

Summary
Posttranslational arginylation mediated by arginyltransferase (ATE1) plays an important role in cardiovascular development, cell motility and regulation of cytoskeleton and metabolic enzymes. This protein modification was discovered decades ago, however, the arginylation reaction and the functioning of ATE1 remained poorly understood due to the lack of good biochemical models. Here we report the development of an in vitro arginylation system, in which ATE1 function and molecular requirements can be tested using purified recombinant ATE1 isoforms supplemented with a controlled number of components. Our results show that arginylation reaction is a self-sufficient, ATP-independent process that can affect different sites in a polypeptide, and that arginyltransferases form different molecular complexes in vivo, associate with components of the translation machinery, and have distinct, partially overlapping subsets of substrates, suggesting that these enzymes play different physiological functions.
Introduction
Arginylation is a poorly understood protein modification that consists of posttranslational addition of arginine to proteins (Kaji et al., 1963a; Kaji et al., 1963b) and is mediated by arginyltransferase (ATE1) (Balzi et al., 1990; Kwon et al., 1999; Rai and Kashina, 2005). Ate1 is an essential mouse gene, whose deletion causes embryonic lethality and severe cardiovascular defects (Kwon et al., 2002; Rai et al., 2008). It has been previously shown that arginylation affects a large number of proteins (Kaji, 1976; Lamon and Kaji, 1980; Soffer and Mendelsohn, 1966; Wang and Ingoglia, 1997; Wong et al., 2007; Xu et al., 1993), and that it regulates in vivo functions of such essential proteins as actin (Karakozova et al., 2006; Rai et al., 2008), regulators of G-protein signaling (RGS) (Lee et al., 2005), and calreticulin (Decca et al., 2006), however the underlying molecular mechanisms that drive arginylation reaction and modulate ATE1 function are poorly understood.
It has been previously hypothesized that arginylation in mammals can occur only on the N-terminally exposed alpha amino groups of Asp, Glu and Cys, and that such arginylation targets proteins for degradation by the N-end rule pathway that relates the half-life of a protein to the identity of its N-terminal residue (Bachmair et al., 1986). Other groups have reported that N-terminal Arg facilitates protein recognition by the ubiquitin conjugation machinery (Elias and Ciechanover, 1990), however later studies suggested that not all protein substrates in vivo undergo arginylation-dependent degradation, and that the relationship between arginylation and degradation may be more complex (Karakozova et al., 2006; Wong et al., 2007). A recent discovery that arginylation can occur in vivo on the acidic side chain of glutamic acid (Eriste et al., 2005) makes it evident that the arginylation mechanisms are even more complicated than previously believed and opens up an exciting possibility that arginylation can also occur on the side chain of other amino acid residues.
Several past studies reported successful reconstitution of arginylation reaction in vitro (Ciechanover et al., 1988; Kwon et al., 2002; Soffer, 1970), however these reactions have been performed in crude cell extracts or partially purified preparations without controlling for the arginyltransferase purity, making it impossible to determine ATE1's specificity and requirements for cofactors or to fully address the mechanism of ATE1 action. A discovery that mammalian Ate1 gene generates a subset of highly homologous but distinct isoforms led to controversial reports about these isoforms' activities and substrate specificities (Hu et al., 2006; Rai and Kashina, 2005; Rai et al., 2006), further suggesting that the arginylation reaction in vivo may be more complex than it appears. To add to the mystery, recent identification of a large number of arginylated proteins in vivo (Wong et al., 2007) raised a multitude of possibilities about the arginylation reaction mechanisms and function. It was found that while arginylation affects unique sites on the surface of the folded proteins – and thus is highly likely to constitute a true regulatory modification – it has an apparently low specificity for the primary sequence around the arginylation site, suggesting that additional recognition cofactors may be required to direct ATE1 to its appropriate protein targets. Arginylation frequently affects amino acid residues located not only on the N-terminus, as previously suggested, but also in the middle of the polypeptide chain, leading to a likely possibility that ATE1 might be coupled with translation and/or proteolysis, by forming transient or stable complexes with other proteins in vivo.
To resolve some of these questions and to develop a system in which arginylation-related mechanisms can be tested directly, we have expressed and purified functional mouse ATE1 isoforms and set up an in vitro system to analyze their activity, requirement for cofactors, and potential relationship to other posttranslational modifications, such as protein acetylation. We have combined these studies with an in vivo analysis of ATE1's intracellular interactions that confirmed the earlier hypotheses about the potential involvement of distinct functional classes of proteins, including the components of the translation machinery, in the in vivo arginylation reaction. Our results constitute the first detailed analysis of the arginyltransferase function in vitro and in vivo and demonstrate that ATE1 is a self-regulating, ATP-independent enzyme forming distinct molecular complexes in vivo that facilitate arginylation of multiple protein substrates.
Results
In vitro arginylation assay
It has been previously established that arginylation reaction occurs by ATE1-mediated transfer of Arg from the charged tRNA to the protein substrate (Soffer, 1970), suggesting that the minimal components for this reaction should include the ATE1 enzyme, protein substrate, free Arg, tRNA, arginyl-tRNA synthetase (RRS), and ATP (essential at least for the formation of charged Arg-tRNA). It has also been found that bovine serum albumin (BSA) can serve as an efficient substrate for ATE1, since the removal of the signal peptide from the BSA's N-terminus exposes the aspartic acid for the arginyl addition (Elias and Ciechanover, 1990). We based our initial experiments on this information. For the initial setup of the in vitro system for ATE1 activity assay, we expressed and purified His-tagged recombinant mouse ATE1 isoforms from E. coli expression system by Ni-NTA affinity chromatography and confirmed the high yield, solubility and purity of the resulting preparation (Fig. 1 and S1a). A 70 kDa co-purifying band that could not be removed by either higher stringency washes or by ion exchange chromatography was found in the preparations of ATE1-3 or 1-4. Mass spectrometry identified this band as bifunctional polymyxin resistance protein arnA (NCBI accession # ARNA_ECOLI). Since no obvious structural or functional connection could be found between this protein and arginyltransferase, we assumed this to be a non-specific contaminant.
Fig. 1. Characterization of the arginylation reaction.

a. Time course of the incorporation of [3H]-Arg into BSA mediated by ATE1-2 (blue diamonds); control curves show arginylation reaction with no added ATE1 (pink squares), no RRS (red triangles), and no BSA substrate (green crosses). Inset shows a Coomassie-stained gel of ATE1-2 preparation used in the experiments; ATE1-2 appears on the gel as a major band of approximately 60 kDa. Curves were normalized to the last time point on the BSA arginylation curve, taken as ‘1’. Numbers and error bars represent mean±SEM, n=3. b. Time course of [3H]-Arg into BSA from pre-synthesized [3H]-Arg-tRNA, performed in the presence or absence of RRS (curves marked as + or − RRS, respectively) or ATP (marked as + or − ATP). Curves were normalized to the last time point on the −RRS- ATP curve, taken as ‘1’. Numbers and error bars represent mean±SEM, n=5.
For the other components of the in vitro arginylation reaction, we used bacterially expressed E. coli RRS (Fig. S1), Arg-specific tRNA from E.coli, [3H]-Arg, and non-acetylated BSA as the substrate. The reaction was supplemented with ATP in the presence of K+ and Mg2+.
Since it has been previously reported that out of the four mouse ATE1 isoforms, ATE1-1 and ATE1-2 appear to have higher activity and more universal substrate specificity (Rai and Kashina, 2005; Rai et al., 2006), and ATE1-2 is the most ubiquitous ATE1 isoform in different mouse tissues (Rai and Kashina, 2005), we used ATE1-2 for the initial arginylation assay setup and characterization of the arginyl transfer reaction. We found that mixing purified ATE1-2 with BSA and the components described above results in rapid and efficient incorporation of [3H]-Arg into BSA (Fig. 1a and S1b, c). Control experiments showed that while RRS used in this reaction could efficiently catalyze the formation of [3H]-Arg-tRNA (Fig. S1c), its activity without ATE1 present in the reaction did not result in arginylation of the BSA substrate (Fig. 1a and S1b, c). Therefore, mixing purified ATE1 with the protein substrate and Arg-tRNA synthesis components results in a rapid and efficient arginylation of the protein substrate.
To estimate the efficiency of arginylation reaction, we used the estimation of the RRS activity shown in Fig. S1 and found that under these conditions approximately 10% of the synthesized Arg-tRNA is consumed in the arginylation reaction at any given moment of time in the linear range of the reaction.
Protein arginylation reaction is RRS and ATP independent
It has been previously suggested (Ciechanover et al., 1988) that in addition to its role in synthesizing the charged Arg-tRNA for the arginylation reaction, RRS is also essential for ATE1 activity by forming a complex with ATE1 during the reaction. It was also suggested that arginylation reaction is energy-dependent and ATP hydrolysis is required for the ATE1-mediated formation of protein-Arg bond. To test these two hypotheses, we used a ‘two-step’ arginylation assay, where Arg-tRNA was first synthesized by RRS in the presence of ATP, then purified away from the protein, ATP, and buffer components, and used in the in vitro arginylation by ATE1-2 in the presence or absence of ATP. As seen in Fig. 1b, no significant difference was detected between the arginylation reactions performed in the absence and presence of RRS and/or ATP. Control experiments using mass spectrometry and luminescence ATP assays showed that no RRS or ATP detectable by the assays were present in the purified Arg-tRNA preparations. Therefore, the arginylation reaction does not require the formation of an ATE1-RRS complex or the presence of ATP.
Arginyltransferase is able to modify N-terminal and internal residues of a protein substrate
Among the biggest questions in recent arginylation studies have been the discoveries that arginylation can happen on the amino acid side chain (Eriste et al., 2005; Wong et al., 2007) and in the middle of a protein (Wong et al., 2007), leaving open the question of whether such Arg incorporation can be performed by the same enzyme, and if yes – whether it requires additional cofactors in vivo. To address these questions and to identify the sites for BSA arginylation in the in vitro reaction described here, we performed arginylation as described above using non-radioactive heavy isotope labeled [13C, 15N]-Arg, and subjected the resulting arginylated BSA protein to mass spectrometry to detect the arginylated sites (Wong et al., 2007; Xu et al., 2009). The results of this analysis (Fig. S2 and Supplemental Table 2) showed that while arginylation did happen with high efficiency on the N-terminal Asp of BSA, additional internal BSA sites were also arginylated, suggesting that in addition to the N-terminally exposed residue arginyltransferase is also able to transfer Arg to internal residues in a protein substrate. Arginylation on two of these sites, Leu 139 and Cys 301 (Fig. S2c,d), has to occur by ‘conventional’ addition of Arg to the N-terminally exposed alpha amino group and therefore must involve additional pre-processing of the substrate protein, possibly by partial proteolysis. Arginylation at Asp 135 (Fig. S2b) could conceivably occur on the carboxy group of the side chain of the aspartic acid, similar to the chemistry previously described for the glutamic acid side chain (Eriste et al., 2005). Proteolytic exposure of internal alpha amino groups in a protein chain could happen either in vivo, e.g. by a previously proposed mechanism of proteolytic processing of folded proteins that could expose internal arginylation sites (Wong et al., 2007), or non-specifically during the preparation and handling of commercial BSA. The latter case is unlikely for Leu139, which was not arginylated by the in vitro reaction, but was been found to be pre-arginylated in the BSA preparation (as evidenced by the addition of non-heavy stable isotope endogenous Arg rather than Arg labeled with the heavy stable isotope, Fig. S2c), suggesting that this site has been arginylated in vivo prior to BSA isolation by the supplier.
Four ATE1 isoforms have different activity in vitro
Unlike lower organisms, the Ate1 gene in mammals encodes a total of four ATE1 isoforms produced by different combinations of four alternatively spliced exons (Kwon et al., 1999; Rai and Kashina, 2005). Previous studies using yeast complementation assays reported that two of these isoforms, ATE1-1 and ATE1-2 are highly active on substrates containing N-terminal Asp- and Glu- (Hu et al., 2006; Kwon et al., 1999; Rai and Kashina, 2005). Activity reports using this same assay, however, have been controversial for ATE1-3 and ATE1-4, shown to be high by one group (Hu et al., 2006), and low by the other with the use of a different yeast expression system (Rai and Kashina, 2005; Rai et al., 2006). To resolve the controversy and characterize the comparative activity of four ATE1 isoforms, we performed a direct comparison of the in vitro arginylation activity of these isoforms (Fig. 2). Using BSA as a substrate, we found that while the activities of ATE1-1 and ATE1-2 were high and relatively similar to each other, with ATE1-2 being slightly more active (Fig. 2a), the activity of ATE1-3 and ATE1-4 compared to the other two isoforms appeared negligibly low or absent. Plotting the activity of ATE1-3 and ATE1-4 on a smaller scale compared to negative control (Fig. 2b) showed that these two isoforms appeared to have very weak activity with the BSA substrate, amounting to 10% (ATE1-3) and 4% (ATE1-4) of the activity of ATE1-1 in the same assay.
Fig. 2. Four ATE1 isoforms have different activity toward substrates containing N-terminal Asp or Glu.

a. Time course of [3H]-Arg incorporation into BSA in the presence of individual ATE1 isoforms, and no-ATE1 control. Curves in all three panels were normalized to the last time point on the ATE1-1 curve, taken as ‘1’. ATE1-1/2 have much higher activity than ATE1-3/4. Numbers and error bars represent mean±SEM, n=5. b. Three lower curves from a plotted on a different scale to show the activity of ATE1-3 and 4. Numbers and error bars represent mean±SEM, n=5. c. Time course of [3H]-Arg incorporation into α-lactalbumin in the presence of individual ATE1 isoforms, and no-ATE1 control. Differences in the ATE1 isoform activity are consistent for different substrates. Numbers and error bars represent mean±SEM, n=3.
To test whether this difference in activity between the four ATE1 isoforms is specific to BSA, we used a different substrate, bovine α–lactalbumin, previously shown to be arginylated on the N-terminal Glu (Elias and Ciechanover, 1990) (Fig. 2c). In this assay, four ATE1 isoforms exhibited a similar pattern to that with BSA substrate: higher activity was detected with ATE1-1 and 1-2, and weaker activity was detected with 1-3 and 1-4. Therefore ATE1-3 and ATE1-4 are indeed much less active than ATE1-1/2 at least with some of the substrates containing N-terminal Asp or Glu.
Four ATE1 isoforms have distinct substrate specificity
The observed differences in activity between the four ATE1 isoforms suggest that ATE1-3 and ATE1-4 may be less enzymatically active than the other two isoforms. However, it has been previously found that these two isoforms are as capable as ATE1-1 and ATE1-2 to induce the in vivo degradation of RGS4, an N-end rule substrate containing N-terminal Cys (Davydov and Varshavsky, 2000; Lee et al., 2005; Rai and Kashina, 2005), suggesting that at least in some cases these isoforms may be as active as the other two. Therefore, it appears more likely that ATE1-3/4 are in fact as active in vivo as ATE1-1/2, but arginylate substrates different from BSA and α-lactalbumin used in our in vitro assays. To test this possibility, we performed the in vitro arginylation assay using Ate1-/- cell extract as a substrate instead of BSA, and subjected the resulting mixture to SDS-PAGE followed by autoradiography (Fig. 3 and S3a) to detect the gel positions of the proteins that incorporate labeled Arg in the presence of each ATE1 isoform. 1D-gel analysis showed that in the presence of cell extracts all four ATE1 isoforms arginylated a large number of bands, some of which were similar for different isoforms, including a prominent 90 KDa band (Fig. 3). Some other bands were arginylated in the presence of only some but not other isoforms (e.g., a prominent ∼80 kDa band, detectable only in ATE1-1 and ATE1-3-arginylated extracts, Fig. 3). To resolve this further and estimate the numbers of overlapping and isoform-specific ATE1 substrates, we subjected arginylated proteins from each reaction mixture to 2D gel electrophoresis followed by autoradiography. Computerized comparison of the positions of the arginylated protein spots on each of the four gels corresponding to individual ATE1 isoforms (Table 1, Fig. S3a, and Supplemental Table 1) showed that among the total of 144 protein spots found to be arginylated under these conditions, the majority (59) were shared between all four ATE1 isoforms, while the remaining arginylated spots were either isoform-specific or shared between some but not other ATE1 isoforms. The ATE1 isoform arginylating the largest number of protein spots was ATE1-2 (108 protein spots on 2D gels); this isoform was also previously found to be the most ubiquitously expressed in different mouse tissues (Rai and Kashina, 2005) and apparently the most active one in the in vitro assays (Fig. 2). The other three isoforms arginylated 74, 92, and 90 protein spots (seen on 2D gels with ATE1-1, 3, and 4, respectively; see also Table 1 for the spot number breakdown). These results suggest that despite the observed differences in vitro and in yeast complementation assays using protein substrates with N-terminally exposed Asp and Glu, all four ATE1 isoforms likely have high activity in vivo and arginylate partially overlapping but distinct subsets of substrates.
Fig. 3. Four ATE1 isoforms arginylate distinct, partially overlapping pools of substrates.
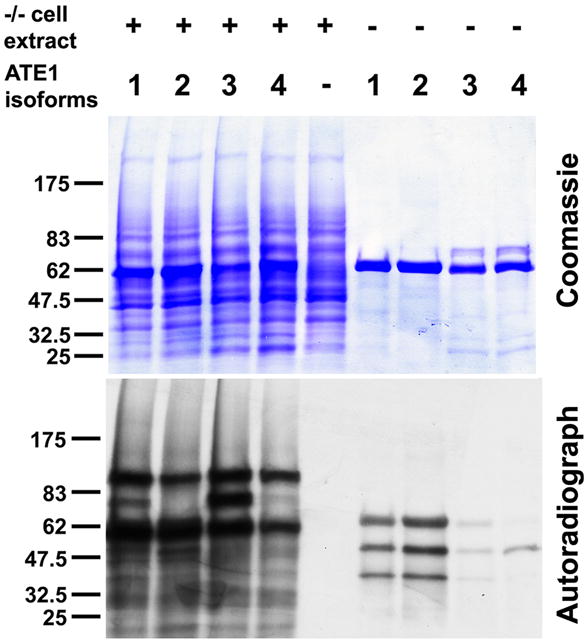
Images of a Coomassie-stained gel (top) and autoradiograph (bottom) of arginylation reactions using Ate1-/- cell extracts as substrate (left five lanes) or buffer as control (right four lanes). Numbers 1-4 represent ATE1 isoforms. Middle lane had no ATE1 added, resulting in no incorporation of radioactive Arg. See Fig. S3a for 2D gel comparisons of samples similar to those used in the left four lanes and Supplemental Table 1 for the results of computerized comparison of the gels. See Table 1 for the complete count of protein spots arginylated by different ATE1 isoforms.
Table 1. Number of protein spots arginylated in cell extracts by different ATE1 isoforms.
| Isoform | # of spots |
|---|---|
| ATE1-1 | 2 |
| ATE1-2 | 39 |
| ATE1-3 | 11 |
| ATE1-4 | 6 |
| ATE1-1/2 | 2 |
| ATE1-1/4 | 2 |
| ATE1-2/4 | 1 |
| ATE1-3/4 | 6 |
| ATE1-2/3/4 | 7 |
| ATE1-1/3/4 | 9 |
| ATE1-1/2/3/4 | 59 |
| Total | 144 |
Differential activity of ATE1 isoforms is defined by cofactors from the cell extracts
While the experiments described above suggest that ATE1-3/4 may be as active as the other two ATE1 isoforms with a certain subset of substrates, it is still unclear whether their inability to efficiently arginylate BSA and α–lactalbumin in vitro reflects the situation in vivo. It has been previously suggested that ATE1 enzymes by themselves have low specificity for the arginylation sites, and their specificity in vivo is determined by other cofactors bound to these enzymes (Wong et al., 2007). To test whether endogenous protein or non-protein factors could aid ATE1-3/4, we performed the in vitro α-lactalbumin arginylation assay in the presence of Ate1-/- cell extracts and compared the arginylation efficiency of α-lactalbumin band in the absence and presence of the extracts by autoradiography.
While, consistent with the assays shown in Fig. 2, ATE1-3/4 in the absence of cell extracts showed very poor ability to arginylate α–lactalbumin (Fig. 4, right), the activity of these two isoforms in the presence of cell extracts appeared to increase (Fig. 4, left), suggesting that added cell extract does have an effect on the ability of ATE1-3/4 to arginylate α–lactalbumin. Addition of increasing doses of cell extract to the reaction mixture (Fig. S4) showed that at lower concentrations Ate1-/- extract was indeed able to ‘rescue’ the activity of ATE1-3/4 toward α–lactalbumin, however the increase of the extract: substrate ratio negated this effect, possibly due to the competition between α–lactalbumin and the endogenous substrates found in the cell extract. These experiments suggest that additional cofactors in the cell extract are needed to increase the activity of ATE1 isoforms toward unfavorable substrates, either via direct interaction or via additional level of posttranslational regulation, supporting the hypothesis that substrate recognition and/or catalysis by ATE1 in some cases is aided by other proteins in vivo.
Fig. 4. ATE1-3 and ATE1-4 are partially aided by factors from cell extracts in arginylating α-lactalbumin.
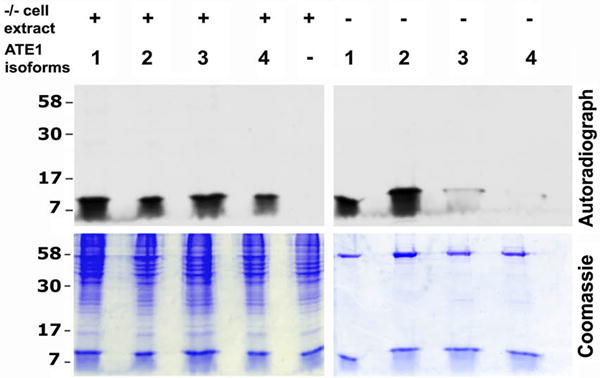
Top, autoradiographs of the gels on the bottom, showing incorporation of [14C]-Arg into α-lactalbumin by each of the ATE1 isoforms (labeled 1-4 on top) in the presence (left) or absence (right) of the added cell extract. Control lane shows arginylation reaction without the addition of ATE1. Consistent with the results of the scintillation counting (Fig. 2), ATE1-3/4 have much weaker activity toward α-lactalbumin than ATE1-1/2. Addition of cell extracts enhances the ability of ATE1-3/4 to arginylate α-lactalbumin. See also Fig. S4.
ATE1 is capable of self-arginylation
During the autoradiography to detect the activity of individual ATE1 isoforms in the presence of cell extracts (Fig. 3), in vitro arginylation reactions without the added extracts were used as a control (right lanes on the gel and autoradiograph in Fig. 3). Remarkably, significant Arg incorporation into the ATE1 gel bands was detected in these experiments, suggesting that in the absence of other substrates, ATE1 isoforms undergo self-arginylation. Arginylated protein bands corresponding to ATE1 molecular weight were also detected in cell extracts (Fig. 3, left lanes), suggesting that self-arginylating activity of ATE1 isoforms is independent of the presence of other substrates and cell extract-specific cofactors. In addition to the full length ATE1 (∼60 kDa), minor 50 kDa and 40 kDa bands were also significantly arginylated in purified ATE1 preparations; control immunoblots with ATE1 antibody suggest that these bands may represent truncated ATE1 fragments, since minor bands corresponding to these ATE1 fragments were present in the His-tag-purified ATE1 preparation (Fig. S3b).
To test whether ATE1 can indeed undergo self-arginylation, we performed a separate arginylation assay using purified ATE1 isoforms without added substrates, and [13C, 15N]- heavy isotope labeled Arg for the transfer reaction, and analyzed ATE1 isoforms in the reaction mixture by mass spectrometry to detect the arginylated sites (Fig. 5, Figs. S5, and Supplemental Table 2). This assay revealed several heavy isotope-labeled arginylation sites in each isoform, located on the N-terminus and internal amino acid residues (as marked in Fig. 5). In addition to the heavy isotope-labeled sites, a number of sites, especially in ATE1-3/4, were labeled by ‘light’ Arg, indicating that arginylation on these sites happened during ATE1 expression in E. coli. Some sites (A2, S3, D117/D110, S251/S244 and N485/N478) were conserved between two or more isoforms, others (Y54 and G411) appeared to be ATE1-4 specific. Their identity may partially underlie the different activity of ATE1 isoforms. While method limitations precluded us from accurately determining the stoichiometry of arginylation, a crude estimation by the number of detected peptides in isoforms ATE1-1/2 suggested that arginylation was the most abundant on residues 2 and 3 from the N-terminus. Similar sites in both isoforms were also found acetylated, suggesting that arginylation and acetylation may both be involved in this previously unknown N-terminal processing of ATE1. This result suggests that ATE1 enzymes undergo self-arginylation, a process that may be essential for their in vivo function.
Fig. 5. ATE1 is capable of self-arginylation in vitro and in vivo.
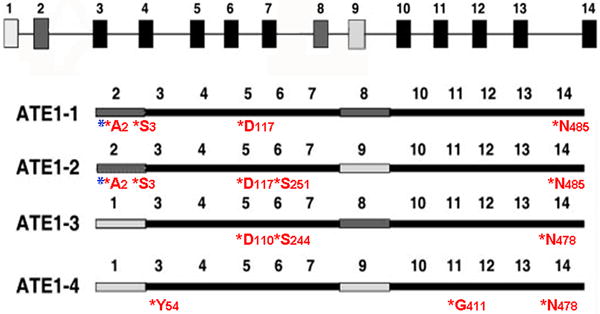
Top, schematic representation of Ate1 gene, with the alternatively spliced exons shaded in dark and light gray. Bottom, exon maps of four ATE1 isoforms, showing the location of the arginylated sites. For each site, red star indicates arginylation, letters and numbers represent amino acid residue and its position within the polylpeptide. Blue stars represent the sites that can also undergo acetylation. See Fig. S5 for the arginylated peptide mass spectra, and Supplemental Table 2 for the peptide parameters.
ATE1 forms different functional complexes in vivo
To gain insights into the molecular interactions of ATE1 isoforms in vivo, we fractionated crude cell extracts on a wide-range sucrose density gradient (0-70%) and analyzed the ATE1 distribution between different intracellular fractions (Fig. 6a and S6a). Under these conditions, ATE1 sedimented as two peaks – one at the top of the gradient corresponding to the soluble cytosolic pool, and one in the middle of the gradient, corresponding to the mid-size cytoplasmic organelles (smaller than mitochondria and larger than soluble protein oligomers). Probing the same fractions with different intracellular markers suggested that ATE1 fully co-localized with the elongation factor eEF1B2 (a component of the translation machinery), and its higher density peak co-localized with the microsomal fraction probed by the endoplasmic reticulum maker cytochrome P450 oxidoreductase (CYPOR). In addition, co-localization of ATE1 with the ribosomal protein L7a was also observed in most fractions, excluding the nucleus and the mitochondria. No significant co-localization of ATE1 with mitochondria (probed by anti-cytochrome C oxidase, COX IV) was observed. Despite its demonstrated ability to modify actin cytoskeleton, ATE1 was not associated with the major pool of actin filaments (found in the bottom fractions co-fractionating with the mitochondria and the nuclei), however it partially co-localized with the tubulin/microtubule pool. Fractionation of only the soluble ATE1 peak (high-speed supernatant fraction of the cell lysate, Fig. S6b) confirmed the co-localization of ATE1 with eEF1B2, and the absence of stable association with the tubulin pool. Thus, ATE1 in vivo exists in two different pools that likely reflect its involvement in two molecular complexes, one soluble, and one at least partially associated with the translation machinery, ribosomes and the endoplasmic reticulum.
Fig. 6. ATE1 in cells forms pools that co-localize with different intracellular fractions.
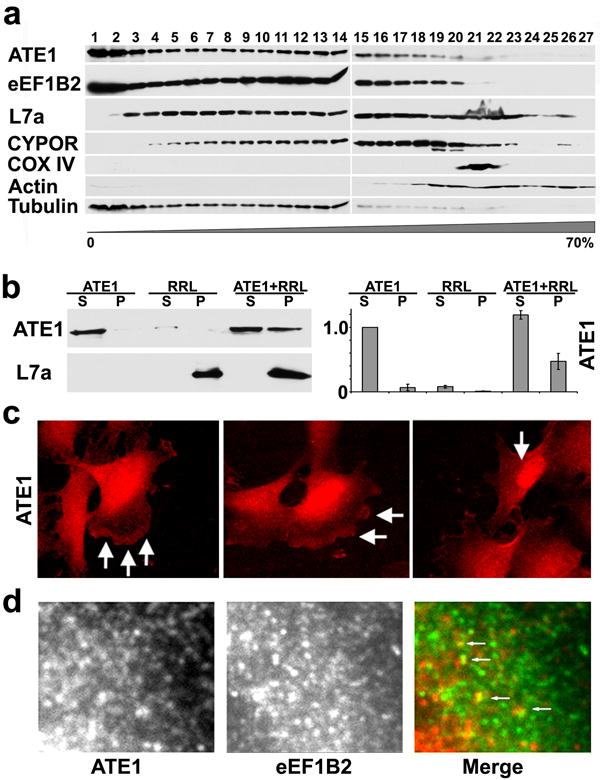
a. 0-70% sucrose gradient fractions of the whole cell lysate probed with antibodies to different intracellular markers, including ATE1, component of the translation machinery eEF1B2, ribosomal protein L7a, microsomal/ER marker CYPOR, mitochondrial marker COX IV, and cytoskeletal markers actin and tubulin. ATE1 localizes in two distinct pools, one soluble and one associated with ER and the translation machinery. b. Co-sedimentation of ATE1 from mouse liver extracts with the ribosomes from rabbit reticulocyte lysates (RRL). Substantial amounts of ATE1 are found in the pellets in the presence but not the absence of the ribosomes. c. Intracellular localization of ATE1 using rat monoclonal anti-ATE1 recognizing all four isoforms shows distinct localization patterns in the nucleus and cytoplasm, with enrichment at the cell leading edge (arrows in middle and left image) and in the nucleus (arrows in right image). d. ATE1 in the cytoplasm forms dots and punctate patterns that partially co-localize with eEF1B2 (arrows in right image show colocalization of ATE1 (red) and eEF1B2 (green)).
To further test whether ATE1 can associate with the ribosomes, we performed cosedimentation assays between ATE1 from mouse liver cell extracts, and the ribosomes from rabbit reticulocyte lysates (RRL) (Fig. 6b). While a small amount of ATE1 sedimented through the sucrose cushion in the absence of the ribosomes (Fig. 6b, left lanes), suggesting this enzyme's ability to aggregate, a much larger fraction of ATE1 was found when it was pelleted in the presence of the ribosomes (Fig, 6b, right lanes), indicating its ability to bind to the ribosomes in this assay. We also used ATE1 antibody to study the intracellular localization of ATE1 protein in cultured mouse embryonic fibroblasts (Fig. 6c). It has been previously found that overexpressed ATE1-GFP fusion proteins localize in distinct intracellular patterns, diffuse in the cytoplasm with or without enrichment at the cell leading edge, in some cases highly concentrated in the nucleus (Kwon et al., 1999; Rai and Kashina, 2005). Consistent with these observations, ATE1 in mouse embryonic fibroblasts localized diffusely, with prominent enrichment at the leading edge (arrows in the left two panels in Fig. 6c) and in some cases – in the nucleus (arrow in the rightmost panel in Fig. 6c). ATE1 in the cytoplasm localized in distinct dots, which partially overlapped with the dots stained by anti-eEF1B2 (Fig. 6d), confirming the results of the sucrose gradient fractionation and suggesting that ATE1 in cells partially associates with the ribosomes and the translation machinery in addition to its ribosome-independent localization in the cytoplasmic pool.
Discussion
The results described above constitute the first detailed study of arginyltransferase in vitro and in vivo, providing insights into the complexity of protein arginylation and some of the mechanisms that ensure efficient ATE1-dependent protein regulation. Our data show that ATE1 requires nothing else but a charged Arg-tRNA and a protein substrate to conduct the reaction, and that this enzyme undergoes self-arginylation in vitro and in vivo, which may constitute a previously unknown step in the regulation of its activity and in vivo function. Our data also demonstrate that different ATE1 isoforms can arginylate distinct, partially overlapping subsets of substrates, suggesting that these isoforms perform different functions in vivo, and that ATE1 enzymes are aided by in vivo cofactors and form distinct molecular complexes associated with different subcellular structures.
It has been previously found (Eriste et al., 2005; Wong et al., 2007) that posttranslational addition of Arg can happen not only on the N-terminally exposed amino acid residue, but also on the internal residues, presumably via a novel chemistry that couples Arg amino group to the carboxyl groups of the side chains of Asp and Glu (Eriste et al., 2005). Our data with in vitro arginylation of the internal Asp in BSA suggest that this reaction may be mediated by ATE1 itself, without additional cofactors or enzymes, suggesting that ATE1 can employ different mechanisms for Arg addition and that arginylation on the side chains of acidic residues seen in vivo may also be also conducted by the same enzyme.
Previous characterization of ATE1 isoforms resulted in controversial reports, where two independent groups showed that ATE1-3 and ATE1-4 can have different activity toward the same substrates in different experimental systems (Hu et al., 2006; Rai and Kashina, 2005; Rai et al., 2006). Our data, showing that ATE1-3 and ATE1-4 have low activity toward some substrates in vitro but are apparently aided by intracellular cofactors to achieve efficient Arg transfer onto the same substrates in cell extracts, resolves this controversy, suggesting that the discrepancies observed by the two groups are due most likely to the action of specific cofactors found in different experimental systems.
It has been previously suggested (Ciechanover et al., 1988) that ATE1 forms a hexamer containing 3 molecules of ATE1 and 3 molecules of RRS, and that the formation of this complex is required for ATE1's function. Our data, however, suggest that RRS is not a required component of arginyl transfer reaction.
Our results that ATE1 can undergo self-arginylation in vitro and in vivo suggest that this enzyme may self-regulate its activity in application to its diverse biological functions. Our in vitro Arg incorporation data (Fig. 1a) suggest that the levels of ATE1 self-arginylation are negligibly low, indistinguishable from the background. These data may reflect the low level of self-arginylation in vivo. An alternative intriguing possibility, however, is that ATE1 self-arginylation levels are actually high, but the majority of it happens during its expression in E.coli, blocking the arginylation sites that could have otherwise been utilized in the in vitro reaction. In support of this, we find that some ATE1 sites are modified by ‘light’ Arg and not the heavy-isotope-labeled Arg derivative used in our in vitro reaction. Overall, the question of the abundance and the exact role of ATE1 self-arginylation is an intriguing subject for further investigation.
Based on our results, we propose a model of ATE1 molecular interactions (Fig. 7). While a small portion of ATE1 under certain conditions localizes to the nucleus (not shown), a prominent subset of ATE1 is found in the soluble, cytosolic fraction, where it presumably forms complexes with different cofactors. Another subset is associated with ribosomes and/or ER. While this pool of ATE1 can form complexes with the same proteins and cofactors, it is possible that its intracellular role includes co-translational arginylation of proteins on the ribosomes, accounting for the arginylation on the internal sites of the polypeptides and the side chains of certain amino acid residues. Finally, under some physiological conditions, a subset of ATE1 localizes in the nucleus, where it may mediate arginylation of specific nuclear proteins. Biological role of these different ATE1 pools and the identity of arginylated proteins constitute an exciting direction of further study.
Fig. 7. Model of ATE1 molecular interactions.

ATE1 enzyme forms two different cytoplasmic pools, one cytosolic and one ribosome-bound, and can arginylate protein N-termini and side chains. Its association with ribosomal proteins facilitates co-translational arginylation, allowing ATE1 to effectively compete with acetylation and to modify proteins in the most efficient way.
Significance
Our study reports the first characterization of arginyltransferase using in vitro arginylation reaction, that followed nearly 40 years of attempts to determine which factors are essential to conduct this enigmatic protein modification. Using this system, we found for the first time that arginylation requires no additional factors besides the ATE1 enzyme, the charged Arg-tRNA, and the protein substrate. We also found that ATE1 can transfer Arg not only to the N-terminus, but also to the internal sites in a protein substrate, and that it is capable of self-arginylation which is likely involved in its in vivo regulation. Our system enabled us to address a recent controversy about the differences in mammalian ATE1 isoforms and show that these isoforms have partially overlapping but distinct subsets of substrates and that they are aided by intracellular cofactors that facilitate their functions. Finally, we found that ATE1 can form different in vivo complexes and associate with the components of the translation machinery. These data constitute the first mechanistic insights into the arginylation reaction and are an essential first step that would enable further studies to understand the molecular components and mechanisms involved in this poorly understood posttranslational modification.
Experimental Procedures
Cells and Escherichia coli strains
For immunostaining and cell extract preparation, immortalized mouse embryonic fibroblasts derived from wild type and Ate1-/- mouse embryos as previously described (Karakozova et al., 2006) were used. For protein expression, BL21-CodonPlus® (DE3)-RIL and BL21(DE3) were purchased from Stratagene (USA).
Expression and purification of the recombinant mouse ATE1 isoforms and E. coli Arg-tRNA synthetase
cDNAs encoding mouse ATE1 isoforms (Rai and Kashina, 2005) were subcloned into pET29a (Novagen) containing C-terminal His6 tag followed by TEV cleavage site. ATE1 isoforms were expressed in BL21-CodonPlus® (DE3)-RIL E. coli strain using LB medium supplemented with 50mg/ml kanamycin at 37 °C. When the optical density (OD 600nm) of the bacterial culture reached 0.4-0.5, the culture was cold shocked on ice for 30 min, and then induced with 0.8 mM IPTG (isopropyl-1-thiod-galactopyranoside) for 18 h at 16°C. For ATE1 purification, cells were collected by centrifuging at 6,000 rpm for 30 min, resuspended in lysis buffer 50 mM Tris, 5 mM imidazole, pH7.5, containing 0.5 M NaCl, 1mM MgCl2, 10 mM β-mercaptoethanol, and 1 mM PMSF and lysed using a French press at 4 °C. The lysate was centrifuged at 20,000 rpm for 30 min, and the supernatant was loaded to the Ni-NTA agarose column (Qiagen) equilibrated with the lysis buffer, followed by washing in 50 mM Tris, 25 mM imidazole, pH 7.5 containing 1M NaCl, 1mM MgCl2, and 10 mM β-mercaptoethanol. Protein was eluted with 50mM Tris, 0.5M imidazole pH 7.5, containing 0.5 M NaCl, 1 mM MgCl2, and 10 mM β-mercaptoethanol, and dialyzed against 50 mM HEPES, pH 7.5, containing 2 mM DTT, 1M NaCl, 0.2 mM EDTA, 2 mM MgCl2 overnight at 4 °C. E. coli arginyl-tRNA synthetase was subcloned into pTYB1 (New England Biolabs) and expressed in BL21(DE3) in LB supplemented with 100mg/ml ampicillin at 37 °C, followed by induction with 0.5 mM IPTG for 16 hrs at 37 °C. Cells were collected by centrifugation at 6000 rpm for 30 min and resuspended in lysis buffer (0.5 M NaCl, 20 mM Tris, 1mM EDTA, 0.1% Triton X -100, 1 mM PMSF, pH 8.5), followed by lysis using a French press at 4 °C. Cell lysates were clarified by centrifugation at 20,000 rpm for 30 min and loaded onto chitin column (New England Biolabs) equilibrated with the lysis buffer. The column was quickly flushed with lysis buffer containing 30 mM DTT and left at 4°C overnight for on-column cleavage. Protein was eluted using 3 column volumes of lysis buffer without DTT. Final preparations of ATE1-1, 2, 3, and 4 were mixed with equal volume of glycerol and kept at -80°C. Dialysis and storage of purified E. coli RRS were the same as ATE1. The estimated purity of each purification product was > 90%; about 5 mg of ATE1 forms or 2 mg of RRS was obtained per liter of LB, respectively.
Assay of ATE1 activity
In vitro assay of ATE1 was derived and modified from (Ciechanover et al., 1988). L-[2,3-3H]-Arginine (Sigma; 40 Ci/mmol), L-[2,3,4,5-3H]-Arginine (GE Healthcare; 54 Ci/mmol), L-[14C(U)]-Arginine (Moravek Biochemicals; 310 mCi/mmol), and L-[13C,15N]-Arginine (Pierce) dissolved in water to 100 μM were used throughout the project. A typical reaction was performed in 50 μl volume, containing 50 mM Hepes, pH 7.5, 25 mM KCl, 15 mM MgCl2, 0.1 mM DTT, 2.5 mM ATP, 12.5 uM [3H]-Arginine, 40 uM tRNAArg, 2 μg RRS, 1 μg ATE1 and 8.3 μM BSA as a substrate. Reaction was mixed and kept on ice for 0 min time point, then started by placing into a heat block at 37 °C, and a 10 μl aliquot was taken out at each time point (usually at 0, 10, 20, 30 and 40 min). For scintillation counting, samples at each time point were immediately quenched into 40 μl of 20% Trichloroacetic acid (TCA) containing 1 mM of unlabeled Arg and kept at room temperature for at least 10 min, followed by heating at 95°C for 15 min (to destroy the excess of the labeled Arg-tRNA), cooling down on ice for 20 min, and spinning at 13,000 rpm for 30 min at room temperature to collect the pellets containing precipitated proteins. Pellets were washed 3× with 5% cold TCA and once with cold acetone, air dried, and counted in a liquid scintillation counter. Control reactions excluding ATE1, RRS, or BSA, plus a control reaction for RRS activity (no ATE1, no heating) were used as indicated in the figures. ATE1 activity was evaluated as counts per minute (cpm) of [3H]-Arg at each time point. For autoradiography, [14C]-Arg instead of [3H]-Arg was used in the reaction. Samples at each data point were mixed 1:1 with 2× SDS sample buffer and boiled. For mass spectrometry, [13C,15N]-Arg was used in the reaction. Samples after 40 min of arginylation were TCA precipitated and processed as described in (Wong et al., 2007; Xu et al., 2009). Different substrates, including non-acetylated BSA, α-lactalbumin (Sigma), or Ate1-/- cell extract (prepared by the lysis of Ate1-/- fibroblasts followed by centrifugation at 67,917g and addition of 100 μg/ml of cycloheximide to block the potential translation) were used as indicated in the text and figures. For RRS and ATP-independent assays shown in Fig. 1b, the arginylation reaction was performed in two steps. First, [3H]-Arg was mixed with all the reaction components except ATE1 and incubated at 37 °C for 60 min for RRS-mediated coupling to tRNA. Then 0.2 U/ul hexokinase and glucose-6-phosphate dehydrogenase (Sigma),10 mM glucose, and 10 mM ß-nicotinamide adenine dinucleotide phosphate (Sigma) were added to the reaction and incubated at 37 °C for 30 min to deplete the ATP. The resulting [3H]-Arg-tRNA was extracted 3× with chloroform, and further purified with RNesay Mini Kit(Qiagen) with slight modifications: 700 μl 100% ethanol was added to each 100μl of Arg-tRNA solution: 350 μl buffer RLT mixture instead of the 250 μl recommended by the manufacturer. 1/10 volume of [3H]-Arg-tRNA was taken for liquid scintillation counting to monitor the efficiency of the RRS reaction. Absense of the RRS and ATP contamination in the final preparation was confirmed by mass spectrometry and FLAAM ATP assay detection kit (Sigma) using Luminoskan Ascent(Thermo scientific), respectively. As the second step of the reaction, the purified [3H]-Arg-tRNA was used as donor for the ATE in vitro assay.
2D gel electrophoresis and comparison
Two-dimensional electrophoresis was performed according to the carrier ampholine method (O'Farrell, 1975) by Kendrick Labs, Inc. (Madison, WI) as described in the Supplemental Experimental Procedures.
Computerized Comparisons
Films from each sample were scanned with a laser densitometer (Model PDSI, Molecular Dynamics Inc, Sunnyvale, CA). The scanner was checked for linearity prior to scanning with a calibrated Neutral Density Filter Set (Melles Griot, Irvine, CA). The images were analyzed using Progenesis Same Spots software (version 3.2, Nonlinear Dynamics) and Progenesis PG240 software (version 2006), Nonlinear Dynamics. The general method of computerized analysis for each included image warping followed by spot finding, background subtraction (average on boundary), matching, and quantification in conjunction with detailed manual checking. Spot percentage equal to spot integrated density above background (volume) was expressed as a percentage of total density above background of all spots measured. Difference was defined as fold-change of spot percentages.
MW and pI Measurements
Approximate isoelectric point (pI) measurements were made based on the pH gradient plot included with the gels for this batch of ampholines for conditions of 9M urea and room temperature of 22°C. The molecular weight and pI values for each spot were determined from algorithms applied to the reference image.
Sucrose gradient fractionation of cell lysates
Livers from wild type mice were removed immediately following euthanasia, washed with ice cold lysis buffer (100mM KCl, 50 mM Tris, pH 7.0), minced with scissors, mixed with 1:1 (w/v) lysis buffer supplemented with 20 μM mammalian Protease Inhibitor Cocktail (Sigma), and homogenized in a Potter-Elvehjem homogenizer with a motor-driven Teflon pestle using 6-8 strokes at 2,500 rpm. Liver homogenates were filtered through four layers of cheese cloth. For whole cell fractionation, filtered homogenates were loaded onto 0-70% linear sucrose gradient in lysis buffer with protease inhibitors, and centrifuged at 77,175g for 1.5 hours at 4°C using SW 41 Ti rotor (Beckman). After centrifugation, 400 μl fraction each was collected from the top and 10 μl aliquot from each fraction was taken for gel analysis. For analysis of the soluble ATE1 pool, filtered homogenates were centrifuged sequentially at 1,500g, 16,000g, and 67,917g at 4°C for 30 min. At each step, gel samples of supernatant and resuspended pellet were collected for SDS PAGE. 500 μl supernatant from the last centrifugation step was loaded onto 5-20% linear sucrose gradient in lysis buffer with protease inhibitors, and centrifuged at 84,168g for 14 hours at 4°C using SW 50.1 rotor (Beckman). After centrifugation, 250 μl fractions were collected from the top and a 10 μl aliquot from each fraction was taken for SDS PAGE. Fractions from both gradients were analyzed by Western blots using rat monoclonal anti-ATE1 antibodies raised by Absea, Inc. against purified His6-tagged mixture of ATE1-1 and ATE1-2 that recognize all four ATE1 isoforms (Kurosaka et al., 2010); rabbit polyclonal antibodies to eEF1B2 (Abcam); rabbit polyclonal antibodies to the Ribosomal Protein L7a (Cell Signaling Technology); anti-actin (Cytoskeleton, Inc.); mouse anti-β-Tubulin (Sigma); mouse anti-cytochrome c oxidase subunit 4 isoform (COX IV) (Mitosciences); mouse anti- cytochrome P450 oxidoreductase (CYPOR) (Santa Cruz Biotechnology); and mouse anti- dynein intermediate chain (DIC) (Fisher Scientific). Western blot quantifications were performed using ImageQuant TL (GE Healthcare).
Ribosome cosedimentation assays were performed by ultracentrifugation as described in the Supplemental Experimental Procedures.
Mass spectrometry and database searches was previously described (Wong et al., 2007; Xu et al., 2009). See Supplemental Experimental Procedures for the detailed description.
Highlights.
Developed pure in vitro arginylation assay and characterized mammalian arginyltransferases
ATE1 is ATP-independent and capable of self-arginylation
ATE1 activity is modulated by intracellular cofactors
ATE1 isoforms have different subsets of substrates, suggesting different functions
ATE1 associates with the translation machinery and forms two distinct complexes in vivo
Supplementary Material
Acknowledgments
We are grateful to Dr. Roberto Dominguez and Dr. Francois Ferron for their help in developing the purification and expression systems for ATE1 and E. coli RRS, Kendrick Laboratories, Inc. for performing the 2D gel analysis of the ATE1 isoform substrates, Dr. Claire Mitchell and Jason Lim for help with ATP detection assays, Chao-Xing Yuan and the University of Pennsylvania proteomics core facility for the mass spectrometry analysis of Arg-tRNA preparations, Dr. J. Pehrson and Dr. S. Kurosaka for helpful discussions throughout the project, and Dr. N. G. Avadhani and Dr. S.Y. Fuchs for critical reading of the manuscript. This work was supported by NIH grant 5R01HL084419, W.W.Smith Charitable Trust, and Philip Morris Research Management Group awards to A.K, American Heart Association award 0560027Z to A.W., and NIH grant P41 RR011823 to JRY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- Balzi E, Choder M, Chen WN, Varshavsky A, Goffeau A. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J Biol Chem. 1990;265:7464–7471. [PubMed] [Google Scholar]
- Ciechanover A, Ferber S, Ganoth D, Elias S, Hershko A, Arfin S. Purification and characterization of arginyl-tRNA-protein transferase from rabbit reticulocytes. Its involvement in post-translational modification and degradation of acidic NH2 termini substrates of the ubiquitin pathway. J Biol Chem. 1988;263:11155–11167. [PubMed] [Google Scholar]
- Davydov IV, Varshavsky A. RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J Biol Chem. 2000;275:22931–22941. doi: 10.1074/jbc.M001605200. [DOI] [PubMed] [Google Scholar]
- Decca MB, Carpio MA, Bosc C, Galiano MR, Job D, Andrieux A, Hallak ME. Post-translational arginylation of calreticulin: A new isospecies of calreticulin component of stress granules. J Biol Chem. 2006 doi: 10.1074/jbc.M608559200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias S, Ciechanover A. Post-translational addition of an arginine moiety to acidic NH2 termini of proteins is required for their recognition by ubiquitin-protein ligase. J Biol Chem. 1990;265:15511–15517. [PubMed] [Google Scholar]
- Eriste E, Norberg A, Nepomuceno D, Kuei C, Kamme F, Tran DT, Strupat K, Jornvall H, Liu C, Lovenberg TW, Sillard R. A novel form of neurotensin post-translationally modified by arginylation. J Biol Chem. 2005;280:35089–35097. doi: 10.1074/jbc.M502567200. [DOI] [PubMed] [Google Scholar]
- Hu RG, Brower CS, Wang H, Davydov IV, Sheng J, Zhou J, Kwon YT, Varshavsky A. Arginyltransferase, its specificity, putative substrates, bidirectional promoter, and splicing-derived isoforms. J Biol Chem. 2006;281:32559–32573. doi: 10.1074/jbc.M604355200. [DOI] [PubMed] [Google Scholar]
- Kaji A, Kaji H, Novelli GD. A soluble amino acid incorporating system. Biochem Biophys Res Commun. 1963a;10:406–409. doi: 10.1016/0006-291x(63)90546-1. [DOI] [PubMed] [Google Scholar]
- Kaji H. Amino-terminal arginylation of chromosomal proteins by arginyl-tRNA. Biochemistry. 1976;15:5121–5125. doi: 10.1021/bi00668a027. [DOI] [PubMed] [Google Scholar]
- Kaji H, Novelli GD, Kaji A. A Soluble Amino Acid-Incorporating System from Rat Liver. Biochim Biophys Acta. 1963b;76:474–477. [PubMed] [Google Scholar]
- Karakozova M, Kozak M, Wong CC, Bailey AO, Yates JR, 3rd, Mogilner A, Zebroski H, Kashina A. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science. 2006;313:192–196. doi: 10.1126/science.1129344. [DOI] [PubMed] [Google Scholar]
- Kurosaka S, Leu NA, Zhang F, Bunte R, Saha S, Wang J, Guo C, He W, Kashina A. Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS Genet. 2010;6:e1000878. doi: 10.1371/journal.pgen.1000878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW, Du F, Varshavsky A. An essential role of N-terminal arginylation in cardiovascular development. Science. 2002;297:96–99. doi: 10.1126/science.1069531. [DOI] [PubMed] [Google Scholar]
- Kwon YT, Kashina AS, Varshavsky A. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol Cell Biol. 1999;19:182–193. doi: 10.1128/mcb.19.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamon KD, Kaji H. Arginyl-tRNA transferase activity as a marker of cellular aging in peripheral rat tissues. Exp Gerontol. 1980;15:53–64. doi: 10.1016/0531-5565(80)90023-6. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Tasaki T, Moroi K, An JY, Kimura S, Davydov IV, Kwon YT. RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc Natl Acad Sci U S A. 2005;102:15030–15035. doi: 10.1073/pnas.0507533102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- Rai R, Kashina A. Identification of mammalian arginyltransferases that modify a specific subset of protein substrates. Proc Natl Acad Sci U S A. 2005;102:10123–10128. doi: 10.1073/pnas.0504500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Mushegian A, Makarova K, Kashina A. Molecular dissection of arginyltransferases guided by similarity to bacterial peptidoglycan synthases. EMBO Reports. 2006;7:800–805. doi: 10.1038/sj.embor.7400747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Wong CC, Xu T, Leu NA, Dong DW, Guo C, McLaughlin KJ, Yates JR, 3rd, Kashina A. Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development. 2008;135:3881–3889. doi: 10.1242/dev.022723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soffer RL. Enzymatic modification of proteins. II. Purification and properties of the arginyl transfer ribonucleic acid-protein transferase from rabbit liver cytoplasm. J Biol Chem. 1970;245:731–737. [PubMed] [Google Scholar]
- Soffer RL, Mendelsohn N. Incorporation of arginine by a soluble system from sheep thyroid. Biochem Biophys Res Commun. 1966;23:252–258. doi: 10.1016/0006-291x(66)90537-7. [DOI] [PubMed] [Google Scholar]
- Wang YM, Ingoglia NA. N-terminal arginylation of sciatic nerve and brain proteins following injury. Neurochem Res. 1997;22:1453–1459. doi: 10.1023/a:1021998227237. [DOI] [PubMed] [Google Scholar]
- Wong CCL, Xu T, Rai R, Bailey AO, Yates JR, Wolf YI, Zebroski H, Kashina A. Global Analysis of Posttranslational Protein Arginylation. PLoS Biology. 2007;5:e258. doi: 10.1371/journal.pbio.0050258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu NS, Chakraborty G, Hassankhani A, Ingoglia NA. N-terminal arginylation of proteins in explants of injured sciatic nerves and embryonic brains of rats. Neurochem Res. 1993;18:1117–1123. doi: 10.1007/BF00978361. [DOI] [PubMed] [Google Scholar]
- Xu T, Wong CCL, Kashina A, Yates JR., III Identification of posstranslationally arginylated proteins and peptides by mass spectrometry. Nature Protocols. 2009;43:325–332. doi: 10.1038/nprot.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.