

Abstract
Profound astrogliosis coincident with neuronal cell loss is universally described in human and animal models of temporal lobe epilepsy (TLE). In the kainic acid-induced status epilepticus (SE) model of TLE, astrocytes in the hippocampus become reactive soon after SE and before the onset of spontaneous seizures. To determine if astrocytes in the hippocampus exhibit changes in function soon after SE, we recorded from SR101-labeled astrocytes using the whole-cell patch technique in hippocampal brain slices prepared from control and kainic-acid treated rats. Glutamate transporter-dependent currents were found to have significantly faster decay time kinetics and in addition, dye coupling between astrocytes was substantially increased. Consistent with an increase in dye coupling in reactive astrocytes, immunoblot experiments demonstrated a significant increase in both glial fibrillary acidic protein (GFAP) and connexin 43, a major gap junction protein expressed by astrocytes. In contrast to what has been observed in resected tissue from patients with refractory epilepsy, changes in potassium currents were not observed shortly after KA-induced SE. While many changes in neuronal function have been identified during the initial period of low seizure probability in this model of TLE, the present study contributes to the growing body of literature suggesting a role for astrocytes in the process of epileptogenesis.
Keywords: Astrocytes, temporal lobe epilepsy, gap junction, glutamate transport
INTRODUCTION
Temporal lobe epilepsy (TLE) is a debilitating seizure disorder that is often resistant to drug therapy. Patients with TLE have complex-partial seizures that can generalize, are prone to increase in severity over time, and are often associated with the development of cognitive impairments (Engel, 1996). One of the most robust findings, universally described in human and animal models of TLE, is neuronal cell loss coupled with profound astrogliosis throughout the temporal lobe (Binder and Steinhauser, 2006). This astrogliosis is characterized by hypertrophy of primary astrocyte processes, dramatic increases in the expression of the intermediate filament protein glial fibrillary acidic protein (GFAP) and, in some cases, a disruption in domain organization (Oberheim et al., 2008).
Astrocytes are intimately linked to the function of neurons and can limit their excitability, in part, by maintaining low concentrations of extracellular potassium ([K+]o) and glutamate (Binder and Steinhauser, 2006; Seifert et al., 2006; Wetherington et al., 2008). Astrocytes spatially buffer [K+]o via inwardly rectifying potassium (KIR) channels and by moving K+ though a gap junction coupled syncytium (Kofuji and Newman, 2004; Orkand et al., 1966; Wallraff et al., 2006) (Gutnick and Prince, 1981; Newman, 1993; Sontheimer, 1994). Astrocytes also express the Na+-dependent glutamate transporters GLAST and GLT-1, which are responsible for the majority of glutamate uptake in the brain (Lehre et al., 1995; Rothstein et al., 1994) (Marcaggi et al., 2003; Tzingounis and Wadiche, 2007). Alterations in the protein expression/function of KIR channels and potassium buffering (Bordey and Sontheimer, 1998; Bordey and Spencer, 2004; Buono et al., 2004; Gabriel et al., 1998b; Kivi et al., 2000), gap junction coupling (Naus et al., 1991), and glutamate transport (Hoogland et al., 2004; Mathern et al., 1999; Proper et al., 2002) have been reported to be associated with epilepsy. Each of these alterations are hypothesized to influence hyperexcitability once epilepsy is established, but it is difficult to ascertain from these studies whether they were present during the process of epileptogenesis.
To determine which properties of astrocytes are altered early in the development of epilepsy, we recorded from astrocytes in the acute hippocampal brain slice preparation obtained from rats soon after systemic treatment with kainic acid (KA). The KA model of status epilepticus (SE) is a well-characterized animal model of TLE (Ben-Ari and Cossart, 2000; Hellier et al., 1998; Williams et al., 2009). Following the initial insult to the CNS that results from the acute KA-induced SE, there is a period during which astrogliosis and cell death is observed, but the probability of behavioral seizures is exceedingly low (Williams et al., 2009). The chronic occurrence of spontaneous convulsive and non-convulsive seizures of temporal lobe origin increases in both frequency and severity over time in this animal model of TLE. The mean duration until the first convulsive seizure is observed is 18 days after SE (Williams et al., 2009). The present experiments, performed after SE during a period of low seizure probability, reveal significant increases in the extent of gap junction coupling of astrocytes and a concomitant increase in connexin 43 expression. In addition, we observed a substantial decrease in the time course of decay of the glutamate transport current, and surprisingly, no change in the KIR current. This pattern of changes observed in hippocampal astrocytes in this status model of TLE differ markedly from that which is observed during the latent period in an albumin model of focal epilepsy (David et al., 2009). These observations suggest that there are fundamental changes in astrocytes early in the epileptic process that may be different depending upon the type of epilepsy studied. Thus, astrocyte function should be carefully considered within theoretical frameworks of both the development and treatment of epilepsy.
MATERIALS AND METHODS
Animals
All experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the University of Utah. For KA-induced SE, male Sprague-Dawley rats 150 – 180 g were injected with kainic acid (Tocris Bioscience, Ellisville, MO) as described previously (Hellier et al., 1998; Smith et al., 2007; Williams et al., 2009). Briefly, injections of kainic acid (5 or 10 mg/kg, i.p.) occurred once every hour until animals exhibited a behavioral stage 4 or 5 seizure, as defined by the Racine scale (Racine, 1972). As described previously, each animal used in the study received its own individual dose of KA so that similar behavioral seizures could be observed (Hellier et al., 1998; Smith et al., 2007) For some control animals, saline (0.9%) was substituted for kainic acid. Following the onset of the first stage 4 or 5 seizure, the time and stage of subsequent seizures were observed. To be included in the study, rats had to exhibit at least one stage 4 or 5 seizure every hour during the subsequent 3.5 hours. At the conclusion of behavioral monitoring, rats were given an injection of 0.9% saline (1 ml, s.c.) to prevent dehydration, but were not given any compounds to prevent further seizures (e.g. diazepam). Rats were then housed individually in a temperature- and light-controlled (12 h light/dark cycle) environment and allowed access to food and water ad libitum until experimentation. Other than the initial seizures following KA injection, none of the rats used in the experiments were observed to have a seizure prior to sacrifice.
Immunoblotting
Crude membrane preparations were prepared from rats 7 days following KA-induced SE, or from saline-injected/weight-matched controls (200-225 g). The tissue was homogenized in ice-cold lysis buffer (0.32 M sucrose, 10 mM Tris-HCl, 1 mM EDTA, supplemented with a mammalian protease arrest protease inhibitor cocktail; G-Biosciences, St. Louis, MO) using glass-Teflon tissue grinders. The homogenate was centrifuged (10 min, 1000 x g, at 4°C) to remove nuclei and debris. The resulting supernatant was centrifuged (15 min, 22,000 x g, 4°C). The pellets were resuspended in suspension buffer (10 mM TRIS-HCl, 1 mM EDTA, supplemented with a mammalian protease arrest protease inhibitor cocktail (G-Biosciences, St. Louis, MO) and frozen (−80°C) until use. Protein analysis was performed using the Bradford assay (Bio-Rad, Hercules, CA). Samples were diluted into loading buffer (final concentration: 2.25% SDS, 18% glycerol, 180 mM Tris base (pH 6.8), and bromophenol blue). Western blot analysis was preformed as described previously (Alex et al., 2006; Riddle et al., 2002). Briefly, equal amounts of protein were loaded onto SDS-polyacrylamide gels. After electrophoresis, samples were transferred to polyvinylidene difluoride membranes (PerkinElmer Life Sciences, Boston, MA) and blocked in StartingBlock blocking buffer (Pierce, Rockford, IL). Membranes were then probed with primary antibodies against GFAP (#556328, BD Pharmingen), connexin-43 (#35-5000, Invitrogen), connexin-30 (#71-2200, Invitrogen), GLT-1 (#AB1783, Chemicon), and GLAST (#AB1782, Chemicon) overnight and then washed in Tris buffered saline with Tween (0.05% Tween 20). Membranes were then incubated for 1 hour with species-specific secondary antibodies conjugated with horseradish peroxidase. Antigen-antibody complexes were visualized by chemiluminescence (PerkinElmer Life Sciences, Boston, MA). Bands were quantified with an Alpha-Innotech FluorChem SP digital imager with AlphaEase software. As a lane loading control, blots were stripped and re-probed with an anti-actin antibody (#sc-1616, Santa Cruz Biotechnology).
Slice Preparation and Labeling with SR101
Acute brain slices were prepared from rats 7-16 days following KA-induced SE, or from saline-injected/weight-matched controls (200-225 g) in a manner similar to previously described methods (Otto et al., 2006; West et al., 2007). Briefly, rats were deeply anesthetized with sodium pentobarbital (25mg/kg, i.p.) and brains were rapidly dissected and placed in ice-cold (4 °C) oxygenated sucrose Ringer’s solution (95% O2/5% CO2) containing (in mM): 200 sucrose, 26 NaHCO3, 10 glucose, 3 KCl, and 1.4 NaH2PO4. Brains were then trimmed, cut down the midline, and one hemisphere was glued dorsal side down to the mounting disk of a Vibratome tissue slicer (Vibratome, St. Louis, MO). Horizontal brain slices (300 μm) containing both the medial entorhinal cortex and hippocampus were cut in oxygenated sucrose Ringer’s solution (4 °C). Following sectioning, slices were transferred to a holding chamber that contained 0.5 μM SR101 (Sigma-Aldrich, St. Louis, MO) in normal oxygenated Ringer’s solution (34 °C, also referred to in this study as artificial cerebrospinal fluid or ACSF) containing (in mM): 126 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 1.4 NaH2PO4, 2 CaCl2, and 1 MgCl2. Following incubation in SR101-ACSF (15 min maximum), slices were transferred to a separate holding chamber containing oxygenated Ringer’s solution without SR101 (34 °C) for 45 min (for a total recovery period of 1 hr) and at room temperature thereafter until experimentation. The SR101 labeling protocol was adopted from Kafitz et al. (Kafitz et al., 2008). The normal Ringer’s solution was continuously bubbled with 95% O2/5% CO2, the pH was adjusted to between 7.35 and 7.40 with HCl or NaOH, and the osmolarity was adjusted to between 300 and 305 mOsm with double distilled water. For experiments using BaCl2, NaH2PO4 was omitted from the Ringer’s solution to prevent precipitation. For low bicarbonate/low pH gap junction blocking experiments, Ringer’s solution was made with 3.6 mM NaHCO3, and osmolarity (~300 mOsm) and pH (~6.4) were corrected by adding appropriate amounts of NaCl and HCl, respectively.
Electrophysiology
Brain slices were transferred to an immersion-style recording chamber (Warner Instruments, Hamden, CT, USA) and perfused with oxygenated room temperature Ringer’s solution at a rate of ~3.0 ml/min. Patch electrodes were pulled from borosilicate glass capillaries (Schott #8250 glass, World Precision Instruments, Sarasota, FL) using a micropipette electrode puller (P-97, Sutter Instruments, Novato, CA) and had impedances of 5-7 MΩ when filled with an internal solution containing (in mM): 140 K-gluconate, 20 HEPES, 10 EGTA, 3 Na2ATP, 1 MgCl2, 0.3 NaGTP, and 0.2% biocytin. Osmolarity of internal solution was adjusted to 285 - 290 mOsm with double distilled water and the pH was adjusted to 7.28 - 7.3 with 1 M KOH.
The stratum radiatum of CA1 was visualized with a 40x water immersion objective (NA 0.8, Carl Zeiss, Thornwood, NY) using infrared differential interference contrast (IR-DIC) microscopy on an upright Axioskop2 microscope (Carl Zeiss, Thornwood, NY). Images were captured using a CCD camera (ASI Imaging, Eugene, OR) and video capture card (FG-7, Scion Corporation, Frederick, MD). Only cells displaying SR101 fluorescence (excitation 586 nm, emission 605 nm) were targeted for electrophysiology. Whole-cell patch-clamp recordings were obtained in the voltage-clamp configuration using an Axopatch 200B amplifier and the CLAMPEX 9 software package interfaced to a Digidata 1322A data acquisition board (Axon Instruments, Union City, CA). Signals were acquired at 10 kHz and filtered at 2 kHz for off-line analysis using Clampfit 9 or Origin 8 (Originlab Corporation, Northampton, MA). Series resistance was not compensated. Resting membrane potential was determined in the I = 0 mode and periodically monitored during the experiment. Cells with variations in resting membrane potential (RMP) greater than ± 10 mV (before drug application) were omitted from study. Membrane (Rm) and access (Ra) resistances were measured using the “membrane test” protocol built into CLAMPEX 9 and only stable recordings were included in the study.
Astrocytes were voltage-clamped at −80 mV and easily distinguished from neurons by their hyperpolarized membrane potential (close to EK), low input resistance (less than ~50 MΩ), and lack of voltage gated action currents at depolarized potentials. Only one astrocyte was patched per slice.
Schaffer collateral stimulation and induction of synaptically activated glutamate transporter-dependent currents (STCs)
Whole-cell glutamate transporter-dependent currents were induced in astrocytes using a bipolar tungsten electrode placed in the Schaffer collateral pathway ~300-400 μm away from the recording electrode. Input-output curves of stimulation evoked currents were determined for each astrocyte. Stimulation strength for each experiment was set at 2.5 times the threshold stimulation amplitude, unless otherwise indicated. STCs were pharmacologically isolated from the whole-cell current induced in normal Ringer’s by blocking KIR dependent currents with 200 μM BaCl2 and blocking post-synaptic receptors with 100 μM APV, 20 μM CNQX, and 10 μM bicuculline, as previously described (Bergles and Jahr, 1997; Diamond and Jahr, 1997; Luscher et al., 1998). The amplitude and time course of STCs were determined using Mini Analysis (Synaptosoft, Fort Lee, NJ). From the time of peak current, the rise time was calculated as the time it took for the current to rise from baseline to peak and the decay time as the time it took for the current to decay from 70% of the peak to 30% of the baseline. Halfwidth was calculated as the time period occurring between the 50% values of rise and decay times.
Drugs
Drugs were bath applied by a gravity fed perfusion system and recordings were made only after complete solution exchanges as determined through experimentation (time was >5 min). All drugs were purchased from Sigma-Aldrich (St. Louis, MO) or Tocris Biosciences, unless otherwise indicated.
Statistics
Data are expressed as mean ± SEM, unless otherwise indicated. Statistical significance was determined with Student’s t test for independent samples (unpaired), unless otherwise indicated.
Intracellular labeling and immunohistochemistry
Following electrophysiology experiments, slices were fixed in freshly prepared 4% paraformaldehyde for 24 hours at 4 °C, and subsequently stored in 1x phosphate buffered saline (PBS) for 24 to 48 hours. For whole mount processing of biocytin and GFAP, slices were rinsed twice in 1x PBS and blocked with 4.5% normal goat serum in 0.5% PBT (1x PBS + 0.5 % triton x-100) for one hour. Slices were then incubated overnight at room temperature in blocking buffer containing streptavidin conjugated to Alexa 546 (1:500, Invitrogen, Carlsbad, CA) and GFAP conjugated to Alexa 488 (1:500, Chemicon/Millipore, Billerica, MA). Slices were then rinsed in 1x PBS and incubated in 4′,6-diamidino-2-phenylindole (DAPI, 0.1 μg/ml, Santa Cruz Biotechnology, Santa Cruz, CA) for 30 min, followed by additional wash steps in 1x PBS. To promote tissue clearing, slices were immersed in 95% glycerol/5% PBS for several minutes before being cover slipped in Prolong gold anti-fade reagent (Invitrogen, Carlsbad, CA).
Confocal imaging
Images were captured with an Olympus FV1000 confocal microscope (IX81 inverted microscope, Olympus America, Center Valley, PA) using a 20x/NA 0.75 or 60x/NA 1.35 oil immersion objective. Initially, positively labeled cells were identified using a 100W halogen lamp and a filter set for TRITC. Confocal laser scanning mode was then used to image in the z-axis. Laser output, photomultiplier, gain, and offset settings were corrected to minimize saturated pixels and maximize signal with respect to noise. Each fluorescent channel was excited and captured sequentially to minimize bleed through.
Cell counts
Two dimensional z-projections (30 to 40 z-sections, 1.5 μm steps) were analyzed with ImageJ (NIH, Bethesda, MD) and the “colocalization” plugin (http://rsb.info.nih.gov/ij/plugins/colocalization.html). The colocalization plugin allowed the use of the particle analysis function in Image J to count only the pixels representing areas of colocalization between two channels (see Results).
RESULTS
Astrogliosis is Present in the Hippocampus Soon After SE
One of the prominent hallmarks of astrogliosis is a change in the immunoreactivity for the astrocyte specific intracellular filament marker GFAP. Astrogliosis is known to be present in epileptic and injured tissue, but we sought to first determine if astrogliosis was present during the latent period, when spontaneous seizures are relatively rare, but the initial damage to the hippocampus has already occurred (Hellier et al., 1998; Smith et al., 2007). We routinely observed immunoreactivity for GFAP throughout the hippocampus from both control and KA-induced SE rats (Figure 1 A and B), however, seven days following KA-administration, GFAP positive cells displayed characteristics typical of reactive astrocytes including hypertrophied morphology and increased immunoreactivity (Figure 1B). Immunoblots also demonstrated a significant increase in GFAP expression in the KA-treated animals (Figure 1 C and D). Furthermore, a similar degree of astrogliosis was also observed up to 5 weeks after SE (data not shown). Therefore, reactive gliosis occurs early in the process of epileptogenesis in this animal model and GFAP expression remains elevated well after the initial injury.
Figure 1. Reactive astrocytes are present in the hippocampus one week following KA-induced SE.

(A) Immunoreactivity for GFAP is observed throughout the hippocampus and cortex in horizontal brain sections from both control and KA-treated rats. dg = dentate gyrus, ecx = medial entorhinal cortex. (B) Close up of CA1 and stratum radiatum from the area indicated by the white box in (A). Sections from KA-treated rats (right) show characteristics typical of reactive astrocytes including a hypertrophied morphology and an increase in the immunoreactivity for GFAP. Scale bar, 50 μm. (C) Western blot of crude hippocampal membrane fractions from control (Ct) and kainate treated (Kt) animals probed with an antibody directed against GFAP. Actin served as a lane loading control. (D) Densitometry values of immunoreactivity for GFAP from kainate treated samples (n = 6) were found to be significantly increased compared to control (n = 8) samples (** = p < 0.0001, student’s t test).
SR101 Identifies Hippocampal Astrocytes In Situ
The photo-stable fluorescent dye SR101 has been shown to label cortical astrocytes in vivo (Nimmerjahn et al., 2004) and astrocytes in hippocampal slice preparations from adult rats (Kafitz et al., 2008). We used SR101 fluorescence to target astrocytes in the stratum radiatum of hippocampus for whole cell patch clamp recordings because visualizing astrocytes in the sclerotic hippocampus from older animals (P50 – P60) with traditional IR-DIC microscopy proved difficult. With a 40x water immersion lens and fluorescent illumination (~565 nm excitation light), numerous SR101 labeled cells were observed throughout the stratum radiatum and hippocampus. Consistent with previous results (Kafitz et al., 2008), SR101 positive cells had several faintly labeled processes that emanated from a small cell body. These cells were targeted for electrophysiology in brain slices from both control and KA-treated rats (Figure 2A). To categorically define the cell type labeled with SR101, we determined their electrophysiological, immunohistochemical, and morphological properties.
Figure 2. SR101 labels astrocytes.

(A)A combination of SR101 immunofluorescence and IR-DIC microscopy was used to identify and patch astrocytes in the stratum radiatum (arrow points to one example). SR101 (middle) identified astrocyte cell bodies that were nearly invisible with IR-DIC microscopy (left) alone. Inset shows a typical response of a patched astrocyte to membrane depolarization in a ramp-like manner. Note the absence of voltage dependent action currents. (B) Patched cells were filled with biocytin during recording. Post-hoc analysis demonstrated that biocytin (left) diffused into numerous neighboring cells, indicating coupling by gap junctions. The astrocyte specific marker GFAP (center) was used in co-labeling studies (right, GFAP in green, biocytin in red), and indicated that biocytin positive cells were also GFAP positive. Scale bar, 20 μm. (C) No significant difference was observed in the resting membrane potential or input resistance of astrocytes from control (n = 27) and kainate treated (KA, n = 29) animals.
Passive membrane properties of SR101 positive cells were determined by patching the cell body in the whole cell voltage clamp configuration with the membrane potential clamped at −80 mV. Action currents were not observed during seal formation or when depolarizing the membrane in a ramp-like manner from −160 mV to +70 mV (Figure 2A). Recordings were made from 56 SR101 positive cells (27 from control rats and 29 from KA-treated rats). No statistically significant differences were found in the resting membrane potential (−79.9 ± 0.6 mV for control vs. −78.7 ± 0.5 mV for KA-treated) or input resistance (28.8 ± 3.1 MΩ for control vs. 28.2 ± 3.9 MΩ for KA-treated) between astrocytes from either group (Figure 2C). The electrophysiological properties of a low input resistance, a membrane potential near EK, and a near-linear current voltage relationship are most closely aligned with the subset of glial cells described in the literature as passive astrocytes, or classical protoplasmic astrocytes (see discussion).
Cell-fills and immunohistochemistry for the astrocyte specific marker GFAP were used to further characterize the identity of SR101 positive cells that displayed the electrophysiological criteria of passive astrocytes (Figure 2B). During recording, the cell was dialyzed with an intracellular patch solution containing 0.2% biocytin, which allowed the identification of biocytin positive cells with the use of fluorescently-conjugated streptavidins at the conclusion of the experiment. Although only one cell was recorded from any given brain slice, biocytin labeling was found in many cells presumably due to its diffusion through gap junctions, a characteristic typical of biocytin labeling in astrocytes (Konietzko and Muller, 1994; Wallraff et al., 2004). Immunoreactivity for GFAP was observed throughout the hippocampus and co-labeling studies (Figure 2B) determined that biocytin positive cells were immunoreactive for GFAP (n = 15 control slices, n = 12 KA-treated slices). Together, the data demonstrates that SR101 reliably identified astrocytes in our slice preparation as it labeled cells characterized by passive membrane currents, GFAP expression, and gap junctional coupling.
KA-induced SE Increases Gap Junction Coupling and Protein Expression in Astrocytes
Gap junctions allow the exchange of small molecules between cells. Gap junctional coupling among astrocytes is thought to promote tissue homeostasis by trafficking glucose from the bloodstream, spatially buffering extracellular potassium, and redistributing glucose (Rouach et al., 2008; Theis et al., 2005; Theis et al., 2003; Wallraff et al., 2006). Therefore, it was important to determine if there were changes in the extent of coupling among astrocytes from KA-treated animals. Post-hoc analysis revealed that biocytin diffused into many neighboring cells during the course of recording from a single astrocyte (Figure 3A). The cell nuclear counterstain DAPI clearly labeled all of the cell nuclei in the slice, including the densely packed CA1 pyramidal cell layer, and double labeling studies with biocytin and DAPI allowed the visualization of the astrocyte syncytium within the hippocampus. Consistent with previous reports, the extent of connected cells in the present study, from both control and KA-treated brain slices, were not confined to the stratum radiatum and in several cases the syncytium crossed stratum moleculare and into stratum oriens (Houades et al., 2006; Konietzko and Muller, 1994). Triple labeling studies with DAPI and GFAP again confirmed that the coupled cells were GFAP positive, confirming their identity as astrocytes.
Figure 3. Increased gap junction coupling and protein expression in astrocytes from KA-treated rats.

(A)After recording from a single astrocyte in a brain slice, processing for biocytin identified numerous gap junction coupled cells (far left, shown, a brain slice from a control animal). DAPI (blue, middle panels) clearly labeled the CA1 pyramidal cell layer and helped to identify the stratum radiatum and the extent of the astrocytic syncytium. Co-labeling with DAPI (blue), GFAP (green), and biocytin (red) determined the spatial extent of gap junction coupling and confirmed that biocytin positive cells were also GFAP positive. Colocalization for biocytin and DAPI is indicated in white (far right image), and these colocalized regions were used for cell counts. Scale bar, 50 μm. (B) When comparing the extent of coupling among astrocytes, slices from kainate treated animals had a significantly higher number of coupled astrocytes compared to controls (control, n = 10 slices, kainate treated, n = 9 slices, p < 0.01). (C) A significant correlation existed between recording duration and the number of coupled astrocytes from KA-treated brain slices, but not from controls. (D) Western blot analysis of crude hippocampal membrane fractions from control (Ct) and kainate treated (Kt) animals using an antibody directed against the astrocytic gap junction protein Connexin 43 (Cx43). A lower molecular mass product was also observed at ~ 25 kDa. Actin served as a lane loading control. (E) Densitometry values of Cx43 immunoreactivity from kainate treated (n = 6) samples were found to be significantly increased compared to control (n = 8) samples (** = p < 0.05, student’s t test). (F) Western blot analysis of crude hippocampal membrane fractions from control (Ct) and kainate treated (Kt) animals using an antibody directed against the astrocytic gap junction protein Connexin 30 (Cx30). Actin served as a lane loading control. (G) Densitometry valuesof Cx30 immunoreactivity from kainate treated (Kt) animals (n= 8) were found to be significantly reduced compared to control (n=8) (** p <0.05, student’s t test).
To determine the extent of gap junction coupled cells, we took advantage of the differential labeling patterns of DAPI and biocytin. Whereas biocytin labeled the entire cell (including the fine processes and cell soma), DAPI only labeled the cell nuclei. This allowed the use of the co-localization plugin developed for ImageJ by P. Bourdoncle (http://rsb.info.nih.gov/ij/plugins/colocalization.html) to highlight the areas of overlap between these 2 markers (see methods); biocytin labeled processes were thus ignored while the cell somas that labeled for both DAPI and biocytin were included in cell counts (shown in white, Figure 3A, right). This technique was important because unequivocally differentiating two adjacent astrocytes was sometimes difficult with biocytin alone. After biocytin positive astrocytes were identified in this manner, the “analyze particle” function allowed for automated and unbiased cell counts. Using this technique, an average of 100 ± 11 astrocytes showed dye coupling in hippocampal slices from KA-treated rats (n = 9), whereas in slices from control rats (n = 10), only an average of 59 ± 8 astrocytes were dye coupled (Figure 3B, p < 0.01, minimum recording duration = 10 minutes).
To determine if recording duration had an effect on the number of coupled cells, a correlation analysis was performed (Figure 3C). We hypothesized that longer recording durations might allow more time for biocytin to diffuse throughout the astrocyte syncytium. Surprisingly, there was no statistically significant correlation in the recording duration to the number of coupled cells from control slices (n = 10, p > 0.05, Spearman). However, when analyzing the number of coupled cells from KA-treated slices as a function of recording time, we found a statistically significant positive correlation (n = 9, p < 0.05, Spearman). The data suggests that biocytin readily diffused into the entire syncytium of control cells in a relatively short amount of time (minimum recording duration was 10 minutes) and that our initial cell counts may have underestimated the full extent of gap junction coupling in KA-treated slices because we included cells with relatively shorter recording durations.
To confirm that biocytin was diffusing through gap junctions and not actively transported into cells from the extracellular space, we pre-incubated control slices in a low bicarbonate/low pH (3.6mM NaHCO3/pH 6.4) solution to block gap junctions (Wallraff et al., 2004). Patching a SR101 labeled astrocyte after this pre-incubation resulted in biocytin diffusing into a single cell with a prominent ‘bushy’ pattern (n = 6, see Supplementary Figure 1). We confirmed that these cells were astrocytes by double labeling with GFAP and establishing that they had passive membrane currents. In agreement with previous studies (Schools et al., 2006; Wallraff et al., 2004), we found that blocking gap junctions did not alter the passive membrane current profile, indicating that it is an intrinsic property of this cell type and is not due to the degree of coupling. We also observed that pre-incubation in the low bicarbonate/low pH solution resulted in these cells (n = 6) having a slightly higher input resistance (75 ± 23 MΩ), as shown by Schools et al. (Schools et al., 2006). We further confirmed that biocytin was not taken up passively during the approach to the cell by placing a patch electrode extruding biocytin (from positive pressure) next to an SR101 positive cell for several minutes. In this configuration, intracellular biocytin labeling was never observed (n = 5, data not shown).
Connexin 43 (Cx43) and connexin 30 (Cx30) are the primary gap junction proteins expressed in astrocytes. To determine if there were any changes in the expression of these proteins following KA-treatment, we performed immunoblot experiments evaluating the expression of Cx43 and Cx30 in the hippocampus at 7 days following KA-treatment. Consistent with the dye coupling experiments suggesting an enhanced coupling between astrocytes, Cx43 expression was significantly increased in the hippocampus of KA-treated rats (Figure 3 D, E). While Cx30 expression was slightly reduced (Figure 3 F), the dramatic enhancement of Cx43 suggests that overall, gap junction protein expression was increased in the KA-treated rats.
Glutamate Transport Currents Have Faster Decay Kinetics in Slices From Kainate-Treated Rats
Another primary function of astrocytes is to remove extracellular glutamate by active transport. Therefore, synaptically-activated, glutamate transporter-mediated currents (STCs) (Diamond and Jahr, 2000) were recorded in astrocytes located in the stratum radiatum to investigate if glutamate transport function was altered in slices from KA-treated animals (Bergles and Jahr, 1997; Diamond et al., 1998; Luscher et al., 1998). Electrical stimulation of the CA3 Schaffer collaterals (SC) elicits action potentials in, and the release of glutamate from, the axons of CA3 neurons and induces a concomitant large inward current in stratum radiatum astrocytes that is due primarily to local changes in [K+]o (Orkand et al., 1966). However, a fast underlying glutamate transport current can also be observed after pharmacological isolation (Bergles and Jahr, 1997; Luscher et al., 1998; Meeks and Mennerick, 2007). Therefore, STCs were isolated by blocking the potassium dependent inward current with extracellular Ba2+ (200 μM) and by eliminating post-synaptic potassium fluxes (from neurons) with APV (100 μM), CNQX (20 μM), and bicuculline (10 μM) (to block NMDA, AMPA/kainate, and GABAA receptors, respectively), as demonstrated previously (Bergles and Jahr, 1997). This pharmacological isolation resulted in a fast inward current in response to SC stimulation that was sensitive to blockade by the potent non-selective competitive glutamate transport blocker dl-threo-β-benzyloxyaspartate (TBOA, 100 μM) (Shimamoto et al., 1998), consistent with the hypothesis that the current was dependent on glutamate transporters (Figure 4A). For each experiment detailed below, the STC was first isolated from fluxes in [K+]o in this manner.
Figure 4. STCs have faster decay kinetics in astrocytes from slices obtained from KA-treated rats.

(A) Isolation of the STC (synaptically activated glutamate transport current). Electrical stimulation of the Schaffer collaterals while recording from a stratum radiatum astrocyte in the presence of 200 μM BaCl2 and post-synaptic antagonists (100 μM APV, 20 μM CNQX, and 10 μM bicuculline) resulted in a fast, TBOA-sensitive, inward current. (B) No difference was observed in the STC peak current amplitude between astrocytes in slices from control (120.3 ± 36.4 pA, n = 8) and KA-treated (128.9 ± 16.45 pA, n = 9) rats. (C) Comparing the STC from control (black trace) and KA-treated animals (gray trace) by normalizing to the peak current demonstrated a change in the current kinetics. For (A) and (C), vertical scale bars: 25 pA, horizontal scale bars: 50 ms. (D) Quantification of the STC kinetics revealed that STCs in astrocytes from KA-treated animals (n = 9) had significantly faster decay and halfwidth, but not rise times, compared to controls (n = 8). (E) A western blot analysis of crude hippocampal membrane fractions from control (Ct) and kainate treated (Kt) animals using an antibody directed against GLT-1. The GLT-1 band resolved ~55 kDa. Higher molecular weight products (~100 – 150 kDa) were also observed. Actin served as a lane loading control. (F) Densitometry values of GLT-1 from kainate treated (n = 6) samples were not significantly different compared to control (n = 8) samples. (G) A western blot analysis of crude hippocampal membrane fractions from control (Ct) and kainate treated (Kt) animals using an antibody directed against GLAST. The GLAST band resolved ~55 kDa. Higher molecular weight products were also observed. Actin served as a lane loading control. (H) Densitometry values of GLAST from kainate treated (n = 8) samples were not significantly different compared to control (n = 8) samples.
We next sought to compare the current amplitude and kinetics of the STC between astrocytes in brain slices from control and KA-treated animals. The STC amplitude reflects the amount of glutamate released at the synapse, whereas the STC time course reflects the rate of glutamate clearance from the cleft (Diamond, 2005; Diamond and Jahr, 2000; Tzingounis and Wadiche, 2007). There was no significant difference between the average peak current amplitude of STCs between astrocytes recorded in slices from control (n = 8) and KA-treated (n = 9) rats when stimulated at 2.5 times the threshold for current induction, as described in the methods (Figure 4B, control: 120.3 ± 36.4 pA, KA-treated: 128.9 ± 16.45 pA, p = 0.83).
The rise phase of the STC reflects the concerted uptake activity of the transporters expressed on the astrocyte membrane in response to glutamate while the decay phase is proportional to the decrease in the amount of extracellular glutamate molecules available for transport (Diamond, 2005; Diamond and Jahr, 2000; Tzingounis and Wadiche, 2007). Analysis of the rise times of STCs did not reveal a statistically significant difference in astrocytes from control (n = 8) and KA-treated (n = 9) brain slices (control: 14.71 ± 0.66 ms vs. reactive 13.30 ± 0.44 ms, p = 0.08). In contrast, the decay times of STCs in astrocytes from KA-treated slices (n = 9) had a significantly faster average decay time of 19.87 ± 0.86 ms, compared to control astrocytes (27.34 ± 1.15 ms, n = 8, p < 0.0001). Likewise, STCs from control astrocytes (n = 8) had a halfwidth of 37.10 ± 1.6 ms, whereas STCs from the KA-treated group (n = 9) had a significantly shorter halfwidth of 27.92 ± 1.11 ms (Figure 4D, p = 0.0003). Normalizing the peak amplitudes of the STCs from the two populations of astrocytes illustrates the faster decay and halfwidth time course in the astrocyte recorded in slices obtained from KA-treated rats (Figure 4C). Therefore, STCs from astrocytes in KA-treated brain slices had significantly faster decay kinetics and halfwidth times that indicate a faster rate of clearance of synaptic glutamate.
Previous work has demonstrated that the time course of STCs is not affected by changes in stimulus strength, the probability of release, the spatial clustering of transporters, or the diffusion kinetics of glutamate (Diamond, 2005; Diamond and Jahr, 2000). To determine if these properties were consistent in brain slices obtained from KA-treated rats, we evoked STCs with different stimulus intensities and frequencies. Increasing the stimulus strength across a range of intensities increased the current amplitude of STCs in astrocytes from both control and KA-treated rats (Figure 5A). Normalizing the peak amplitude of STCs measured in the same astrocyte for two different current intensities (0.6 mA and 2.1 mA) demonstrated that the STC time course was not altered by increasing stimulus strengths (Figure 5B). We measured and compared these observations in both KA-treated astrocytes (shown, n = 4) and control astrocytes (n = 2). Therefore, in slices from KA-treated animals, consistent with previously published results, larger stimulus intensities resulted in proportionally larger STC current amplitudes (with similar kinetics), presumably due to increased glutamate release from a larger number of recruited Shaffer collaterals.
Figure 5. Enhancing glutamate release does not change STC kinetics.
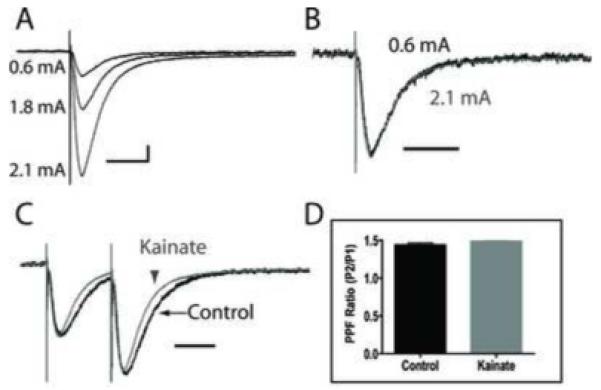
(A)STCs were elicited in a reactive astrocyte with varying stimulus strengths. Stronger stimuli resulted in larger STC current amplitudes. (B) Normalizing the peak amplitudes of STCs elicited in response to 0.6 mA (black trace) and 2.1 mA (gray trace) stimulation revealed that changes in stimulus strength (and hence, larger STC currents) do not alter the time course of STCs in astrocytes from KA-treated animals. (C) STCs from both a control astrocyte, and an astrocyte from a kainate treated animal, in response to paired-pulse-stimulation, with 80 ms separation, revealed a larger STC amplitude following the second stimulation, indicating enhanced glutamate release via paired pulse faciliatation. However, when normalizing the current amplitude in response to the first stimulation between groups, there was no change in the faster STC kinetics during the second pulse (ie. during enhanced glutamate release) from astrocytes in KA-treated slices. (D) A comparison of the pared pulse facilitation ratios from control (n = 4) and reactive (n = 5) astrocytes indicate no difference in the properties of glutamate release between slices from control and KA-treated rats. Vertical scale bars: 25 pA, horizontal scale bars: 50 ms.
To determine if changes in the probability of presynaptically released glutamate could alter the STC kinetics, we used a paired pulse stimulation protocol of the SC (Figure 5C) with an 80 msec pulse separation. When comparing the peak amplitudes of the STC in response to the first and second pulse, paired pulse facilitation (PPF) resulted in a larger STC peak amplitude in response to the second pulse, consistent with enhanced glutamate release (Figure 5C). To calculate PPF-dependent ratios, we divided the peak STC amplitude of the second pulse by the peak STC amplitude of the first pulse. The PPF ratio was 1.44 ± 0.03 for control astrocytes (n = 4), and 1.49 ± 0.01 in astrocytes from KA-treated (n = 5) rats (Figure 5D). The difference was not statistically significant (p = 0.13), and suggests similar presynaptic release probabilities of neuronal glutamate in both control and KA-treated slices. Importantly, when the peak amplitude from the first pulse was normalized between traces obtained from control and KA-treated astrocytes, the STCs from the KA-treated group maintained the faster decay and halfwidth kinetics for both the first and second pulse (Figure 5C). This result suggests that the faster kinetics were not due to changes in the release probability of presynaptic glutamate or the amount of glutamate in the synaptic cleft and raises the possibility that they could be due to intrinsic differences of the glutamate transporters expressed on the reactive astrocytes.
It has been demonstrated that glutamate transport current decay kinetics can become faster during development, most likely as a consequence of changes in expression of transporter proteins (Diamond, 2005). Therefore, to determine if KA-treatment resulted in altered expression of GLT-1 or GLAST, the main glutamate transporters expressed by mature astrocytes, we performed immunoblot experiments in the hippocampus at 7 days following KA-treatment (Figure 4E and F). Surprisingly, there was no significant difference between the control and KA-treated groups, suggesting that changes in the expression of GLT-1 and/or GLAST (Bergles and Jahr, 1997) did not underlie the faster STC decay times found in this study.
Inwardly Rectifying Potassium Currents Are Not Altered Following SE
Astrocytes buffer [K+]o, in part, by expressing highly permeable KIR channels. Because several studies suggest that K+ buffering is altered in tissue resected from TLE patients and in several different animal models of epilepsy and neuronal injury (see discussion), we investigated whether it was also altered during the period of low seizure probability following KA-induced SE.
KIR channels maximally activate at hyperpolarized membrane potentials and are sensitive to blockade by extracellular barium (Ba2+) (Ballanyi et al., 1987; Makhina et al., 1994; Ransom and Sontheimer, 1995). Therefore, to study KIR currents we utilized a voltage step protocol to maximally activate KIR channel-dependent currents and then pharmacologically isolated them by blocking KIR channels with Ba2+ (Figure 6A) (D’Ambrosio et al., 1999). To maximally activate KIR channels, the astrocyte membrane was first depolarized to a pre-pulse potential of 0 mV for 250 ms to inactivate any outward currents before stepping the membrane voltage to more negative potentials ranging from −150 mV to −50 mV (Ransom and Sontheimer, 1995). In normal Ringer’s solution (ACSF), large sustained inward currents were observed at the most hyperpolarized potentials in astrocytes from control rats (n = 6 cells, 5 animals) (Figure 6A, top trace, ACSF). The application of 200 μM Ba2+ attenuated these large hyperpolarization-dependent currents (Figure 6A, ACSF + BaCl2), consistent with published results (D’Ambrosio et al., 1999). Plots of the sustained current for each holding potential recorded in ACSF (black squares) and in the presence of extracellular Ba2+ (gray circles) demonstrate the Ba2+-dependent block of the hyperpolarization-dependent inward current (Figure 6A, I-V curves). Current subtraction revealed the Ba2+ sensitive current to show rectification, typical of KIR channels (Figure 6A, difference current). In six astrocytes recorded in brain slices from 6 KA-treated rats, we obtained similar results (Figure 6A, bottom traces); inward currents recorded in ACSF at hyperpolarized potentials were blocked by 200 μM Ba2+ and current subtraction revealed the Ba2+ sensitive component to show rectification. Therefore, astrocytes from both control and KA-treated brain slices displayed intact KIR currents.
Figure 6. Astrocytes recorded in slices obtained from KA-treated rats display Ba2+ sensitive currents.

(A) Comparison of the current voltage relationship between astrocytes in brain slices from control and KA-treated animals. Astrocyte membranes were depolarized to 0 mV for 250 ms from a holding potential of −80 mV, before applying voltage steps from −150 mV to −50 mV. This voltage step protocol was performed in normal ACSF (ACSF, left) and later, after the addition of 200 μM extracellular Ba2+ (ACSF + BaCl2). Plotting the current-voltage relationship (I-V curve) before and after Ba2+ (gray circles) shows the Ba2+ sensitive component. Current subtraction (difference current) reveals the Ba2+ sensitive component as an inwardly rectifying current. These Ba2+ sensitive currents were found in slices from both the control (n = 6 cell, 5 animals) and kainate treated (n = 6 cells, 6 animals) groups. Scale bars, 500 pA/10 msec. (B) Whole cell current traces in response to Shaffer collateral stimulation in ACSF (black trace), and in the presence of extracellular BaCl2 (gray trace) demonstrate that astrocytes from both groups express a Ba2+ sensitive inward current, and thus are most likely able to buffer extracellular potassium. Scale bars, 30 pA/ 0.5 sec (C) Quantification of the peak potassium uptake current (black trace in B) in astrocytes from control and kainate treated rats demonstrate no significant difference.
The extensive gap junction coupling and low input resistance of astrocytes limits our ability to have an adequate space clamp during voltage steps. Therefore, electrical stimulation of the CA3 Shaffer collaterals (SC) while recording from astrocytes in the stratum radiatum further tested the ability of astrocytes to buffer extracellular K+ without changing the membrane potential. Astrocytes from both control and KA-treated brain slices displayed similar responses to SC stimulation (Figure 6B). In normal ACSF, SC stimulation resulted in a large, slow inward current that took several seconds to return to baseline. A significant portion of this inward current was blocked by application of 200 μM BaCl2 in astrocytes from both groups, indicating it as a potassium channel dependent current. Determination of the peak amplitude of the K+ dependent current in control astrocytes (n = 7, 53.2 ± 13.6 pA) and astrocytes in slices from KA-treated rats (n = 9, 58.4 ± 9.8 pA) did not reveal a statistically significant difference (Figure 6C, p = 0.75). Therefore, astrocytes recorded in slices obtained from rats soon after SE did not appear to be compromised in their ability to detect changes in [K+]o nor in their ability to buffer extracellular K+.
DISCUSSION
In this study, astrocytes were investigated early in the epileptogenic process of the KA-induced SE model of TLE for changes in three intrinsic properties: gap junction coupling, glutamate uptake, and potassium buffering. Our data suggests that the astrocytes from this period of low seizure probability (1) form a larger gap junction coupled syncytium (2) express STCs with significantly faster decay times and halfwidths that suggest a faster rate of glutamate clearance, and (3) express Ba2+-sensitive KIR currents and are probably not compromised in their ability to buffer extracellular potassium. Taken together, the evidence suggests that astrocytes may continue to offer considerable protection from hyperexcitable conditions in the hippocampus soon after KA-induced SE.
SR101 Identifies Hippocampal Astrocytes
SR101 labeling was instrumental in recording from astrocytes (Kafitz et al., 2008; Nimmerjahn et al., 2004). In addition to microglia and oligodendrocytes, the rodent hippocampus contains at least two additional types of glia that are differentiated by either “passive” (time- and voltage-independent) or “complex” (time- and voltage-dependent) membrane currents (D’Ambrosio et al., 1998; Jabs et al., 1997; McKhann et al., 1997; Steinhauser et al., 1994). Passive cells in the hippocampus (i.e. the prototypical astrocyte) usually express glutamate transporter dependent currents, GFAP, and couple to one another via gap junctions. In contrast, complex cells usually express AMPA-type glutamate receptor currents, the chondroitin sulfate proteoglycan NG2 (neuron/glial antigen 2), and are not coupled by gap junctions (Ge et al., 2006; Jabs et al., 2005; Matthias et al., 2003; Wallraff et al., 2004). In the present study, SR101 labeled cells that expressed passive currents, GFAP, KIR currents, STCs, and formed a gap junction coupled syncytium and the data presented in this study were obtained from cells with these properties. Therefore, in our experiments, SR101 reliably identified the “typical” protoplasmic astrocytes in the stratum radiatum of hippocampus in slices from both control and KA-treated rats. However, it should be noted that these “typical” characteristics may differ widely depending on brain region, developmental time point, pathology, and in different organisms (for example, see D’Ambrosio et al., 1998; McKhann et al., 1997; Oberheim et al., 2009). Furthermore, it is unknown at this time how much subtle heterogeneity in structure and function exists within each of the major glial subclasses (astrocytes, oligodendroyte, NG2 cell, microglia). Therefore, while SR101 appears to be a reliable marker for astrocytes in the hippocampus of mature rodents, care should be taken when using SR101 in different organisms, brain regions, etc. to categorically define the cell population studied.
KA-treatment Increases Gap Junction Coupling
Gap junctions are intercellular membrane channels that link the cytoplasm of adjacent cells and allow the direct exchange of ions and small molecules up to ~1 kDa (Simon and Goodenough, 1998). The channel is formed from two closely opposed (< 4 nm) connexin hemichannels, with each hemichannel formed from six transmembrane proteins encoded by connexin (Cx) genes (Musil and Goodenough, 1993). Astrocytes express Cx43 (connexin protein of 43 kDa) and Cx30, and represent the largest gap junction coupled network in the CNS (for reviews, see (Nagy and Rash, 2000; Theis et al., 2005)). Numerous studies point to changes in astrocytic gap junction coupling or protein expression in neurological disorders. Cx43 protein was found to be upregulated in reactive astrocytes from hippocampus resected from patients with MTLE (Fonseca et al., 2002). Interestingly, an increase in Cx43 expression has also been observed with glial scarring after ischemic injury (Haupt et al., 2007) and with chemically induced lesions in the striatum and thalamus (Ochalski et al., 1995), suggesting that increased expression of astrocytic connexins may be associated with reactive astrocytes in general. Increased functional gap junction coupling has been observed in cultured astrocytes from human epileptic tissue (Lee et al., 1995), following epileptiform burst activity in slices from control rats (Rouach et al., 2008), and in reactive astrocytes from an animal model of cortical dysplasia (Bordey et al., 2001). In the present study, the number of coupled astrocytes in brain slices from KA-treated rats was also found to be significantly increased early in the process of epileptogenesis, at a time when seizure probability is quite low (Williams et al., 2009).
Astrocyte coupling is thought to underlie spatial buffering and the redistribution of potassium and glutamate. A conditional knock-out of the main astrocyte gap junction protein Cx43 decreases coupling and enhances the velocity of spreading depression (Theis et al., 2003). Spreading depression is a pathophysiological phenomenon in the CNS characterized by a combined reaction of neurons and glia in the form of a propagating wave of neuronal depolarization and subsequent inactivation. This suggests that astrocyte coupling is critical for the redistribution of ions to minimize spreading depression and abnormal neuronal depolarization.
Another important role of the astrocytic syncytium, however, is to provide a metabolic link between neurons and the vasculature. Astrocytes in the hippocampus contact blood vessels (Bushong et al., 2002) and traffic glucose and lactate through gap junctions (Giaume et al., 1997). This trafficking is essential for the maintenance of both normal and epileptiform activity (Rouach et al., 2008). Therefore, enhanced coupling could paradoxically support hyperexcitability by increasing the ability of neurons to receive the metabolic substrates required for seizure activity.
Astrocytes in the normal brain have discrete spatial domains with minimal overlap and interdigitation (Bushong et al., 2002). Gap junction proteins are presumably localized at the distal processes where astrocytes contact one another. Interestingly, while this was not investigated in the present set of experiments, in several animal models of epilepsy including KA-induced SE, reactive astrocytes lose their discrete domain organization and begin to interdigitate extensively (Oberheim et al., 2008). Therefore, increased interdigitation might lead to an enhanced astrocyte-astrocyte contact and hence, more coupling. In addition, such interdigitation, if it is also accompanied by an increase in synapse ensheathment, may lead to faster glutamate uptake. While further experiments are necessary to determine if interdigitation occurs soon after KA-induced SE, the observed changes in Cx43 expression, the increase in gap junctionally-coupled astrocytes, and the increase in the rate of glutamate uptake that have been observed in these studies would be consistent with an increase in overlapping astrocytic domains.
Glutamate Transport Currents Have Faster Decay Kinetics in Brain Slices from KA-Treated Rats
The STC recorded in astrocytes is due to the coupled transport into the cell of 1 glutamate molecule, 3 Na+, and 1 H+ and the efflux of 1 K+ (Tzingounis and Wadiche, 2007). The STC rise phase is hypothesized to reflect the activity of the transporters in response to glutamate while the amplitude reflects the number of activated transporters (and hence the number of glutamate molecules). The decay phase reflects the proportional decrease in extracellular glutamate levels due to clearance/diffusion (Diamond, 2005; Diamond and Jahr, 2000; Tzingounis and Wadiche, 2007). In this study, SC stimulation elicited STCs from astrocytes in KA-treated slices with similar rise times and amplitudes as controls, but with significantly faster decay times and shorter halfwidths. The faster STC decay kinetics in reactive astrocytes were not altered with changes in stimulation strength nor with PPF, suggesting that they were not influenced by amount of presynaptic glutamate release. The lack of a change in the rise time of the currents suggest that changes in filtering properties as a consequence of increased coupling do not underlie the observed changes in decay time in the astrocytes recorded in slices obtained from KA-treated rats. These results suggest that glutamate clearance occurred more rapidly in brain slices from KA-treated rats and given the fact that protein expression of either GLT-1 or GLAST was not altered, our data suggests that changes in STC kinetics may be due to changes in intrinsic properties of the astrocytes in KA-treated brain slices.
Astrocyte ensheathment of hippocampal synapses is non-uniform and can dictate the rate of diffusion of glutamate out of the synapse (Ventura and Harris, 1999). Structural relationships between astrocytes and neurons increase during development (Pomeroy and Purves, 1988) and are correlated with faster glutamate clearance (Bergles and Jahr, 1997; Diamond, 2005). Astrocytic ensheathment is also dynamic and responds to neuronal activity (Genoud et al., 2006; Haber et al., 2006) and is enhanced in kindling models of epilepsy (Hawrylak et al., 1993). Therefore, enhanced ensheathment could underlie the faster STC decay kinetics observed in our study. Additionally, increased ensheathment would place transporters closer to the synapse, thus providing greater access to synaptic glutamate and facilitating uptake. Such alterations in the spatial relationship between astrocytes and neurons in epileptic tissue might represent compensatory mechanisms to counteract hyperexcitability by enhancing glutamate clearance and limiting abnormal excitability.
Numerous studies have investigated changes in protein expression of glutamate transporters in epileptic models. For example, in both lithium pilocarpine induced SE rats (Crino et al., 2002) and amygdala kindled rats (Miller et al., 1997), GLT-1 expression remained unchanged while EAAC1 (the predominant neuronal glutamate transporter) expression increased. Contrasting these results, however, EAAC1 expression was found to be downregulated following KA-induced SE, while the expression of astrocytic transporters modestly increased (Simantov et al., 1999). Recently, EAAC1 was shown to dictate the rate of clearance of glutamate by astrocytic transporters (Scimemi et al., 2009). Our STC time course and protein expression results are consistent with a downregulation in the neuronal glutamate transporters without a change in astrocytic transporters, but further experiments are necessary to confirm this hypothesis.
KIR-Mediated Currents Are Present in Reactive Astrocytes
Rapid neuronal depolarizations during heightened activity results in the extrusion of K+ with each action potential repolarization (Dichter et al., 1972). Even small amounts extruded from neurons can quickly elevate [K+]o because of the small extracellular space volume (Nicholson and Sykova, 1998). The concentration of [K+]o must be kept low to maintain the driving force for K+ efflux from neurons. Otherwise, unchecked rises in [K+]o will depolarize neuronal membranes and lead to hyperexcitability (Traynelis and Dingledine, 1988).
Astrocytes are perfectly suited to buffer [K+]o. They express the highly permeable KIR channel KIR4.1 (Higashi et al., 2001; Kalsi et al., 2004; Poopalasundaram et al., 2000) which has a high open probability at rest (Ransom and Sontheimer, 1995) and whose channel conductance increases with rises in [K+]o (Newman, 1993; Sakmann and Trube, 1984). In addition, the astrocyte syncytium is hypothesized to allow the movement of K+ from areas of high concentration to areas of lower concentrations in a process called spatial buffering (Kofuji and Newman, 2004; Orkand et al., 1966). Pharmacological blockade of KIR will raise [K+]o (Gabriel et al., 1998b; Kivi et al., 2000) and promote synchronous firing in CA1 (Janigro et al., 1997), while conditionally knocking-out KIR4.1 results in behavioral ataxia, seizures, and early lethality (Djukic et al., 2007).
The results from the present study demonstrate that KIR continues to be functionally present in astrocytes early in the process of epileptogenesis. Astrocytes from both groups displayed Ba2+-sensitive inwardly rectifying currents with membrane hyperpolarization and Ba2+-sensitive uptake currents in response to SC stimulation. This is in contrast to studies from resected human TLE tissue and pilocarpine-treated rats (many weeks post-insult) which suggests a deficiency in KIR function (Bordey and Spencer, 2004; Gabriel et al., 1998a; Hinterkeuser et al., 2000; Kivi et al., 2000). A recent study in an albumin model of focal epilepsy showed rapid changes in gene expression that predicted a reduction in KIR and glutamate transporter expression soon after albumin exposure and prior to the development of focal seizures (David et al., 2009). While such a deficiency occurring before the onset of spontaneous seizures would certainly be considered pro-epileptogenic, we did not observe these changes in the reactive astrocytes from KA-treated rats. This raises the interesting possibility that reactive astrocytes, though morphologically similar, may nevertheless have unique functional properties that are specific to the nature of injury or epilepsy.
Conclusion
Gliosis is of particular interest to the study of epilepsy, given its prevalence in human TLE, in animal models of epilepsy, and because seizures often initiate in or near gliotic tissue (McKhann et al., 2000). Here, we report intrinsic functional differences in astrocytes soon after KA-induced SE. These data suggest that some of the early functional changes observed in astrocytes soon after an insult may serve to prevent hyperexcitability in damaged regions of brain. It will be important to determine if there are additional changes in astrocyte function that could be exploited therapeutically during periods of high seizure frequency in TLE (e.g., months after KA-induced SE (Williams et al., 2009)) and future work combining chronic video EEG recordings with brain slice recordings will further contribute to our understanding of the varied functions of astrocytes in epilepsy.
Research Highlights.
Following kainic acid induced status epilepticus, astrocytes were found to:
-
○
Have significantly faster decay time kinetics of glutamate transporter-dependent currents
-
○
Have increased dye coupling and expression of connexin 43
-
○
Have no changes in potassium currents
Supplementary Material
Supplementary Figure 1. Low bicarbonate/low pH blocks gap junctions and the diffusion of biocytin.
(A)Representative recording from an astrocyte in a brain slice obtained from a control animal in a low bicarbonate/low pH (3.6mM NaHCO3/pH 6.4) solution results in the blockage of biocytin diffusion into neighboring cells (left). Co-labeling for biocytin and GFAP (middle, right) was used to confirm the identity of the recorded cell. The recorded cell is indicated by the arrow, and is GFAP positive. Scale bars, 50 μm (B) GFAP positive astrocytes, with gap junctions blocked, also displayed time- and voltage-independent passive membrane currents in response to a voltage step protocol
Acknowledgements
This work was funded by an Epilepsy Foundation Predoctoral Fellowship (DKT), an Epilepsy Foundation Health Science Summer Fellowship (JRV), NIH NS066774 (JRV) NIH NS044210 (KSW), and NIH NS062419 (KSW). The authors wish to thank the Anticonvulsant Drug Development Program, Dr. C. Rodesch, of the University of Utah Microscopy Core Facility, Dr. K. Kafitz for providing us with the SR101 protocol, Dr. J. Huguenard for comments on the manuscript, and Dr. Annette Fleckenstein and Greg Hadlock for use of equipment and advice on western blots. In addition, the authors wish to thank Gerald Saunders for help with kainic acid injections.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alex AB, et al. Effect of Conantokin G on NMDA receptor-mediated spontaneous EPSCs in cultured cortical neurons. J Neurophysiol. 2006;96:1084–92. doi: 10.1152/jn.01325.2005. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, et al. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol. 1987;382:159–74. doi: 10.1113/jphysiol.1987.sp016361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends in Neuroscience. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54:358–68. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- Bordey A, et al. Electrophysiological characteristics of reactive astrocytes in experimental cortical dysplasia. J Neurophysiol. 2001;85:1719–31. doi: 10.1152/jn.2001.85.4.1719. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 1998;32:286–303. doi: 10.1016/s0920-1211(98)00059-x. [DOI] [PubMed] [Google Scholar]
- Bordey A, Spencer DD. Distinct electrophysiological alterations in dentate gyrus versus CA1 glial cells from epileptic humans with temporal lobe sclerosis. Epilepsy Res. 2004;59:107–22. doi: 10.1016/j.eplepsyres.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Buono RJ, et al. Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res. 2004;58:175–83. doi: 10.1016/j.eplepsyres.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Bushong EA, et al. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–92. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, et al. Increased expression of the neuronal glutamate transporter (EAAT3/EAAC1) in hippocampal and neocortical epilepsy. Epilepsia. 2002;43:211–8. doi: 10.1046/j.1528-1157.2002.35001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, et al. Impaired K(+) homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J Neurosci. 1999;19:8152–62. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, et al. Functional specialization and topographic segregation of hippocampal astrocytes. J Neurosci. 1998;18:4425–38. doi: 10.1523/JNEUROSCI.18-12-04425.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David Y, et al. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009;29:10588–99. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–16. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, et al. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron. 1998;21:425–33. doi: 10.1016/s0896-6273(00)80551-6. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–87. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–43. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- Dichter MA, et al. Silent cells during interictal discharges and seizures in hippocampal penicillin foci. Evidence for the role of extracellular K+ in the transition from the interictal state to seizures. Brain Res. 1972;48:173–83. doi: 10.1016/0006-8993(72)90177-1. [DOI] [PubMed] [Google Scholar]
- Djukic B, et al. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–65. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J., Jr. Clinical evidence for the progressive nature of epilepsy. Epilepsy Res Suppl. 1996;12:9–20. [PubMed] [Google Scholar]
- Fonseca CG, et al. Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res. 2002;929:105–16. doi: 10.1016/s0006-8993(01)03289-9. [DOI] [PubMed] [Google Scholar]
- Gabriel S, et al. Effects of barium on stimulus induced changes in extracellular potassium concentration in area CA1 of hippocampal slices from normal and pilocarpine-treated epileptic rats. Neurosci Lett. 1998a;242:9–12. doi: 10.1016/s0304-3940(98)00012-3. [DOI] [PubMed] [Google Scholar]
- Gabriel S, et al. Effects of barium on stimulus-induced changes in [K+]o and field potentials in dentate gyrus and area CA1 of human epileptic hippocampus. Neurosci Lett. 1998b;249:91–4. doi: 10.1016/s0304-3940(98)00420-0. [DOI] [PubMed] [Google Scholar]
- Ge WP, et al. Long-term potentiation of neuron-glia synapses mediated by Ca2+-permeable AMPA receptors. Science. 2006;312:1533–7. doi: 10.1126/science.1124669. [DOI] [PubMed] [Google Scholar]
- Genoud C, et al. Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol. 2006;4:e343. doi: 10.1371/journal.pbio.0040343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, et al. Metabolic trafficking through astrocytic gap junctions. Glia. 1997;21:114–23. [PubMed] [Google Scholar]
- Gutnick MJ, Prince DA. Dye coupling and possible electrotonic coupling in the guinea pig neocortical slice. Science. 1981;211:67–70. doi: 10.1126/science.7444449. [DOI] [PubMed] [Google Scholar]
- Haber M, et al. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J Neurosci. 2006;26:8881–91. doi: 10.1523/JNEUROSCI.1302-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt C, et al. Up-regulation of Connexin43 in the glial scar following photothrombotic ischemic injury. Mol Cell Neurosci. 2007;35:89–99. doi: 10.1016/j.mcn.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Hawrylak N, et al. Astrocytic and synaptic response to kindling in hippocampal subfield CA1. II. Synaptogenesis and astrocytic process increases to in vivo kindling. Brain Res. 1993;603:309–16. doi: 10.1016/0006-8993(93)91253-o. [DOI] [PubMed] [Google Scholar]
- Hellier JL, et al. Recurrent spontaneous motor seizures after repeated low-dose systemic treatment with kainate: assessment of a rat model of temporal lobe epilepsy. Epilepsy Research. 1998;31:73–84. doi: 10.1016/s0920-1211(98)00017-5. [DOI] [PubMed] [Google Scholar]
- Higashi K, et al. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–31. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- Hinterkeuser S, et al. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12:2087–96. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- Hoogland G, et al. Synaptosomal glutamate and GABA transport in patients with temporal lobe epilepsy. J Neurosci Res. 2004;76:881–90. doi: 10.1002/jnr.20128. [DOI] [PubMed] [Google Scholar]
- Houades V, et al. Shapes of astrocyte networks in the juvenile brain. Neuron Glia Biol. 2006;2:3–14. doi: 10.1017/S1740925X06000081. [DOI] [PubMed] [Google Scholar]
- Jabs R, et al. Qualitative analysis of membrane currents in glial cells from normal and gliotic tissue in situ: down-regulation of Na+ current and lack of P2 purinergic responses. Neuroscience. 1997;81:847–60. doi: 10.1016/s0306-4522(97)00207-8. [DOI] [PubMed] [Google Scholar]
- Jabs R, et al. Synaptic transmission onto hippocampal glial cells with hGFAP promoter activity. J Cell Sci. 2005;118:3791–803. doi: 10.1242/jcs.02515. [DOI] [PubMed] [Google Scholar]
- Janigro D, et al. Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. J Neurosci. 1997;17:2813–24. doi: 10.1523/JNEUROSCI.17-08-02813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafitz KW, et al. Developmental profile and properties of sulforhodamine 101--Labeled glial cells in acute brain slices of rat hippocampus. J Neurosci Methods. 2008;169:84–92. doi: 10.1016/j.jneumeth.2007.11.022. [DOI] [PubMed] [Google Scholar]
- Kalsi AS, et al. Kir4.1 expression by astrocytes and oligodendrocytes in CNS white matter: a developmental study in the rat optic nerve. J Anat. 2004;204:475–85. doi: 10.1111/j.0021-8782.2004.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivi A, et al. Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci. 2000;12:2039–48. doi: 10.1046/j.1460-9568.2000.00103.x. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–56. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konietzko U, Muller CM. Astrocytic dye coupling in rat hippocampus: topography, developmental onset, and modulation by protein kinase C. Hippocampus. 1994;4:297–306. doi: 10.1002/hipo.450040313. [DOI] [PubMed] [Google Scholar]
- Lee SH, et al. Human epileptic astrocytes exhibit increased gap junction coupling. Glia. 1995;15:195–202. doi: 10.1002/glia.440150212. [DOI] [PubMed] [Google Scholar]
- Lehre KP, et al. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–53. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, et al. Monitoring glutamate release during LTP with glial transporter currents. Neuron. 1998;21:435–41. doi: 10.1016/s0896-6273(00)80552-8. [DOI] [PubMed] [Google Scholar]
- Makhina EN, et al. Cloning and expression of a novel human brain inward rectifier potassium channel. J Biol Chem. 1994;269:20468–74. [PubMed] [Google Scholar]
- Marcaggi P, et al. The role of glial glutamate transporters in maintaining the independent operation of juvenile mouse cerebellar parallel fibre synapses. J Physiol. 2003;552:89–107. doi: 10.1113/jphysiol.2003.044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, et al. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:453–72. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- Matthias K, et al. Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J Neurosci. 2003;23:1750–8. doi: 10.1523/JNEUROSCI.23-05-01750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, 2nd, et al. Heterogeneity of astrocyte resting membrane potentials and intercellular coupling revealed by whole-cell and gramicidin-perforated patch recordings from cultured neocortical and hippocampal slice astrocytes. J Neurosci. 1997;17:6850–63. doi: 10.1523/JNEUROSCI.17-18-06850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, 2nd, et al. Intraoperative hippocampal electrocorticography to predict the extent of hippocampal resection in temporal lobe epilepsy surgery. J Neurosurg. 2000;93:44–52. doi: 10.3171/jns.2000.93.1.0044. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Astrocyte membrane responses and potassium accumulation during neuronal activity. Hippocampus. 2007;17:1100–8. doi: 10.1002/hipo.20344. [DOI] [PubMed] [Google Scholar]
- Miller HP, et al. Alterations in glutamate transporter protein levels in kindling-induced epilepsy. J Neurochem. 1997;68:1564–70. doi: 10.1046/j.1471-4159.1997.68041564.x. [DOI] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–77. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Rash JE. Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev. 2000;32:29–44. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- Naus CC, et al. Gap junction gene expression in human seizure disorder. Exp Neurol. 1991;111:198–203. doi: 10.1016/0014-4886(91)90007-y. [DOI] [PubMed] [Google Scholar]
- Newman EA. Inward-rectifying potassium channels in retinal glial (Muller) cells. J Neurosci. 1993;13:3333–45. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–15. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, et al. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1:31–7. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009;29:3276–87. doi: 10.1523/JNEUROSCI.4707-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, et al. Loss of astrocytic domain organization in the epileptic brain. J Neurosci. 2008;28:3264–76. doi: 10.1523/JNEUROSCI.4980-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochalski PA, et al. Astrocytic gap junction removal, connexin43 redistribution, and epitope masking at excitatory amino acid lesion sites in rat brain. Glia. 1995;14:279–94. doi: 10.1002/glia.440140405. [DOI] [PubMed] [Google Scholar]
- Orkand RK, et al. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- Otto JF, et al. A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J Neurosci. 2006;26:2053–9. doi: 10.1523/JNEUROSCI.1575-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeroy SL, Purves D. Neuron/glia relationships observed over intervals of several months in living mice. J Cell Biol. 1988;107:1167–75. doi: 10.1083/jcb.107.3.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poopalasundaram S, et al. Glial heterogeneity in expression of the inwardly rectifying K(+) channel, Kir4.1, in adult rat CNS. Glia. 2000;30:362–72. doi: 10.1002/(sici)1098-1136(200006)30:4<362::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Proper EA, et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2002;125:32–43. doi: 10.1093/brain/awf001. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–94. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol. 1995;73:333–46. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- Riddle EL, et al. Differential trafficking of the vesicular monoamine transporter-2 by methamphetamine and cocaine. Eur J Pharmacol. 2002;449:71–4. doi: 10.1016/s0014-2999(02)01985-4. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, et al. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–25. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Rouach N, et al. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–5. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Trube G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. J Physiol. 1984;347:641–57. doi: 10.1113/jphysiol.1984.sp015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schools GP, et al. Development of gap junctions in hippocampal astrocytes: evidence that whole cell electrophysiological phenotype is an intrinsic property of the individual cell. J Neurophysiol. 2006;96:1383–92. doi: 10.1152/jn.00449.2006. [DOI] [PubMed] [Google Scholar]
- Scimemi A, et al. Neuronal transporters regulate glutamate clearance, NMDA receptor activation, and synaptic plasticity in the hippocampus. J Neurosci. 2009;29:14581–95. doi: 10.1523/JNEUROSCI.4845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert G, et al. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, et al. DL-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- Simantov R, et al. Changes in expression of neuronal and glial glutamate transporters in rat hippocampus following kainate-induced seizure activity. Brain Res Mol Brain Res. 1999;65:112–23. doi: 10.1016/s0169-328x(98)00349-0. [DOI] [PubMed] [Google Scholar]
- Simon AM, Goodenough DA. Diverse functions of vertebrate gap junctions. Trends Cell Biol. 1998;8:477–83. doi: 10.1016/s0962-8924(98)01372-5. [DOI] [PubMed] [Google Scholar]
- Smith MD, et al. Phenytoin- and carbamazepine-resistant spontaneous bursting in rat entorhinal cortex is blocked by retigabine in vitro. Epilepsy Res. 2007;74:97–106. doi: 10.1016/j.eplepsyres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Sontheimer H. Voltage-dependent ion channels in glial cells. Glia. 1994;11:156–72. doi: 10.1002/glia.440110210. [DOI] [PubMed] [Google Scholar]
- Steinhauser C, et al. Properties of voltage-activated Na+ and K+ currents in mouse hippocampal glial cells in situ and after acute isolation from tissue slices. Pflugers Arch. 1994;428:610–20. doi: 10.1007/BF00374585. [DOI] [PubMed] [Google Scholar]
- Theis M, et al. Emerging complexities in identity and function of glial connexins. Trends Neurosci. 2005;28:188–95. doi: 10.1016/j.tins.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Theis M, et al. Connexin43 is not expressed in principal cells of mouse cortex and hippocampus. Eur J Neurosci. 2003;18:267–74. doi: 10.1046/j.1460-9568.2003.02740.x. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–76. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–47. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallraff A, et al. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–47. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallraff A, et al. Distinct types of astroglial cells in the hippocampus differ in gap junction coupling. Glia. 2004;48:36–43. doi: 10.1002/glia.20040. [DOI] [PubMed] [Google Scholar]
- West PJ, et al. Differential contribution of kainate receptors to excitatory postsynaptic currents in superficial layer neurons of the rat medial entorhinal cortex. Neuroscience. 2007;146:1000–12. doi: 10.1016/j.neuroscience.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington J, et al. Astrocytes in the epileptic brain. Neuron. 2008;58:168–78. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, et al. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–12. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Low bicarbonate/low pH blocks gap junctions and the diffusion of biocytin.
(A)Representative recording from an astrocyte in a brain slice obtained from a control animal in a low bicarbonate/low pH (3.6mM NaHCO3/pH 6.4) solution results in the blockage of biocytin diffusion into neighboring cells (left). Co-labeling for biocytin and GFAP (middle, right) was used to confirm the identity of the recorded cell. The recorded cell is indicated by the arrow, and is GFAP positive. Scale bars, 50 μm (B) GFAP positive astrocytes, with gap junctions blocked, also displayed time- and voltage-independent passive membrane currents in response to a voltage step protocol