAPs are beginning to be investigated. As small GTPases can be regulated by multiple GEFs and GAPs under different conditions, deciphering the mechanisms of activation and the intracellular targeting of GEFs and GAPs will significantly strengthen our understanding of small GTPase function.
The small GTPase Rap is a Ras-family small GTPase that is a key regulator of cell differentiation, growth, polarity, and adhesion (Bos, 2005; Stork, 2003). Recent studies have begun to uncover the role of Rap signaling in neurons. The Rap family of small GTPases is composed of two proteins, Rap1 and Rap2. Activation of Rap proteins, examined using mutant constructs in hippocampal slice cultures, is required for NMDA-dependent endocytosis of short-tailed AMPA receptors (e.g. GluR2) during LTD and depotentiation (Zhu et al., 2002; Zhu et al., 2005). This small GTPase has also been implicated in the regulation of NMDA receptor currents (Imamura et al., 2003), dendritic development (Chen et al., 2005), neuronal excitability, early- and late-phase LTP, storage of spatial memory, and cAMP-dependent LTP (Morozov et al., 2003). Further evidence for the regulation of synapses by Rap emerged when Rap1 was shown to exert bidirectional control of dendritic spine morphology and AMPA receptor content (Xie et al., 2005). This study also revealed important clues regarding Rap regulation in spiny neurons as Rap1 activity was controlled by synaptic activity. More recently it has been shown that activation of Rap1 by the neurosteroid estrogen results in a novel form of spine plasticity (Srivastava et al., 2008). In this scenario, estrogen can act through Rap-dependent mechanisms to “prime” a neuron to respond to subsequent synaptic-activity stimuli with greater efficacy, by acutely modulating dendritic spine morphology and functional plasticity in neural circuits.
Signaling pathways involving Rap are prominent candidates for regulating synapse destabilization and depression, because Rap promotes spine shrinkage and AMPA receptor endocytosis (Xie et al., 2005; Zhu et al., 2002). However, little is known about the endogenous regulators of Rap in neurons. Inhibition of Rap signaling through the Rap-GAPs SPAR (Pak et al., 2001) or RAP-GAP1 (Xie et al., 2005) results in larger spine heads in neuronal culture. Interestingly, active Rap is required for cAMP-dependent plasticity and memory, suggesting a key role for cAMP-dependent Rap-GEFs in the regulation of synapse structure and function (Morozov et al., 2003).
Recently, a study examining transgenic mice expressing constitutively active Rap2 (Rap2V12) revealed that Rap2V12 expression resulted in fewer and shorter dendritic spines in CA1 hippocampal neurons, and enhanced LTD at CA3-CA1 synapses (Ryu et al., 2008). Furthermore, behavioral analysis demonstrated that these mice displayed impaired spatial learning and deficits in extinction of contextual fear conditioning. These data support a role for Rap2 signaling in regulating synapse destabilization and depression in vivo, and further implicate Rap2 signaling in hippocampal-based learning and extinction of contextual fear conditioning in vivo (Ryu et al., 2008). However, the pathways that result in Rap activation in neurons have not been well described.
Epac2 is a PKA-independent target of cAMP and activator of Rap
Epac2 (Exchange protein activated by cAMP; also known as cAMP-GEFII or RapGEF4) is a GEF for the small GTPase Rap (Fig. 2) (Bos, 2003; Kawasaki et al., 1998). Different genes encode the two isoforms of Epac proteins, Epac1 and 2 (Fig. 2A) (Gloerich and Bos, 2010). These proteins feature complementary tissue distribution: Epac1 is present in most tissues but has very low expression in brain; Epac2, the larger of the two isoforms, is highly enriched in brain and adrenals, but is present at very low levels elsewhere (Kawasaki et al., 1998). Thus Epac2 represents one of the only two known PKA-independent cAMP targets, opening many new directions for research as an alternative pathway to PKA (Bos, 2003). Outside of the brain, Epac proteins have been implicated in a number of functions, including mediating cAMP control of cardiac function, regulation of insulin secretion in pancreatic beta cells, and regulation of adhesion molecules at cell-cell junctions in endothelial cells, thereby mediating hormonal regulation of vascular function and permeability (Gloerich and Bos, 2010). Moreover, roles for Epac proteins in renal function and in regulating the immune response of leukocytes have also been suggested (see Gloerich and Bos, 2010 for an extensive review on these topics). Signaling mediated by the ubiquitous second messenger cAMP in pyramidal neurons is crucial for synaptic plasticity and learning and memory (Frey et al., 1993; Silva and Murphy, 1999). Conversely, aberrant cAMP signaling is a component of the pathological plasticity observed in psychiatric disease (Kelley et al., 2008) and drug addiction (Nestler, 2001). Because most previous studies on cAMP signaling in pyramidal neurons have focused on PKA, very little is known about PKA-independent effects of cAMP. However, several studies report that postsynaptic cAMP-dependent but PKA-independent mechanisms induce LTD, depress basal synaptic transmission, and reverse potentiation (Otmakhov and Lisman, 2002).
Figure 2.

Epac2 is a multi-domain protein enriched at excitatory synapses. (A) Schematic of Epac2 domain structure. The location of the four rare non-synonymous variants in the EPAC2 gene are shown. (B) Immunofluorescence staining of endogenous Epac2. Note punctate staining of Epac2 along and offset from the dendrite, consistent with a synaptic localization. Inset demonstrates the co-localization of Epac2 with the excitatory synaptic marker, PSD-95, by endogenous staining. PSD-95 is stained in green; Epac2 is stained in magenta; overlap is identified by white.
Structurally, Epac2 contains two cAMP-binding domains, a Dishevelled, Egl-10 and Pleckstrin (DEP) domain, a Ras-exchanger motif (REM) which interacts with the GEF domain, and a Rap-GEF domain (Fig. 2A). The cAMP-binding domains have been thought to be sufficient to regulate GEF activity in vitro: the catalytic activity of Epac is inhibited by direct interaction between the GEF domain and the cAMP-binding domain in the absence of cAMP; Epac becomes activated by release of this inhibition upon cAMP binding by means of a conformation change in the protein structure (Rehmann et al., 2008; Rehmann et al., 2003) (Fig. 3). Studies using X-ray crystallography and single particle electron microscopy have identified a region term the “hinge” as being critical for the activation of Epac2: upon binding of cAMP to Epac2, Epac2 rotates at the “hinge” domain, thus moving into an active conformation (Rehmann et al., 2008). Epac2 also contains a second N-terminal low-affinity cAMP-binding, and a Ras-binding (RA) domain, absent in Epac1 (Rehmann et al., 2008). A valuable tool for studying Epac function is the synthetic cAMP analog 8-CPT [8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate] which specifically activates Epac, but not PKA, both in vitro and in vivo (Enserink et al., 2002; Woolfrey et al., 2009).
Figure 3.

Schematic of the two mechanisms that may regulate Epac2 function. (1) Regulation of Epac2 function by increased cAMP levels, either through activation of Ca2+-sensitive adenylate cyclase, or by G-protein coupled receptor, for example dopamine D1/5 receptor, activation. Binding of cAMP causes a conformational change in Epac2 structure, resulting in activation of the Epac2 protein and increased binding of GTP by Rap. (2) Recruitment of Epac2 to synapses by neuroligin 3, mediated by the scaffold protein, PSD-95, results in an increase in Epac2 activation and Rap activity.
Synaptic localization of Epac2
In the brain, Epac1 has been reported to be only present in the brain at very early postnatal developmental stages (Ster et al., 2007). Quantitative PCR analysis of mRNA from mature cultured cortical neurons confirmed the enrichment of Epac2 over Epac1, suggesting that the larger isoform is the predominate form found in the adult brain. In non-neuronal cells, Epac2 has been localized to the (sub)plasma membrane, cystolic fractions, actin cytoskeleton and Golgi (Grandoch et al., 2010; Li et al., 2006). In neurons, Epac2, but not Epac1, has been detected in forebrain post-synaptic densities (PSDs), by proteomic studies (Jordan et al., 2004; Peng et al., 2004). Consistent with this observation, Epac2 immunofluorescence revealed punctate structures along the dendrites and in the somata of cultured cortical neurons (Fig.2B) (Woolfrey et al., 2009). Epac2 is enriched at excitatory synapses, as determined by its colocalization with the PSD proteins, PSD-95 and the NR1 subunit of NMDA receptors (Woolfrey et al., 2009) (Fig. 2B). Furthermore, Epac2 co-immunoprecipitated with PSD-95 in rat forebrain homogenates. This suggests that Epac2 and PSD-95 participate within the same synaptic signaling complexes, and that the PSD-95 scaffold protein may mediate the interaction of Epac2 with other synaptic proteins, allowing it to influence synapse structure and function. Interestingly, Epac2 was also found along Tau5-positive processes, indicating that a small amount of Epac2 is present in axons, suggesting a possible presynaptic role. Together, these studies suggest that signaling by Epac2 emerges during synapse maturation, and that it is well placed to regulate central spiny synapses during this period.
Regulation of spine dynamics and turnover by Epac2
The presence of Epac2 at synapses and the ability to influence Rap activity, a known regulator of the actin cytoskeleton, makes Epac2 an excellent candidate to regulate Rap-dependent dendritic spine plasticity. Activation of Epac2 with its specific agonist, 8-CPT, resulted in shrinkage of dendritic spine size in EGFP-expressing mature cultured cortical neurons (Fig. 4A). Consistent with previous reports of bidirectional modulation of dendritic spine size by Rap, specific shRNA knockdown of Epac2 resulted in a increase in dendritic spine size (Woolfrey et al., 2009). Epac2-dependent shrinkage of dendritic spine size also led to a reduction in the overlap between postsynapse and presynapse, concurrent with a reduction in dendritic spine size, a structural correlate of synapse destabilization.
Figure 4.
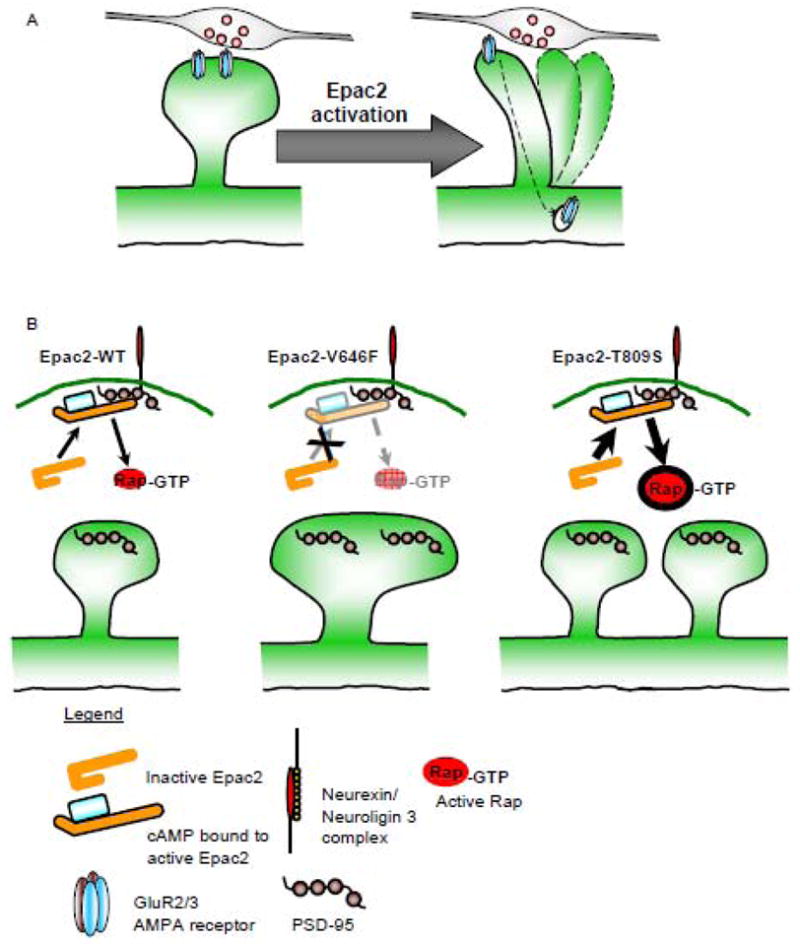
Effect of Epac2 or Epac2 rare non-synonymous variants on dendritic spine remodeling and synaptic function. (A) Activation of Epac2 results in dendritic spine shrinkage, and removal of AMPA GluR2/3 from synapses. These effects are consistent with synapse destabilization. (B) Ectopic expression of Epac2 mutants V646F or T809S result in either an increase in dendritic spine size, accompanied with an increase in PSD-95 content (Epac2-V646F), or an increase in the number of dendritic spines and PSD-95 clusters (Epac2-T809S). These effects on spine density are consistent with previously reported effects of hypo- or hyperactivity of Rap, respectively.
Previous reports have demonstrated that smaller dendritic spines display increased levels of motility. Destabilization of synapses is thought to produce more plastic and labile synaptic connections. In agreement with this idea, time-lapse imaging experiments demonstrated that Epac2 activation not only caused structural destabilization of spines, but also resulted in increased motility and turnover. shRNAi and rescue experiments further demonstrated that Epac2 was specifically required for enhancing dendritic spine dynamics (Woolfrey et al., 2009). Moreover, ectopic expression of mutant Epac2 or Rap dominant-negative constructs revealed that 8-CPT-dependent synapse structural destabilization requires Epac2 GEF activity and Rap activation (Woolfrey et al., 2009).
Previous studies have demonstrated that there is a steady state of dendritic spine morphing and turnover in the adolescent/adult brain (Holtmaat and Svoboda, 2009; Kasai et al., 2010). Interestingly, it has been suggested that neural circuits represent the properties of dendritic spine populations rather than that of a single dendritic spine (Kasai et al., 2010). Indeed, consistent with in vivo studies, in vitro studies have demonstrated that dendritic spine size fluctuate spontaneously over days; that is, dendritic spines grew and shrank in size (Kasai et al., 2010). Despite these long-term changes, termed “intrinsic fluctuations”, the average change in spine size was close to zero, suggesting that populations of spines are in an equilibrium of various sizes (Kasai et al., 2010). It is therefore interesting to speculate that Epac2 activation may promote the dynamic remodeling of such otherwise stable spines, shifting the balance of these fluctuations, resulting in a net decrease in size in a population of spines. The effect of such a destabilization would allow for the active refinement of neural circuits necessary for future remodeling of neural networks. Importantly, as few molecules are known to promote spine shrinkage and increased motility, without elimination, when activated, Epac2 represents a novel molecule that actively achieves this end result.
Removal of AMPA receptors and synaptic depression induced by Epac2
At excitatory synapses, changing the number of synaptic AMPA receptors leads to fine-tuning of synaptic communication (Shepherd and Huganir, 2007). Signaling pathways involved in regulating spine morphology have also been shown to influence AMPA receptor trafficking (Srivastava et al., 2008; Woolfrey et al., 2009; Xie et al., 2007). Previous studies have shown that enlargement of spines results in increased synaptic expression of AMPA receptors and glutamatergic transmission (Kasai et al., 2010; Matsuzaki et al., 2001; Xie et al., 2007). Conversely, shrinkage of dendritic spine size has been linked with removal of the receptor from synapses and a reduction, or depression of AMPA receptor-mediated transmission (Srivastava et al., 2008; Woolfrey et al., 2009; Xie et al., 2005; Zhu et al., 2002; Zhu et al., 2005). In cortical neurons Epac2 was found to co-immunoprecipitate with the GluR2/3, but not with the GluR1, subunits of AMPA receptors. Moreover, specific activation of Epac2 selectively removed GluR2/3 subunit-containing AMPA receptors from synapses (Woolfrey et al., 2009) (Fig. 3, 4A). The remaining synaptic AMPA receptors may thus consist of a larger fraction of GluR1/GluR2-containing receptors, and fewer GluR2/GluR3-containing receptors. It is likely that Epac2 specifically regulates GluR2/3, due to its participation in protein complexes containing the GluR2/3 subunit, mediated in part by Epac2 s interaction with PSD-95. In these complexes, Epac2 can respond to stimuli and activate Rap, which in turn diffuses to nearby GluR2/3 receptors and triggers their internalization.
The functional consequence of AMPA GluR2/3 internalization resulted in reduced amplitude and frequency of AMPA receptor-mediated mEPSCs, indicating Epac2 also depresses glutamatergic transmission. As GluR2/3 is removed from spines, the total amount of functional AMPA receptors is reduced, leading to reduced AMPA receptor mEPSC amplitudes (Woolfrey et al., 2009). Epac2-mediated reduction in mEPSC frequencies could arise from two possibilities: firstly, Epac2 may have a role on the presynaptic side in regulating vesicle release probability, or secondly, that there is an increase in the proportion of silent synapses. Given that knockdown of Epac2 by shRNA in the postsynaptic cell only blocked Epac2-mediated reduction in AMPA receptor mEPSC amplitudes, and the fact that Epac2 is observed in small clusters along the axon (possibly in a subset of presynaptic termini) (Woolfrey et al., 2009), it is likely that this reduction in AMPA receptor mEPSC frequency is driven, in part, by presynapticly located Epac2. Together with the shrinkage of dendritic spine size, Epac2-driven depression of AMPA receptor transmission points to an essential role of Epac2 in the destabilization of functional synapses. Importantly, as Epac2 activation does not induce elimination of synapses, the ability of Epac2 to coordinate both structural and functional destabilization, via a Rap-dependent pathway, provides the most complete cellular pathway underlying this process critical for the refinement of neural circuits (Fig. 3, 4A).
Roles of Epac2 in plasticity and learning
Epac2-dependent changes in dendritic spine size and synaptic GluR2/3 content may contribute to several types of plasticity. A range of effects of 8-CPT incubation have been reported in different neuronal preparations. Short-term and transient presynaptic potentiation has been reported in invertebrate neuromuscular junctions (Cheung et al., 2006; Zhong and Zucker, 2005), the calyx of Held, and in young hippocampal and cortical neurons (Gekel and Neher, 2008). We also found Epac2 immunofluorescence in small puncta along axons, and detected a reduction in mEPSC frequency following treatment with 8-CPT that was not blocked by postsynaptic knockdown of Epac2, both indicative of some presynaptic presence and function. However, the exactly mechanisms by which Epac2 regulates presynaptic function is not known.
Recent studies on Epac signaling in the hippocampus have reported seemingly disparate effects on synaptic potentiation. Using pharmacological activation and blockage of Epac signaling, a recent study found a role for Epac in pituitary adenylate cyclase activating polypeptide- (PACAP-), protein synthesis- and ERK-dependent LTD (Ster et al., 2009). Conversely, another study found Epac activation facilitated β-adrenergic receptor-, high frequency stimulation (HFS)-, protein synthesis-, and ERK-dependent LTP maintenance, without affecting LTP induction (Gelinas et al., 2008). One potential explanation for these diverging effects is that transient destabilization could make synapses more receptive to subsequent activity-dependent potentiating or depressing stimuli, leading to LTP or LTD, respectively. We have previously demonstrated a similar phenomenon by following estrogen treatment of cortical neurons with an activity-like stimulus (acute activation of NMDA receptors) to achieve a form of two-step wiring plasticity (Srivastava et al., 2008). Further exploration of the relationship between Epac2-induced stabilization and subsequent synaptic plasticity-inducing stimuli is warranted.
Modulation of synapses by Epac proteins seems to also affect cognitive functions and behavior. Epac and PKA are jointly required for hippocampal memory retrieval (Ouyang et al., 2008). In addition, Epac activation by 8-CPT enhanced prepulse inhibition of the acoustic startle response (PPI) and short- and long-term and memory in a context-dependent fear conditioning paradigm (Kelly et al., 2009). Together these studies indicate a potential role for Epac2-dependent signaling in synaptic plasticity, and learning, potential via its ability to regulate dendritic spine remodeling.
Dopamine D1/5 receptor regulates synaptic remodeling via Epac2
The classic second messenger cAMP has multiple cellular targets, and substantial work has been devoted to uncovering mechanisms of cAMP signal specificity (Beene and Scott, 2007; Cooper, 2005). Over the past decade, our concept of cAMP signaling has evolved from a simple linear cascade to the realization that multiple factors can influence this signaling pathway. These factors include parallel signaling cascades, compartmentalization/subcellular localization of components that participate in cAMP signaling pathways, and scaffold proteins that facilitate macromolecular protein complexes. The organization of multiple proteins into a macromolecular complex not only allows for signal specificity but signal integration (Cooper, 2005). In cortical pyramidal neurons, dopamine D1/5 receptors (which are Gs coupled and thus signal by increasing cAMP levels) control plasticity bidirectionally, modulating both LTD and LTP (Chen et al., 1996; Huang et al., 2004; Lemon and Manahan-Vaughan, 2006), while applications of cAMP under specific conditions depress synaptic transmission (Frey et al., 1993; Gereau and Conn, 1994). Moreover, postsynaptic cAMP-dependent but PKA-independent mechanisms induce LTD (Yu et al., 2001), depress basal synaptic transmission and reverse potentiation (Otmakhov and Lisman, 2002). Thus, cAMP signaling in general and dopamine signaling in particular have complex implications for synaptic potentiation and depression. The mechanisms underlying the synaptic effects of cAMP signaling are not well understood, but cAMP-dependent GEFs such as Epac2 represent an excellent candidate for mediating cAMP-driven plasticity.
The contribution of dendritic spine remodeling in cAMP-dependent plasticity is not known, and may play an essential role in dopamine-induced synaptic plasticity. As dopamine D1/5 receptors signal by increasing cAMP levels, and cAMP affects LTD in a PKA-independent manner (Yu et al., 2001), one suggestion is that signaling via this receptor may affect both structural and functional plasticity via Epac2. In agreement with this idea, Woolfrey et al. (2009) showed that dopamine D1/5 activation by the specific agonist, SKF-38393, stimulated dendritic Rap signaling. Furthermore, SKF-38393 treatment also caused shrinkage of dendritic spine size, and the removal of surface-expressing AMPA GluR2 (Fig. 3, 4A). Importantly, shRNA specific for Epac2 blocked dopamine D1/5-dependent spine shrinkage and AMPA GluR2 internalization. Epac2 thus mediates neuromodulation by dopamine D1/5, and links dopamine signaling with synapse structural remodeling.
Novel regulation of Epac2 localization and function by Neuroligins
Regulation of Epac2 subcellular localization has been shown to be an important factor in regulating its function (Li et al., 2006). As discussed above, Epac2 participates in a protein complex with PSD-95, which may serve to link it with other signaling proteins or regulators. A recent study has shown that the function of the Rac-GEF, kalirin-7, can be regulated via the adhesion molecule N-cadherin. This interaction is mediated by the scaffold protein afain/AF-6, thus allowing for a macromolecular complex to be formed between these proteins (Xie et al., 2008). This opens up the possibility that Epac2 function may be regulated by such an adhesion protein, mediated by its interaction through PSD-95. One such family of prominent postsynaptic adhesion molecules are the Neuroligins (NL). This family of adhesion molecules, which consist of NL1, NL2, NL3 and NL4, binds to the presynaptic protein neurexin, and are known interactors of PSD-95 (Lise and El-Husseini, 2006). Previous studies have demonstrated that NLs regulate synapse morphology (Lise and El-Husseini, 2006), and the balance between excitatory and inhibitory synapses (Chih et al., 2005; Lise and El-Husseini, 2006). However, the mechanisms by which NLs regulate dendritic spine morphology have not been well described. Owing to the interaction of NLs with PSD-95 and its influence on dendritic spine morphology, it could be posited that NLs are well suited to regulate the activity of GEFs to influence the cytoskeleton. Indeed, in rat forebrain cell lysates, NL3 was found to strongly and specifically interacted with Epac2 (Woolfrey et al., 2009). Epac2 also colocalized with NL3 in spines. Interestingly, Epac2 also associated with NL1 and 2, albeit to a lesser extent than with NL3. An insight into the functional implication of the Epac2 and NL3 interaction was provided using heterologous cells. Ectopic expression of NL3 resulted in the recruitment of Epac2 to the plasma membrane, and enhances its Rap-GEF activity. It is likely that recruitment of Epac2 to the plasma membrane resulted in the Rap-GEF being closer to populations of Rap that can be readily activated (Fig. 3). Binding of Rap to the plasma membrane is required for its proper activation (Li et al., 2006; Takai et al., 2001). As enhanced Epac2 activity promotes spine shrinkage and increases spine dynamics, it suggests that a NL/Epac2/Rap macromolecular complex may offer a pathway that NL3 may act through to modulate dendritic spine remodeling (Fig. 3).
It is of note that previous studies have shown that NLs promotes synapse formation and maturation (Chih et al., 2005; Lise and El-Husseini, 2006). Thus, the ability of NL3 to promote Epac2 function and thereafter spine destabilization is seemingly opposite to previous described function of this protein. One possible answer for this apparent disparity between function of NLs is that signaling via NL/Epac2/Rap serves to promote the dynamic spines that sample presynaptic environment through increased spine motility. Furthermore, NLs may offer a means by which trans-synaptic connections between dendritic spines and newly contacted presynaptic partners may interact. Interestingly, signaling via ephrinB and EphB adhesion molecules have also been shown to perform such opposite functions (Kayser et al., 2008). Importantly, these adhesion molecules promote filopdia motility and motility-dependent synaptogenesis.
Synaptic pathology in autism
Autism spectrum disorders (ASDs) are neurodevelopmental disorders with a strong but complex genetic component and multifactorial etiology (Geschwind, 2008). These disorders are characterized by deficits in social interactions, verbal communication, and the presence of repetitive behavior, and affect 0.9% of children (CDC, 2009). At present, surprisingly little is known about the cellular and circuit level perturbations in autistic brain. Recent evidence from postmortem ASD human brain tissue has revealed an increase in spine density on apical dendrites of pyramidal neurons from cortical layers II and V (Hutsler and Zhang, 2010); an inverse relationship was found between spine density and cognitive function.
To supplement understanding of ASD brain pathology, researchers have turned to diseases that are frequently comorbid with ASDs, including Rett syndrome, fragile X, and tuberous sclerosis (Armstrong, 2005; Irwin et al., 2000; Tavazoie et al., 2005). Work on these disorders benefits from known genetic etiologies and established animal models. A shared feature of all of these disorders is abnormal dendritic spine morphology (Dierssen and Ramakers, 2006; Irwin et al., 2000; Tavazoie et al., 2005). It is important to note that these monogenic diseases do not necessarily reflect the heterogeneity of idiopathic or “pure” autism. Nevertheless, the relatively large data sets acquired from studying these diseases coupled with their exceptionally high rate of coincidence with ASD symptomology may yield important clues for revealing mechanisms of ASD pathology.
Based on genetic, neuropathological and model system studies, a compelling theory suggests that ASDs are primarily synaptic disorders (Geschwind and Levitt, 2007; Walsh et al., 2008a). Several lines of evidence support this view. The onset of autism coincides with a period of intense synapse formation, elimination, and turnover. Indeed, defective synapse remodeling is thought to be a contributing factor to ASDs (Zoghbi, 2003). Secondly, brain circuit miswiring is thought to be hallmark of ASDs (Belmonte et al., 2004; Geschwind, 2008), and synaptic dysfunction could be an important contributor to this malfunction. This notion is supported by the modulatory effect on spine morphology of several autism-associated proteins including the synaptic adhesion proteins NL3 and 4 and the postsynaptic scaffolding proteins Shank2 and 3 (Berkel et al., 2010; Chih et al., 2004; Durand et al., 2007; Tabuchi et al., 2007). How disruptions in the function of these proteins and their synaptic binding partners such as Epac2 may contribute to ASD pathology is explored in the subsequent section.
Implications of Epac2 mutations in autism
The EPAC2 gene resides on the chromosomal region 2q31-32, a locus which putatively encodes genes relevant for ASDs. Four independent studies have concluded that this region is an autism susceptibility locus (Buxbaum et al., 2001; Philippe et al., 1999; Shao et al., 2002). In agreement with these studies, a genome-wide screen identified chromosome 2 as the most significantly linked to autism (IMGSAC, 2001). In addition, anecdotal evidence supports the association of this locus with ASD, as a case study of an autistic patient revealed a 2q31-32 de novo deletion (Gallagher et al., 2003). Collectively, this work established 2q31-32 as a prominent candidate locus for ASD-associated genes.
Guided by these findings, a screen for candidate ASD genes located on chromosome 2q was conducted and resulted in the identification of rare non-synonymous variants in the EPAC2 gene (Bacchelli et al., 2003). These autism-associated variants (M165T, V646F, G706R and T809S) (Fig. 2A) strictly segregated with autistic family members (with the exception of M165T) (Bacchelli et al., 2003). These mutations are located on the first cAMP-binding domain (M165T), the REM domain (V646F), the RA domain (G706R) and the Rap-GEF domain (T809S) of the Epac2 protein (Fig. 2A). However, as these mutations in Epac2 are rare, they do not fully account for the association of this chromosomal region with autism. Nevertheless, rare mutations have been recently implicated in psychiatric disorders (Walsh et al., 2008b), which has lead to the suggestion that an accumulation of these rare variants may alter signaling networks, contributing to the etiology of neuropsychiatric disorders (Walsh et al., 2008b).
As such, these mutations in EPAC2 might offer clues into the role of abnormal synapse remodeling in ASD. Constructs encoding two of these mutant forms of Epac2 elicited synaptic structural phenotypes in primary neuron cultures (Woolfrey et al., 2009). The Epac2-V646F Interestingly mutant increased dendritic spines size (Fig. 4B) and also displayed reduced basal levels of Rap activity in heterologous cells (Woolfrey et al., 2009). On the other hand, the Epac2-T809S mutation resulted in elevated spine density (Fig. 4B), but had no effect on basal Rap levels in heterologous cells (Woolfrey et al., 2009). Interestingly, in the presence of NL3, which displays an ability to activate Epac2 GEF activity, ectopic expression of NL3 failed to activate Rap in the presence of Epac2-V646F, whereas Epac2-T809S and NL3 co-expressed resulted in an increased Rap activation above Epac2-WT and NL3 expression (Woolfrey et al., 2009). Moreover, in cultured cortical neurons, exogenous expression of Epac2-V646F reduced basal levels of phosphorylated B-Raf (a downstream target of Rap signaling), whereas Epac2-T809S increased phosphorylated B-Raf levels, thus mirroring the effects of these mutations in heterologous cells (Woolfrey et al., 2009). When PSD-95 puncta were investigated in the presence of these two mutations, to determine the potential result of altered Rap signaling and altered dendritic spine morphology; Epac2-V646F expression lead to an increase in PSD-95 cluster size, while expression of Epac2-T809S increased the number of PSD-95 clusters (Fig. 4B), paralleling the effects of these mutations on dendritic spine morphology.
Interestingly, examining the location of these mutations may provide an insight into the potential molecular determinates underlying these alterations in Epac2 function. The location of the Epac2-V646F mutation in the REM domain (Fig. 2A) places it close to the “hinge” domain of Epac2; it is conceivable that this mutation may alter the ability of Epac2 to undergo conformation changes into an active state, and thus may reside in an inactivate (hypoactivity) conformation under basal or activated conditions. Conversely, the Epac2-T809S mutation is located in the Rap-GEF domain (Fig. 2A); although there is no alteration in basal activity, under actived conditions, it is possible that this mutation allows for hyperactivation of Epac2 GEF function. Furthermore, we have previously shown that Rap hypoactivity is consistent with enlarged dendritic spines (Xie et al., 2005) while elevated Rap has been linked to increased spine number (Srivastava et al., 2008).
The interaction of Epac2 with NL3 juxtaposes Epac2 with a growing list of synaptic proteins putatively altered in ASDs. Thus, a promising hypothesis is that mutations in one or more of these protein complex members may lead to a disruption in a signaling cascade essential for appropriate synaptic remodeling during development. Mutations of proteins that associate in complexes and that have synaptic modulatory roles may therefore contribute to the pathophysiology of ASDs and may partially explain how heterogeneous genetic alterations can have a common clinical presentation.
Several recent reviews have also highlighted the importance of single-gene mutations in uncovering the mechanisms that contribute to ASD (Geschwind, 2008; Kelleher and Bear, 2008; Sudhof, 2008; Walsh et al., 2008a; Walsh et al., 2008b). NLGN3, NLGN4, SHANK2, SHANK3, NRXN1 and EPAC2 have been implicated in ASD by studies independent from those that examined specific variants. As all these sequence changes/mutations are rare, none of them account for the strong association of their loci with ASD. Larger gene disruptions, such as deletions, chromosomal breakpoints leading to truncations within the gene, or frameshift mutations provide strong association with ASD. These have been detected in SHANK3, NLGN4, and NRXN1, but not in NLGN3 or EPAC2. Rare missense mutations in autistic individuals have been detected in SHANK3, NLGN4, NRXN1, NLGN3 and EPAC2. Missense mutations affect protein function; this has been shown for SHANK3, NLGN3 and now for EPAC2; truncations in NLGN4 and SHANK3 also affect protein function. While NRXN1 mutations and copy number variants have been found in more than one population, several studies on distinct subject pools did not find the originally reported mutations in NLGN3, NLGN4. Similarly, EPAC2 mutations have not been found in another tested proband group. However, this negative result is not surprising given the rare frequency of mutations and the multigenic etiology of the disease.
Potential role of Epac2 in altered cAMP and dopamine signaling in ASD
An alternative theory to abnormal Epac2 signaling being part of a signaling pathway comprising NL3/Epac2/Rap, involves altered cAMP-dependent signaling in autistic brains. Several studies have implicated altered cAMP in fragile X, ASD and fragile X comorbid with ASD (Kelley et al., 2008; Kelley et al., 2007). cAMP signaling is reported to be reduced in fragile X, possible due to regulation of cAMP levels by the FMRP protein (Berry-Kravis and Ciurlionis, 1998). Conversely, some studies have reported elevated cAMP levels in cerebrospinal fluid and blood of autistic patients but not controls (Cook, 1990; Kelley et al., 2008; Winsberg et al., 1980). Further support of a possible dysfunction in cAMP-dependent signaling in ASD comes from recent models that suggest deficits in social cognition in ASD (a core symptom in ASD patients) is characterized by reduced social motivation early in development (Dawson, 2007), which maybe a result of altered dopamine signaling (Neuhaus et al., 2010). Moreover, reduced dopamine signaling has been detected in the frontal cortices by functional imaging studies of ASD patients (Ernst et al., 1997). In addition, pharmacological studies have suggested that both dopamine hyper- and hypo-activation may contribute to the ASD phenotype (Canitano, 2006; Neuhaus et al., 2010; Toda et al., 2006). As Epac2 is emerging as important transducer of dopamine, and cAMP signaling, in cortical neurons, these studies are in support of a theory whereby disruptions in cAMP signaling may play a role in ASDs. Interestingly, suggestions that cAMP levels and dopamine signaling are either hypo- or hyper-active in ASD or comorbid disorders, the effects on cAMP-dependent signaling would very closely mirror the effects of the Epac2-V646F and Epac2-T809S mutations in resulting in hypo- or hyper- Rap activation respectively.
Epac2 in other neuropsychiatric disorders
Alterations in Epac2 signaling are likely to cause defective or excessive synapse destabilization, which may contribute to a variety of CNS disorders. Nicotine self-administration increases Epac2 expression in rat prefrontal cortex, and there is an association of single nucleotide polymorphisms (SNPs) in Epac2 with nicotine dependence (Chen et al., 2004). This is consistent with the crucial role of dopamine receptors and cAMP in addiction, and with the regulation of Epac in non-neuronal cells by D1 receptors (Helms et al., 2006). Recent data also implicated Epac2 and Rap signaling in depression. Postmortem forebrain tissue samples from depressed suicide victims were found to have elevated levels of Epac2 and reduced levels of Rap (Dwivedi et al., 2006). Interestingly, β-adrenergic and serotonergic signaling, two neurotransmitters implicated in depression (Goodnough and Baker, 1994) (Caspi et al., 2003), are both capable of increasing cAMP levels and thus altering the regulation of Epac2. Epac2 also partially mediates signaling by 5-HT2A and 5-HT7A receptors, important in depression (Johnson-Farley et al., 2005). Understanding Epac2 function in spines may expedite the development of treatments for these diseases.
Concluding remarks and future directions
It has become clear that the remodeling of dendritic spines is an essential component of neural circuit refinement, and in vivo imaging studies have linked dendritic spines remodeling with the acquisition and storage of information. Continued efforts to elucidate the molecular mechanisms underlying the remodeling of dendritic spines have not only uncovered essential clues required for normal brain function, but have also revealed that multiple genes associated with neuropathological disorders of the brain encode for proteins that play important roles in dendritic spine remodeling.
In this review, we have described the evidence that Epac2 is a novel cAMP target at synapses (Fig. 2B) that regulates small GTPase signaling in neurons. Activation of an Epac2/Rap pathway leads to both structural and functional destabilization of synapses by means of spine shrinkage accompanied by the removal of synaptic glutamate receptors (Fig. 4A). By showing that Epac2 activation induces spine shrinkage and increased spine motility and turnover, Woolfrey et al. (2009) have identified a protein which actively promotes synapse destabilization. Importantly, Epac2 integrates dopamine D1/5 receptor signaling with synapse destabilization, offering a novel mechanism by which neuromodulators may influence synaptic plasticity.
Interestingly, the identification that Epac2 participates in a macromolecular complex with NL3 may represent an example of a disease-associated signaling pathway important for the pathophysiology of ASDs. Although mutations in single genes may not be sufficient to explain the large number of ASD cases, disruption of a protein function within a signaling cascade may compromise signaling pathways that are essential for formation and maintance of dendritic spines, or for the active remodeling of synaptic structure.
Further work is required to develop a complete understanding of Rap signaling in spines. Whereas the roles of RapGAP such as SAPR and RapGAP1, and the of Rap isoforms, such as Rap2, are beginning to be uncovered, more work is required to understand how these regulators of Rap signaling integrate to control the functions of each isoform of Rap in different regions of the brain.
Future work will have to focus on dissecting the pathways linking Epac2 function with both dendritic spine remodeling and ASD-associated genes. This will reveal novel relationships between genes mutated in complex diseases such as ASD, and further give insight into mechanisms that underlie these neuropathologies. Furthermore, determining the mechanisms underlying Epac2 modulation of cognitive processes and the ability of Epac2 activation to rescue cognitive deficits in disease states will be an important area of future research. Together, such investigations will yield novel therapeutic avenues which could be explored in the development of treatments for ASDs. The development of effective treatments for ASD will be facilitated by a better understanding of the causal links between spine remodeling and socio-cognitive deficits, the availability of better animal models specifically linking spine remodeling with socio-cognitive deficits, and by the identification of novel drug targets based on the links between synapse remodeling and socio-cognitive deficits. While genetic studies in ASD have identified rare mutations in several synaptic proteins, including neuroligin, unfortunately these proteins are difficult to target therapeutically, as they are not readily modulated by small molecules that can be made into orally delivered drugs. To overcome this obstacle, novel druggable downstream targets of neuroligin need to be identified and characterized. Indeed, Epac2 is modulated by neuroligin, is readily targeted by specific “drug-like” cAMP analogs with high likelihood of oral bioavailability and low toxicity in humans, and has been investigated as a drug target in several diseases (Springett et al., 2004).
Acknowledgments
We would like to thank Kelly A. Jones for careful editing of this work. This work was supported by the National Alliance for Autism Research (NAAR), the National Alliance for Research on Schizophrenia and Depression (NARSAD), Alzheimer s Association, NIH grant MH 071316 to P.P., a pre-doctoral American Heart Association (AHA) fellowship to K.M.W., and a post-doctoral AHA fellowship to D.P.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- Bacchelli E, Blasi F, Biondolillo M, Lamb JA, Bonora E, Barnby G, Parr J, Beyer KS, Klauck SM, Poustka A, Bailey AJ, Monaco AP, Maestrini E. Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Mol Psychiatry. 2003;8:916–924. doi: 10.1038/sj.mp.4001340. [DOI] [PubMed] [Google Scholar]
- Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007;19:192–198. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte MK, Allen G, Beckel-Mitchener A, Boulanger LM, Carper RA, Webb SJ. Autism and abnormal development of brain connectivity. J Neurosci. 2004;24:9228–9231. doi: 10.1523/JNEUROSCI.3340-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, Bonin M, Riess A, Engels H, Sprengel R, Scherer SW, Rappold GA. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Ciurlionis R. Overexpression of fragile X gene (FMR-1) transcripts increases cAMP production in neural cells. J Neurosci Res. 1998;51:41–48. doi: 10.1002/(SICI)1097-4547(19980101)51:1<41::AID-JNR4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annu Rev Physiol. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T, Yuste R. Spine motility. Phenomenology, mechanisms, and function. Neuron. 2002;35:1019–1027. doi: 10.1016/s0896-6273(02)00906-6. [DOI] [PubMed] [Google Scholar]
- Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- Bos JL. Linking Rap to cell adhesion. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Bose M, Munoz-Llancao P, Roychowdhury S, Nichols JA, Jakkamsetti V, Porter B, Byrapureddy R, Salgado H, Kilgard MP, Aboitiz F, Dagnino-Subiabre A, Atzori M. Effect of the environment on the dendritic morphology of the rat auditory cortex. Synapse. 2009;64:97–110. doi: 10.1002/syn.20710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Silverman JM, Smith CJ, Kilifarski M, Reichert J, Hollander E, Lawlor BA, Fitzgerald M, Greenberg DA, Davis KL. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet. 2001;68:1514–1520. doi: 10.1086/320588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canitano R. Self injurious behavior in autism: clinical aspects and treatment with risperidone. J Neural Transm. 2006;113:425–431. doi: 10.1007/s00702-005-0337-x. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- CDC. Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ. 2009;58:1–20. [PubMed] [Google Scholar]
- Chen X, Wu B, Kendler KS. Association study of the Epac gene and tobacco smoking and nicotine dependence. Am J Med Genet B Neuropsychiatr Genet. 2004;129:116–119. doi: 10.1002/ajmg.b.30040. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang PY, Ghosh A. Regulation of cortical dendrite development by Rap1 signaling. Mol Cell Neurosci. 2005;28:215–228. doi: 10.1016/j.mcn.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Chen Z, Ito K, Fujii S, Miura M, Furuse H, Sasaki H, Kaneko K, Kato H, Miyakawa H. Roles of dopamine receptors in long-term depression: enhancement via D1 receptors and inhibition via D2 receptors. Receptors Channels. 1996;4:1–8. [PubMed] [Google Scholar]
- Cheung U, Atwood HL, Zucker RS. Presynaptic effectors contributing to cAMP-induced synaptic potentiation in Drosophila. Journal of neurobiology. 2006;66:273–280. doi: 10.1002/neu.20218. [DOI] [PubMed] [Google Scholar]
- Chih B, Afridi SK, Clark L, Scheiffele P. Disorder-associated mutations lead to functional inactivation of neuroligins. Hum Mol Genet. 2004;13:1471–1477. doi: 10.1093/hmg/ddh158. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- Cook EH. Autism: review of neurochemical investigation. Synapse. 1990;6:292–308. doi: 10.1002/syn.890060309. [DOI] [PubMed] [Google Scholar]
- Cooper DM. Compartmentalization of adenylate cyclase and cAMP signalling. Biochem Soc Trans. 2005;33:1319–1322. doi: 10.1042/BST0331319. [DOI] [PubMed] [Google Scholar]
- Dawson GBR, editor. Social brain circuitry in autism. Guilford Press; New York: 2007. [Google Scholar]
- Dierssen M, Ramakers GJ. Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav. 2006;5(Suppl 2):48–60. doi: 10.1111/j.1601-183X.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- DSM-IV. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: 2000. [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Roge B, Heron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Mondal AC, Rizavi HS, Faludi G, Palkovits M, Sarosi A, Conley RR, Pandey GN. Differential and brain region-specific regulation of Rap-1 and Epac in depressed suicide victims. Arch Gen Psychiatry. 2006;63:639–648. doi: 10.1001/archpsyc.63.6.639. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nature cell biology. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D, Cohen RM. Low medial prefrontal dopaminergic activity in autistic children. Lancet. 1997;350:638. doi: 10.1016/s0140-6736(05)63326-0. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Frost NA, Kerr JM, Lu HE, Blanpied TA. A network of networks: cytoskeletal control of compartmentalized function within dendritic spines. Curr Opin Neurobiol. 2010 doi: 10.1016/j.conb.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher L, Becker K, Kearney G, Dunlop A, Stallings R, Green A, Fitzgerald M, Gill M. Brief report: A case of autism associated with del(2)(q32.1q32.2) or (q32.2q32.3) J Autism Dev Disord. 2003;33:105–108. doi: 10.1023/a:1022242807513. [DOI] [PubMed] [Google Scholar]
- Gekel I, Neher E. Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J Neurosci. 2008;28:7991–8002. doi: 10.1523/JNEUROSCI.0268-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas JN, Banko JL, Peters MM, Klann E, Weeber EJ, Nguyen PV. Activation of exchange protein activated by cyclic-AMP enhances long-lasting synaptic potentiation in the hippocampus. Learning & memory (Cold Spring Harbor, NY. 2008;15:403–411. doi: 10.1101/lm.830008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gereau RWt, Conn PJ. Potentiation of cAMP responses by metabotropic glutamate receptors depresses excitatory synaptic transmission by a kinase-independent mechanism. Neuron. 1994;12:1121–1129. doi: 10.1016/0896-6273(94)90319-0. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135:391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- Goodnough DB, Baker GB. 5-Hydroxytryptamine2 and beta-adrenergic receptor regulation in rat brain following chronic treatment with desipramine and fluoxetine alone and in combination. J Neurochem. 1994;62:2262–2268. doi: 10.1046/j.1471-4159.1994.62062262.x. [DOI] [PubMed] [Google Scholar]
- Gorman JM, Docherty JP. A hypothesized role for dendritic remodeling in the etiology of mood and anxiety disorders. J Neuropsychiatry Clin Neurosci. 2010;22:256–264. doi: 10.1176/jnp.2010.22.3.256. [DOI] [PubMed] [Google Scholar]
- Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Gan WB. Two-photon imaging of synaptic plasticity and pathology in the living mouse brain. NeuroRx. 2006;3:489–496. doi: 10.1016/j.nurx.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L710–L722. doi: 10.1152/ajplung.00486.2004. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Simpson E, Kellendonk C, Kandel ER. Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proc Natl Acad Sci U S A. 2004;101:3236–3241. doi: 10.1073/pnas.0308280101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Imamura Y, Matsumoto N, Kondo S, Kitayama H, Noda M. Possible involvement of Rap1 and Ras in glutamatergic synaptic transmission. Neuroreport. 2003;14:1203–1207. doi: 10.1097/00001756-200307010-00003. [DOI] [PubMed] [Google Scholar]
- IMGSAC. A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet. 2001;69:570–581. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Johnson-Farley NN, Kertesy SB, Dubyak GR, Cowen DS. Enhanced activation of Akt and extracellular-regulated kinase pathways by simultaneous occupancy of Gq-coupled 5-HT2A receptors and Gs-coupled 5-HT7A receptors in PC12 cells. J Neurochem. 2005;92:72–82. doi: 10.1111/j.1471-4159.2004.02832.x. [DOI] [PubMed] [Google Scholar]
- Jones KA, Srivastava DP, Allen JA, Strachan RT, Roth BL, Penzes P. Rapid modulation of spine morphology by the 5-HT2A serotonin receptor through kalirin-7 signaling. Proc Natl Acad Sci U S A. 2009;106:19575–19580. doi: 10.1073/pnas.0905884106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, Neubert TA. Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Kayser MS, Nolt MJ, Dalva MB. EphB receptors couple dendritic filopodia motility to synapse formation. Neuron. 2008;59:56–69. doi: 10.1016/j.neuron.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kelley DJ, Bhattacharyya A, Lahvis GP, Yin JC, Malter J, Davidson RJ. The cyclic AMP phenotype of fragile X and autism. Neurosci Biobehav Rev. 2008;32:1533–1543. doi: 10.1016/j.neubiorev.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DJ, Davidson RJ, Elliott JL, Lahvis GP, Yin JC, Bhattacharyya A. The Cyclic AMP Cascade Is Altered in the Fragile X Nervous System. PLoS ONE. 2007;2:e931. doi: 10.1371/journal.pone.0000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MP, Stein JM, Vecsey CG, Favilla C, Yang X, Bizily SF, Esposito MF, Wand G, Kanes SJ, Abel T. Developmental etiology for neuroanatomical and cognitive deficits in mice overexpressing Galphas, a G-protein subunit genetically linked to schizophrenia. Mol Psychiatry. 2009;14:398–415. 347. doi: 10.1038/mp.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon N, Manahan-Vaughan D. Dopamine D1/D5 receptors gate the acquisition of novel information through hippocampal long-term potentiation and long-term depression. J Neurosci. 2006;26:7723–7729. doi: 10.1523/JNEUROSCI.1454-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Sweet RA. Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J Clin Invest. 2009;119:706–716. doi: 10.1172/JCI37335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Asuri S, Rebhun JF, Castro AF, Paranavitana NC, Quilliam LA. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J Biol Chem. 2006;281:2506–2514. doi: 10.1074/jbc.M508165200. [DOI] [PubMed] [Google Scholar]
- Lise MF, El-Husseini A. The neuroligin and neurexin families: from structure to function at the synapse. Cell Mol Life Sci. 2006;63:1833–1849. doi: 10.1007/s00018-006-6061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska AK, Newton JR, Sur M. Remodeling of synaptic structure in sensory cortical areas in vivo. J Neurosci. 2006;26:3021–3029. doi: 10.1523/JNEUROSCI.4454-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron. 2003;39:309–325. doi: 10.1016/s0896-6273(03)00404-5. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Neuhaus E, Beauchaine TP, Bernier R. Neurobiological correlates of social functioning in autism. Clin Psychol Rev. 2010;30:733–748. doi: 10.1016/j.cpr.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Bosch M, Hayashi Y. The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: a potential molecular identity of a synaptic tag? Physiology (Bethesda, Md. 2009;24:357–366. doi: 10.1152/physiol.00029.2009. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Lisman JE. Postsynaptic application of a cAMP analogue reverses long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2002;87:3018–3032. doi: 10.1152/jn.2002.87.6.3018. [DOI] [PubMed] [Google Scholar]
- Ouyang M, Zhang L, Zhu JJ, Schwede F, Thomas SA. Epac signaling is required for hippocampus-dependent memory retrieval. Proc Natl Acad Sci U S A. 2008;105:11993–11997. doi: 10.1073/pnas.0804172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/s0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, Sheng M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem. 2004;279:21003–21011. doi: 10.1074/jbc.M400103200. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, Srivastava DP. Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol. 2008;18:405–413. doi: 10.1016/j.tcb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L, Penet C, Feingold J, Brice A, Leboyer M. Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet. 1999;8:805–812. doi: 10.1093/hmg/8.5.805. [DOI] [PubMed] [Google Scholar]
- Pickett J, London E. The neuropathology of autism: a review. Journal of neuropathology and experimental neurology. 2005;64:925–935. doi: 10.1097/01.jnen.0000186921.42592.6c. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–199. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature. 2008;455:124–127. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Prakash B, Wolf E, Rueppel A, de Rooij J, Bos JL, Wittinghofer A. Structure and regulation of the cAMP-binding domains of Epac2. Nat Struct Biol. 2003;10:26–32. doi: 10.1038/nsb878. [DOI] [PubMed] [Google Scholar]
- Ryu J, Futai K, Feliu M, Weinberg R, Sheng M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal-regulated kinase signaling, enhanced long-term depression, and impaired spatial learning and fear extinction. J Neurosci. 2008;28:8178–8188. doi: 10.1523/JNEUROSCI.1944-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines, synaptic plasticity and neuronal survival: activity shapes dendritic spines to enhance neuronal viability. Eur J Neurosci. 2010;31:2178–2184. doi: 10.1111/j.1460-9568.2010.07270.x. [DOI] [PubMed] [Google Scholar]
- Shao Y, Raiford KL, Wolpert CM, Cope HA, Ravan SA, Ashley-Koch AA, Abramson RK, Wright HH, DeLong RG, Gilbert JR, Cuccaro ML, Pericak-Vance MA. Phenotypic homogeneity provides increased support for linkage on chromosome 2 in autistic disorder. Am J Hum Genet. 2002;70:1058–1061. doi: 10.1086/339765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Murphy GG. cAMP and memory: a seminal lesson from Drosophila and Aplysia. Brain Res Bull. 1999;50:441–442. doi: 10.1016/s0361-9230(99)00124-0. [DOI] [PubMed] [Google Scholar]
- Springett GM, Kawasaki H, Spriggs DR. Non-kinase second-messenger signaling: new pathways with new promise. Bioessays. 2004;26:730–738. doi: 10.1002/bies.20057. [DOI] [PubMed] [Google Scholar]
- Srivastava DP, Woolfrey K, Jones KA, Shum CY, Lash LL, Swanson GT, Penzes P. Rapid enhancement of two-step wiring plasticity by estrogen and NMDA receptor activity. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0801581105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ster J, de Bock F, Bertaso F, Abitbol K, Daniel H, Bockaert J, Fagni L. Epac mediates PACAP-dependent long-term depression in the hippocampus. The Journal of physiology. 2009;587:101–113. doi: 10.1113/jphysiol.2008.157461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ster J, De Bock F, Guerineau NC, Janossy A, Barrere-Lemaire S, Bos JL, Bockaert J, Fagni L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc Natl Acad Sci U S A. 2007;104:2519–2524. doi: 10.1073/pnas.0611031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork PJ. Does Rap1 deserve a bad Rap? Trends in biochemical sciences. 2003;28:267–275. doi: 10.1016/S0968-0004(03)00087-2. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Sudhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- Takashima S, Ieshima A, Nakamura H, Becker LE. Dendrites, dementia and the Down syndrome. Brain & development. 1989;11:131–133. doi: 10.1016/s0387-7604(89)80082-8. [DOI] [PubMed] [Google Scholar]
- Tau GZ, Peterson BS. Normal development of brain circuits. Neuropsychopharmacology. 2010;35:147–168. doi: 10.1038/npp.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Toda Y, Mori K, Hashimoto T, Miyazaki M, Nozaki S, Watanabe Y, Kuroda Y, Kagami S. Administration of secretin for autism alters dopamine metabolism in the central nervous system. Brain Dev. 2006;28:99–103. doi: 10.1016/j.braindev.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- van Spronsen M, Hoogenraad CC. Synapse pathology in psychiatric and neurologic disease. Curr Neurol Neurosci Rep. 2010;10:207–214. doi: 10.1007/s11910-010-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CA, Morrow EM, Rubenstein JL. Autism and brain development. Cell. 2008a;135:396–400. doi: 10.1016/j.cell.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science (New York, NY. 2008b;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Wilbrecht L, Holtmaat A, Wright N, Fox K, Svoboda K. Structural plasticity underlies experience-dependent functional plasticity of cortical circuits. J Neurosci. 2010;30:4927–4932. doi: 10.1523/JNEUROSCI.6403-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsberg BG, Sverd J, Castells S, Hurwic M, Perel JM. Estimation of monoamine and cyclic-AMP turnover and amino acid concentrations of spinal fluid in autistic children. Neuropediatrics. 1980;11:250–255. doi: 10.1055/s-2008-1071393. [DOI] [PubMed] [Google Scholar]
- Woolfrey KM, Srivastava DP, Photowala H, Yamashita M, Barbolina MV, Cahill ME, Xie Z, Jones KA, Quilliam LA, Prakriya M, Penzes P. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nature neuroscience. 2009;12:1275–1284. doi: 10.1038/nn.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Huganir RL, Penzes P. Activity-dependent dendritic spine structural plasticity is regulated by small GTPase Rap1 and its target AF-6. Neuron. 2005;48:605–618. doi: 10.1016/j.neuron.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Xie Z, Photowala H, Cahill ME, Srivastava DP, Woolfrey KM, Shum CY, Huganir RL, Penzes P. Coordination of synaptic adhesion with dendritic spine remodeling by AF-6 and kalirin-7. J Neurosci. 2008;28:6079–6091. doi: 10.1523/JNEUROSCI.1170-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. J Neurosci. 2008;28:5740–5751. doi: 10.1523/JNEUROSCI.3998-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara Y, De Roo M, Muller D. Dendritic spine formation and stabilization. Curr Opin Neurobiol. 2009;19:146–153. doi: 10.1016/j.conb.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Yu TP, McKinney S, Lester HA, Davidson N. Gamma-aminobutyric acid type A receptors modulate cAMP-mediated long-term potentiation and long-term depression at monosynaptic CA3-CA1 synapses. Proc Natl Acad Sci U S A. 2001;98:5264–5269. doi: 10.1073/pnas.091093998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Zucker RS. cAMP acts on exchange protein activated by cAMP/cAMP-regulated guanine nucleotide exchange protein to regulate transmitter release at the crayfish neuromuscular junction. J Neurosci. 2005;25:208–214. doi: 10.1523/JNEUROSCI.3703-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP, Velamoor V, Auberson YP, Osten P, van Aelst L, Sheng M, Zhu JJ. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905–916. doi: 10.1016/j.neuron.2005.04.037. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron. 2005a;46:181–189. doi: 10.1016/j.neuron.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Yang G, Kwon E, Gan WB. Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature. 2005b;436:261–265. doi: 10.1038/nature03715. [DOI] [PubMed] [Google Scholar]