

Abstract
Vascular anomalies are localized defects of vascular development. Most of them occur sporadically, i.e. there is no familial history of lesions, yet in a few cases clear inheritance is observed. These inherited forms are often characterized by multifocal lesions that are mainly small in size and increase in number with patient’s age. On the basis of these inherited forms, molecular genetic studies have unraveled a number of inherited mutations giving direct insight into the pathophysiological cause and the molecular pathways that are implicated. Genetic defects have been identified for hereditary haemorrhagic telangiectasia (HHT), inherited cutaneomucosal venous malformation (VMCM), glomuvenous malformation (GVM), capillary malformation - arteriovenous malformation (CM-AVM), cerebral cavernous malformation (CCM) and some isolated and syndromic forms of primary lymphedema. We focus on these disorders, the implicated mutated genes and the underlying pathogenic mechanisms. We also call attention to the concept of Knudson’s double-hit mechanism to explain incomplete penetrance and the large clinical variation in expressivity of inherited vascular anomalies. This variability renders the making of correct diagnosis of the rare inherited forms difficult. Yet, the identification of the pathophysiological causes and pathways involved in them has had an unprecedented impact on our thinking of their etiopathogenesis, and has opened the doors towards a more refined classification of vascular anomalies. It has also made it possible to develop animal models that can be tested for specific molecular therapies, aimed at alleviating the dysfunctions caused by the aberrant genes and proteins.
Keywords: Angiogenesis, pathogenesis, disorder, disease, genetic, gene, hemangioma, vascular malformation, venous malformation, capillary malformation, arteriovenous malformation, lymphatic malformation, lymphedema, VM, GVM, CM, CM-AVM, AVM, LM, TIE2, GLMN, RASA1, CCM, ENG, ACVRL1, SMAD4, VEGFR3, FOXC2, SOX18, CCBE1
Introduction
Vascular anomalies are histopathologically characterized by a focal increase in the number of vessels that are abnormally tortuous and enlarged. This is likely due to localized defects in vascular development during vasculogenesis and especially during angiogenesis [1]. Vasculogenesis is defined as vessel growth from embryonic cells: hemangioblasts (mesoderm derived precursors) that give rise to angioblasts (endothelial precursors) and hemocytoblasts (blood cell precursors). Angioblast fusion takes place in “vascular islets” inducing the formation of the primary capillary plexus. During angiogenesis, this primary capillary system extends and matures. It involves both endothelial cell proliferation and mural cell recruitment to generate the fully developed and functional vascular and lymphatic trees [2]. The pathophysiological studies of vascular anomalies have been helped by the astounding parallel progress made in understanding the factors and regulation of the development of the lymphatic and vascular systems [1,2].
Several angiogenic factors, such as VEGFs (vascular endothelial growth factors), FGFs (fibroblast growth factors), PDGF-beta (platelet derived growth factor beta) and angiopoietins (ANGPT-1 and ANGPT-2) regulate angiogenesis. They activate precursor cells, and lead to migration, proliferation and differentiation of the primary capillary plexus. Vascular endothelial growth factors, angiopoietins and their endothelial tyrosine kinase receptors are central regulators of vasculogenesis, angiogenesis and lymphangiogenesis [2].
Most vascular anomalies are sporadic, yet rare familial cases have been recorded. These forms have allowed to use molecular genetics for the identification of the underlying causes [3]. This molecular characterization of the inherited forms has subsequently led to hypothesize on the causes of the more common sporadic forms. In the first such extrapolation, this was shown to be true [4]. Thus, there is hope that in the near future, the molecular basis of vascular anomalies will be deciphered, allowing a precise classification of all of them. Moreover, some inherited forms have been proven to follow paradominant inheritance, which may be a general rule [1]. Thus, inhibition of second-hits may play a role in prevention of lesions in the familial forms. Finally, the data have laid the ground to develop specific in vitro and in vivo models of vascular anomalies. These can be used to characterize in detail the pathophysiological mechanisms and to develop novel specific treatments.
Hemangioma
The ethiopathogenesis of hemangioma of infancy (HI) still remains a mystery. Different hypotheses have been proposed, such as Human Papillomavirus Virus-8 infection, abnormal hormonal influence, and chorionic villus sampling [5]. Local hypoxia has also been suggested, as an initiating factor for proliferation [6].
Many angiogenic factors are highly expressed in proliferating hemangiomas [7,8]. In contrast, the vascular endothelial growth factor receptor 1 (VEGFR1; FLT1) is regularly downregulated [7]. The lack of this decoy receptor results in persistent activation of VEGFR2 by VEGF. The VEGFR1 downregulation seems to be due to amino acid substitutions in VEGFR2 and in the tumor endothelial marker-8 (TEM8), an integrin-like receptor, at least in some cases, although these changes are not specific to hemangioma patients. Normalization of VEGFR2 signaling with soluble VEGFR1, or by other means, may become an effective treatment [7].
To date, we know that endothelial cells from proliferating hemangiomas show patterns of X chromosome inactivation suggestive of clonal origin [8,9]. Bischoff and co-workers reported that hemangioma endothelial cells (hemECs) and hemangioma endothelial progenitor cells (Hem EPCs), both present in hemangiomas, are immature and share features with cord blood ECs, and are able to recapitulate hemangiomas in nude mice [10–13]. HemECs express genes that are expressed by placenta, umbilical cord, and bone marrow stem cells. One of them, the glucose transporter protein GLUT-1, has become a marker for histopathological diagnosis of hemangiomas [14]. Thus, it could be that hemangiomas are the result of a localized abnormal proliferation of progenitor cells, which preferentially occurs in children with predisposing genetic variants. Yet, the triggering factor(s) for such growth remain unknown. New clues may come from hemangiomas being part of a polymalformative syndrome, such as PHACES (Posterior fossa malformation, facial Hemangioma, Arterial anomalies, Cardiovascular anomalies, Eye anomalies and Sternal anomalies) [15].
Venous malformations
Venous malformations are classified into sporadic venous malformations (VM), dominantly inherited cutaneomucosal venous malformations (VMCM), and dominantly inherited glomuvenous malformations (GVM). They account for 94%, 1% and 5% of venous anomalies, respectively. No sex preponderance has been observed [3,16].
Sporadic venous malformation is the most common referral to specialized centers for vascular anomalies, as they often cause functional impairment, organ dysfunction, esthetic disfigurement, and can sometimes threaten life [3,4,16]. They are present at birth, never regress and grow proportionately with the child. VMs tend to enlarge during puberty and pregnancy, and can become symptomatic [17]. Pain at awakening, after activity or with temperature variation are commonly experienced [17].
Local intravascular coagulation (LIC), characterized by elevated D-Dimer level with normal fibrinogen level, is present in almost 50% of patients with a venous malformation [18,19]. This coagulopathy can cause acute pain and the formation of thrombi, which often calcify and form pathognomonic phleboliths [18,19]. Severe LIC (elevated D-Dimer level with low fibrinogen level) is often associated with extensive VM of the extremities [19]. This coagulopathy can decompensate into disseminated intravascular coagulopathy (DIC) during surgery and cause severe hemorrhage, if not treated with low molecular weight heparin. Therefore, measurement of this specific biomarker should be done as a routine test for every VM. Moreover, it can help to differentiate VM from GVM and other vascular anomalies with normal D-Dimer level.
VMCMs are clinically characterized by small (less than 5 cm in diameter), multifocal and hemispherical bluish lesions. They mainly involve skin and oral mucosa, but can also invade superficial muscle, GI tract, lungs and brain [17,20–22]. VMCMs are often asymptomatic and family members can be unaware of their lesions. They are inherited as an autosomal dominant trait with high penetrance. Genome-wide scans permitted to identify the mutated gene, TIE2/TEK, located on 9p21-22 [20,21]. The most common mutation causes an Arg849-to-Trp substitution (R849W) in the intracellular kinase domain of the membrane-bound tyrosine kinase receptor [21,22].
Expression analysis of the R849W mutant receptor showed that this mutation causes a 6–10 fold increase in autophosphorylation of the receptor [21]. So far, eight hyperphosphorylating mutations have been identified, R849W being the most common with an incidence of 60% [16,20,22,23]. TIE2 signaling pathway is crucial for angiogenesis and vascular maturation [4,20]. The receptor substitutions affect intracellular signaling thereby altering endothelial migration, vascular sprouting, maturation and stability [16,20]. Because VMCMs are localized, but caused by an inherited predisposition, modifying factors must exist. We hypothesized that this could be a somatic second-hit mutation in the same gene [21]. In one resected VMCM, we identified a somatic second hit in the normal allele of TIE2, in addition to the inherited TIE2 mutation [16]. This is analogous to the Knudson’s double-hit hypothesis for retinoblastoma [24] and suggests that the inherited form of venous malformation (VMCM) needs a somatic alteration to eliminate the protective wild-type TIE2 allele before the inherited mutation can induce the development of a VMCM lesion (Figure 1) [16].
Figure 1. Scheme of paradominant inheritance.

White individual: no mutation, unaffected; Grey individual: carrier of an inherited mutation that predisposes to lesions; Irregular black spots of different size and form: localized vascular anomalies due to somatic second-hit mutations in the same gene.
Encouraged by this finding, we decided to look for implication of somatic TIE2 mutations in sporadic VMs, and identified them in 50% of lesions [4]. These mutations differ somewhat from the inherited ones that cause VMCM. The most common one, L914F, accounts for 85% of lesions, and has not been observed as an inherited mutation, suggesting that it causes lethality, when germline [4,16]. The other 20% are caused by pairs of double mutations that always occur together on the same allele [4]. All the mutations cause hyperphosphorylation of the TIE2 receptor and render TIE2 an interesting target for future therapies [4,16].
Glomuvenous malformations clinically resemble VMs (Figure 2A,B,C) [17]. Most of them (at least 70%) are inherited as an autosomal dominant disorder [3,17,25–29]. Like most inherited lesions, they are multifocal, small and new lesions can occur with time. In contrast to VMCMs, GVMs are more bluish-purple, hyperkeratotic and with a cobblestone appearance [17]. Classically, GVMs involve skin and subcutis, and rarely mucosa. Extremities are often affected. Pain on palpation is pathognomonic [3,17]. No sex preponderance is noted.
Figure 2. Clinical variability of CM-AVM within the same family.
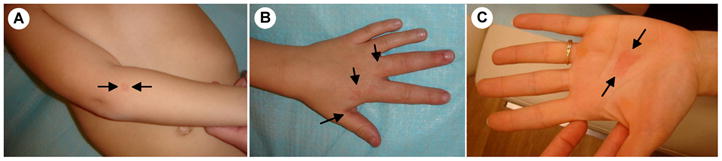
(A) a CM with a white halo on the arm, (B) an AVM of the hand of the same patient, and (C) a CM without a halo on the palm of the mother.
Like for VMCM, there is a large inter-familial and intra-familial variability in size, color and location of lesions, suggesting that the inherited mutation alone is not sufficient for the phenotype to occur. This clinical variability may hinder diagnosis, as some patients may have a single, small lesion, which does not evoke the physician to think of GVM (Figure 2B). Moreover, another family member may have a large lesion covering a whole extremity or for example the thorax, which the patient does not necessarily think of being expression of the same disorder as the tiny blue spot of a relative. Thus, when physicians ask on familial history of the disease, the patients likely answer is: none! In addition, GVM can also present as a large, plaque-like lesion in the childhood, reminiscent of a capillary malformation [25]. Thus, identification of GVM, as all other rare inherited vascular anomalies, needs detailed questioning of family history and evaluation of lesions.
Histologically, GVMs have a variable number of pathognomonic, abnormally differentiated vascular smooth muscle cells (vSMCs), known as “glomus cells” [26,28]. GVM is linked to 1p21-22 and is caused by loss-of-function of glomulin [27,29]. Interestingly, 75% of patients with GVM can be genetically diagnosed by screening one of the eight common glomulin mutations [27–29]. Like for VMCM, a somatic second-hit mutation has been identified in a GVM tissue of a patient, who also carried an inherited glomulin mutation [21]. Therefore, GVM follows paradominant transmission, and a complete localized loss of glomulin needs to occur for GVMs to develop [16,20,21].
Little is known of glomulin function. Glomulin (FAP68) seems to alter vascular smooth muscle cell phenotype, probably via the transforming growth factor beta (TGFb) and/or hepatocyte growth factor signaling pathways, as glomulin has been demonstrated to bind the HGF receptor and one of the TGFb receptor binding proteins [30,31]. In our laboratory, we have generated glomulin+/− and −/− mice. The heterozygotes are phenotypically normal, whereas the homozygous knock-outs are embryonic lethal (Brouillard et al., unpublished). This underscores the paradominant inheritance in families. Patients have homozygous loss-of-function only locally in the lesional cells, whereas the rest of the cells of the patients are heterozygous, and thus phenotypically normal (Figure 1). The glomulin knock-out mice now serve to study the detailed pathophysiological mechanisms, and eventually development of therapies.
Capillary malformations
Capillary malformations (CMs) or “port-wine stains” are flat, reddish lesions, which typically affect the head and neck. The reported incidence is 0,3% of newborns [32]. In most cases, they occur as a sporadic unifocal lesion. Similar birthmarks, called “salmon patch”, “angel’s kiss” and “nevus flammeus neonatorum” are more common and fade progressively in contrast to CMs, which darken with age [3]. Histologically, cutaneous capillary-like vessels are dilated and/or increased in number and size [33].
Inherited CMs have also been identified, leading our group to discover a new distinct sub-entity: capillary malformation - arteriovenous malformation (CM-AVM), characterized by autosomal dominant transmission [34,35]. Clinically, CMs of CM-AVM are different from common CMs, being smaller and multifocal (Figure 3), and often surrounded by a pale halo [35–37](Figure 3A). These lesions are associated in 30% of patients with fast-flow vascular malformations: arteriovenous malformation (Figure 3B), arteriovenous fistula or Parkes Weber syndrome [35–38]. The majority of the fast-flow anomalies (80 %) are located in the head and neck region [16,35–38]. Phenotypic heterogeneity, multifocality of lesions, and incomplete penetrance, which reaches its maximum by about 20 years of age, all suggest additional, local events to play a role; signs of paradominant inheritance.
Figure 3. Clinical variability of GVM within the same family.
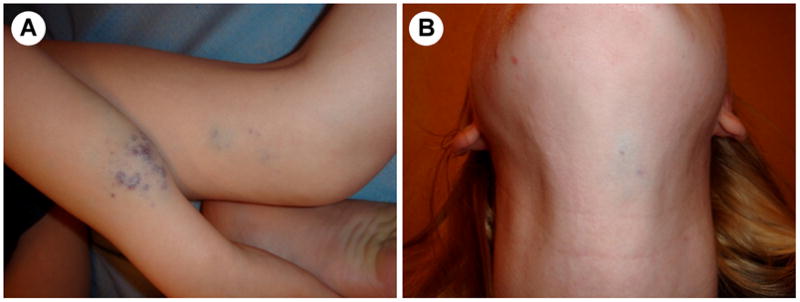
(A) a painful GVM of the arm and the leg of a boy, and (B) a small asymptomatic GVM on the neck of the mother.
We have also identified the causal gene RASA1 on chromosome 5q13-22 [34–38]. Dysfunction of the RASA1 gene, which encodes p120-RasGTPase activating protein (p120-RasGAP), a homologue of the gene causing neurofibromatosis (NF1), modifies the Ras-signaling pathway [39]. In normal circumstances, p120-RasGAP represses Ras/MAPK-signaling pathway [40]. In CM-AVM patients, prolongued Ras/MAPK activation occurs after activation by various receptor tyrosine kinases on the cell surface, leading to altered cellular growth, differentiation and proliferation [40]. With the generation of in vitro and in vivo models for testing, modulators of the Ras-signaling pathway may become future therapies.
Hereditary Hemorrhagic Telangiectasia (HHT) or Osler-Weber-Rendu syndrome is an autosomal dominant disorder [41]. Patients have variable clinical signs and symptoms, but almost invariably spontaneous and recurrent epistaxis secondary to nasal telangiectasias. Cutaneous and mucosal telangiectasias are frequent, and seen in association with arteriovenous malformations (AVM) in the lung (30–50% of patients), the liver (40%), the gastrointestinal tract (15%-30%) and in the brain (5–20%). AVMs in the liver rarely bleed, but are occasionally responsible for heart and liver failure.
Telangiectasias are focal dilatations of post-capillary venules. They are also seen in Cutis Marmorata Telangiectatica Congenita (CMTC), and Ataxia Telangiectasia (AT) [3]. The Curaçao diagnostic criteria were defined by the HHT International Foundation to help the clinician in the diagnosis of HHT. The diagnosis is definite, when 3 or more of the features are present, possible or suspected, when 2 findings are present, and unlikely, with fewer than 2 findings. Genetic diagnostic test can also be helpful [41]. Mutations in three genes have been identified: in endoglin (ENG) (HHT1 on 9q33-34) [42–44], in activin receptor-like kinase1 (ACVRL1) (HHT2 on 12q11-14) [45,46], and in SMAD4, localized on chromosome 5 (JPHT; HHT associated with juvenile polyposis) [47,48]. Two additional chromosomal loci are known, but the genes have not yet been identified (HHT3 on 5q and HHT4 on 7p14) [41]. More than 150 ENG mutations and 120 ACVRL1 mutations have been identified [3,41]. A useful genotype-phenotype correlation exists. HHT1 is more commonly associated with AVMs in the lung, whereas patients with HHT2 likely have hepatic AVMs [41]. However, since genes for HHT3 and HHT4 have not yet been identified, a negative genetic result does not rule out the diagnosis. Moreover, paradominant inheritance, in which the subject inherits from an affected parent a germline mutation, which becomes significant only when a second mutation affects the other allele, may also apply to the multifocal, localized telangiectasias and fast-flow lesions of HHT (Figure 1).
Angioma serpiginosum
Angioma serpiginosum is a progressive, patchy CM [49] characterized by dilated capillaries following Blaschko’s lines, associated with mild nail and hair dystrophy [50]. Typical lesions begin in childhood, involve extremities and are often asymmetric [51]. Clinically, only incomplete blanching is obtained on pressure. In several cases, papillomatosis of the esophagus is found [50]. This rare congenital cutaneous disorder results from X-linked dominant transmission, localized on Xp11.3-Xq12 [50]. This locus includes five genes, PORCN being one of them [52]. Dysfunction in this gene causes Goltz-Gorlin syndrome, characterized by focal dermal hypoplasia, esophageal papillomas, and eye and skeletal anomalies [53]. The resemblance suggests that angioma serpiginosum, in association with papillomatosis of oesophagus, is a variant of Goltz-Gorlin syndrome.
Cerebral cavernous malformation
Cerebral cavernous malformations or cerebral capillary malformations (CCMs) involve the brain, the eye and the spinal cord [54]. Histologically, CCMs are composed of capillary-like vessels and large saccular vessels with fibrotic walls [55]. Clinically, patients can be asymptomatic or complain about headaches and epilepsy, and cerebral bleeding can occur [54,55]. CCMs can be inherited as an autosomal dominant disorder (20% of patients), although the majority are sporadic [55]. Four chromosomal loci have been identified [55]: CCM1 (7q21-q22), with mutations in KRIT-1 (KREV1 interaction trapped 1) [56–58], CCM2 (7p15-p13), with mutations in malcavernin [59,60], and CCM3 (3q25.2-27), with mutations in PDCD10 [61]. A fourth gene has been suggested to exist on the same 3q26.3-27.2 locus (CCM4) [62]. CCM1 represents almost 40% of inherited cerebral cavernous malformations and more than a hundred mutations have been identified in KRIT-1 [55,63]. Interestingly, the vast majority of all CCM mutations lead to a premature termination codon suggesting loss of function of the respective protein. Only four missense mutations have been reported in CCM1 [63].
Cutaneous vascular malformations are seen in 9% of patients with CCM [64–67]. There are three distinct phenotypes: hyperkeratotic cutaneous capillary-venous malformation (HCCVM)(39%), capillary malformation (CM)(34%) and venous malformation (VM)(21%) CCM [64–67]. The high frequency of KRIT1 mutations (87%) in CCM patients with cutaneous vascular malformations suggests that KRIT1 has an important function also in cutaneous angiogenesis [65–67]. No HCCVM, CM or VM were detected in patients with CCM2 mutation, whereas VMs were sometimes observed both in CCM1 and CCM3 patients [65–67]. Therefore, the presence of a cutaneous vascular malformation may be a clue for detecting CCMs, with the specific molecular diagnosis.
CCMs, like VMCMs and GVMs, are likely due to paradominant inheritance. Somatic second-hits have been identified in a small number of lesions for the three CCM genes [68–70], suggesting, localized, complete loss-of-function of one of the CCM proteins to take place. Thus, our hypothesis that the clinical variability in patients with inherited vascular malformations may be explained by Knudson’s double-hit theory [22] seems to be true for a number of them. The difficulty encountered in the identification of the second hits in CCM is likely due to high tissular heterogeneity, as observed for sporadic venous malformations [4].
In vitro and in vivo studies have helped to unravel the function of CCM proteins. CCM1 is expressed in astrocytes, neurons, epithelial and endothelial cells [71], whereas CCM2 is expressed in mesenchymal and parenchymal vessels [72]. CCM proteins act as a scaffold and function in a complex [72–76]. KRIT1 also interacts with the β 1 integrin cytoplasmic domain associated protein 1 (ICAP-1α), which is implicated in the regulation of cell adhesion and migration [72]. In return, ICAP-1α is able to sequester KRIT1 into the nucleus [72]. In contrast, the phosphotyrosine-binding domain of CCM2 is able to sequester KRIT1 in the cytoplasm [73]. Thus, ICAP-1α and CCM2 are implied in the same signaling pathway and both are able to sequester KRIT1 [55,76].
KRIT 1−/− embryos die during gestation due to defective vascular development [77]. Conditional endothelial deletion of CCM2 affects angiogenesis leading to massive heart and blood vessel defects, and to embryonic death [73]. In contrast, neuroglial specific deletion does not lead to cerebrovascular defects [73]. This underscores the endothelial function of the CCM proteins, and the paradominant effect in CCM patients. Interestingly, CCM2 is the human ortholog of OSM, a Osmosensing Scaffold for MEKK3 [75]. OSM interacts with the p38 mitogen activated kinase pathway (p38MAPK) in response to osmolarity stress, which is a crucial mediator of cellular survival. As CCM3 interacts with CCM1 and CCM2, a multifaceted signaling complex is constituted by the three CCM proteins and seems to be involved in several cellular functions, including homeostasis of cell-cell junctions and cytoskeletal remodeling [76].
Whitehead and Li suggested statins for treatment of CCMs to stabilize blood vessels. They based their hypothesis on the observation that without CCM2, vascular endothelium is misformed and leads to dilated, leaky blood vessels. This coincides with, increased Rho activity, which regulates endothelial formation. As Simvastatin injection is an inhibitor of Rho activity, it may become a potential treatment for CCMs [78].
Lymphatic malformations
Lymphatic malformations (LMs) are also focal lesions. Genetic approach has not been possible, as LMs occur sporadically, with no evidence for inheritance. This suggests that the potential genetic causes may be somatic events, which when in germline, are incompatible with life [3]. The etiology remains unknown. In contrast, genetic analysis of inherited primary lymphedema has unraveled key regulators and molecular pathways involved in lymphangiogenesis [3,16].
Lymphedema
Lymphedema is a defect of lymphatic drainage, characterized by the accumulation of lymphatic fluid in the interstitial space, classically involving the lower extremities. It is divided into primary (due to unknown cause) and secondary (due to a known cause, such as infection and surgery). Primary lymphedema is further subdivided according to age at onset, into congenital, pubertal and late-onset lymphedema [79]. Five aberrant genes have been identified [16,80–84].
Familial congenital lymphedema, Nonne-Milroy syndrome, is inherited as an autosomal dominant trait, and maps on 5q34-35. It is caused by dominant missense mutations in the tyrosine kinase domain of the vascular endothelial growth factor receptor 3 (VEGFR3; also known as FLT-4), a crucial regulator of lymphatic development [2,80]. Lymphedema in this syndrome is principally present on lower extremities, from toes upwards to the hips. Most patients have lymphedema limited below the knees and less than 30% have associated hydrocele, large caliber leg veins (23%), cellulitis (20%), curled toenails (10%) or papillomatosis (10%) [85]. This congenital lymphedema seems to evolve slowly.
Interestingly, de novo dominant VEGFR3 mutations have also been identified in patients with congenital lymphedema without family history [86,87]. Thus, clinically the diagnostic criteria of Nonne-Milroy syndrome are not fulfilled. Yet, the molecular cause is the same, demonstrating how molecular classification can help in identifying patients with the same etiopathogenic underpinning, even if clinical criteria are different. Furthermore, dominant VEGFR3 mutations have been identified in patients with sporadic hydrops fetalis/generalized fetal edema [87]. Although associated with a high-risk of mortality, the presence of a VEGFR3 mutation appears to be a good sign for prognosis, as most of the few so-far reported, had in utero resorption of the lymphedema, leading to a limited lower extremity lymphedema present at birth [87].
In addition to Nonne-Milroy syndrome, with dominant transmission, a specific VEGFR3 amino-acid mutation has been found in a family with recessive inheritance of lymphedema [88]. Consanguinity between the healthy parents of the affected child was the clue to the discovery. The large clinical variability in patients with proven VEGFR3 mutations illustrates how important it is for the clinician to be alert of various signs that may lead to the precise, molecular diagnosis, which will likely be the basis for therapeutic choices in the future.
Puberty-onset lymphedema, Meige’s disease becomes evident around puberty and represents 80% of primary lymphedema. One third of the patients have a familial predisposition [89]. Genetic analysis revealed mutations in the FOXC2 gene, located on chromosome 16q24.3 [81]. These mutations cause lymphedema often associated with distichiasis (presence of an extra row of eyelashes), and sometimes with ptosis, yellow nails, syndactyly, cleft palate and cardiac septal defects [3,81].
FOXC2, a transcription factor, has several functions. One of them is to regulate angiogenesis by controlling the expression of target genes, such as Ang-2 [90], integrin β 3 [91], D114 and Hey-2, via interaction with VEGF-Notch signaling pathway in endothelial cells [92]. FOXC2 also inhibits secretion of PDGFβ, which is overproduced by lymphatic collecting ducts in Meige’s disease. This overproduction increases vascular smooth muscle cell recruitment, abnormal mural tone and lymphatic dysfunction [93].
Hypotrichosis-Lymphedema-Telangiectasia (HLT) is a rare condition characterized by sparse hair, lymphedema and cutaneous telangiectasias. Both recessive and dominant transmission is possible. This syndrome is caused by mutations in the SOX18 gene, in 20q13.33 [83]. The dominant nonsense mutation is located in the transactivation domain, whereas the homozygous recessive substitutions are in the DNA-binding domain [83]. Arrival to the diagnosis is not easy, as the lack of hair is not evident at birth, the age at onset of lymphedema is variable, and the telangiectasias may be mild and localized.
Hennekam syndrome is another rare form of lymphedema, characterized by extensive peripheral lymphedema, with visceral involvement, mental retardation and unusual, flat face, hypertelorism and broad nasal bridge [94]. Hydrops fetalis can also been seen. This phenotype is severe causing significant morbidity and mortality. Both autosomal recessive and dominant transmission of the Hennekam syndrome are possible [84].
Genetic analysis in zebrafish identified the Ccbe1 gene (Collagen and calcium-binding EGF domain) to be required for lymphangiogenesis [95]. The zebrafish Ccbe1 mutants are devoid of parachordal lymphangioblasts and lymphatic vessels. Zebrafish Ccbe1 is expressed along the migration routes of endothelial cells, suggesting a role in cellular guidance. Patients with Hennekam syndrome have either homozygous or a combination of two different heterozygous recessive mutations in CCBE1, localized on 18q21.32 [84,96].
Other rare forms of familial lymphedema include Osteoporosis Lymphedema Anhydrotic Ectodermal Dysplasia with Immunodeficiency (OLEDAID), which becomes evident during childhood and results from a premature termination codon mutation in the NFκB essential modulator (NEMO). The gene on Xq28, encodes NEMO, which moderates NFκB activation. So far, only two patients have been reported [82].
Another type of Primary congenital resolving lymphedema was reported in one Pakistani family. This autosomal dominant form of congenital lymphedema with reduced penetrance maps to 6q16.2-q22.1 [97]. Although the gene has not been unraveled, it is clearly a novel regulator of lymphatic function.
Aagenaes Syndrome or Hereditary Lymphedema Cholestasis (HLC) is a recessive disorder, which occurs in families with consanguinity [98,99]. It typically appears during childhood. Extended lymphedema of the lower extremities is associated with malabsorption, growth retardation, rickets, cholestatic jaundice and hepatomegaly. Lymphedema may be complicated by infections. The implicated gene is unknown, but mapped to 15q. Like the one for primary congenital resolving lymphedema, it is likely an important regulator of lymphatic development and/or function [99].
Concluding remarks
Vascular malformations classically present autosomal dominant transmission. However, it is necessary to perform careful clinical questioning of familial history, and examination of the patient and his/her parents for detecting this. There is large clinical variation in signs and symptoms, and lesions can be small enough to be overlooked hindering identification of familial inheritance [17,21,22,35,36,55,86]. Classically, the index patient, the one that seeks medical advice, is the most severely affected, and thus, family members’ insignificant lesions are not even thought to be of the same pathology. This is an important reason for patients to deny familial history of their disorder.
Though dominant inheritance by pedigree analysis, the molecular mechanism often seems to involve paradominance. This means that the inherited mutation is accompanied at various time points of development of the fetus and the child, by secondary mutations on the second allele of the same gene in the dividing cells. Such somatic mutations are common, non-hereditary, and lead to localized, complete abolishment of normal gene function. This phenomenon logically explains why inherited vascular malformations are localized, multifocal, of variable size and increase in number by age [3,4,22,29,68–70]. This discovery has opened the door for unraveling the causes of the non-hereditary vascular malformations as well.
The identification of the underlying genetic mutations in different families and disorders are crucial starting points to better understand the pathophysiologic mechanisms that play a role in the development of vascular anomalies. This also enables more accurate diagnosis, which in time, will lead to a more precise evaluation of prognosis and development of treatments. Moreover, unraveling the pathophysiological mechanisms of vascular anomalies allows to unravel key regulators and pathways implicated in angiogenesis.
TABLE 1.
Vascular anomalies with known genetic mutations, loci, or predisposing factors
| Vascular anomaly | Clinical signs | Linked Locus/Loci | Mutated/Predisposing Gene |
|---|---|---|---|
| Hemangioma | Erythematous macular patch, blanched spot or telangiectasia with rapid postnatal growth. | - | VEGFR2/TEM8, predisposing variants |
| Venous malformation (VM) | Bluish lesion compressible on palpation. | - | TIE2, 40–50% |
| Cutaneomucosal venous malformation (VMCM) | Multiple small punctate bluish spots. | 9p21-22 | TIE2 |
| Glomuvenous malformation (GVM) | Small, multifocal bluish-purple, cobblestone and hyperkeratotic lesions. | 1p21-22 | Glomulin |
| Capillary malformation-Arteriovenous malformation (CM-AVM) | Multifocal capillary malformation with pale halo, AVM, AVF, Vein of Galen, aneurysmal malformation, Parkes Weber syndrome. | 5q13-22 | RASA1 |
| Hereditary Hemorrhagic Telangiectasia (HHT1/HHT2/HHT3/HHT4) | Epistaxis, telangiectasia, AVM (lung, liver, brain, GI tract). | 9q33-34/12q11-14/5q/7p14 | ENG/ACVRL1/?/? |
| HHT Juvenile Polyposis (JPHT) | HHT with juvenile polyposis | 18q21 | SMAD4 |
| Angioma Serpiginosum | Patchy capillary malformation with dilated capillaries following BlaschkoÕs lines, mild nail, hair dystrophy, papillomatosis. | Xp11.3-Xq12 | PORCN ? |
| Cerebral cavernous malformation (CCM1/CCM2/CCM3) | Cerebral capillarovenous malformations, and sometimes cutaneous lesions (HCCVM). | 7q21-Q22/7p15-p13/3q25.2-27 | KRIT1/malcavernin/PDCD10 |
| Nonne-Milroy syndrome | Lymphedema, hydrocele, large caliber leg veins, cellulitis, curled toenails, papillomatosis. | 5q34-35 | VEGFR3 |
| Lymphedema-distichiasis | Lymphedema, distichiasis, ptosis, yellow nails, syndactyly, cleft palate and cardiac septal defects. | 16q24.3 | FOXC2 |
| Hypotrichosis-Lymphedema-Telangiectasia (HLT) | Sparse hair, lymphedema and cutaneous telangiectasias. | 20q13.33 | SOX18 |
| Hennekam syndrome | Peripheral lymphedema, with visceral involvement, mental retardation and unusual, flat face, hypertelorism and broad nasal bridge. | 18q21.32 | CCBE1 |
| OLEDAID | Osteoporosis Lymphedema Anhydrotic Ectodermal Dysplasia with Immunodeficiency. | Xq28 | NEMO |
| Primary Congenital Resolving Lymphedema | Early onset lymphedema with papillomatosis resolving at 30 to 40 year of age | 6q16.2-q22.1 | ? |
| Aagenaes Syndrome or Hereditary Lymphedema Cholestasis (HLC) | Extended lymphedema of lower extremities, malabsorption, growth retardation, rickets, cholestatic jaundice and hepatomegaly. | 15q | ? |
Acknowledgments
The authors thank Ms Liliana Niculescu for expert secretarial assistance. These studies were partially supported by the Interuniversity Attraction Poles initiated by the Belgian Federal Science Policy, network 6/05; Concerted Research Actions (A.R.C.) – Convention No 07/12-005 of the Belgian French Community Ministry; the National Institute of Health, Program Project P01 AR048564; EU FW6 Integrated project LYMPHANGIOGENOMICS, LSHG-CT-2004-503573; the F.R.S.-FNRS (Fonds de la Recherche Scientifique); and la Communauté française de Wallonie-Bruxelles et de la Lotterie nationale, Belgium (all to M.V.).
Footnotes
The authors have nothing to disclose.
Conflict of interest statement. None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brouillard P, Vikkula M. Vascular malformations: localized defects in vascular morphogenesis. Clin Genet. 2003;63:340. doi: 10.1034/j.1399-0004.2003.00092.x. [DOI] [PubMed] [Google Scholar]
- 2.Lohela M, Bry M, Tammela T, et al. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154. doi: 10.1016/j.ceb.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Brouillard P, Vikkula M. Genetic causes of vascular malformations. Hum Mol Genet. 2007;16(Spec No 2):R140. doi: 10.1093/hmg/ddm211. [DOI] [PubMed] [Google Scholar]
- 4.Limaye N, Wouters V, Uebelhoer M, et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet. 2009;41:118. doi: 10.1038/ng.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmes LB. Chorionic villus sampling and hemangiomas. J Craniofac Surg. 2009;20 (Suppl 1):675. doi: 10.1097/SCS.0b013e318193d61a. [DOI] [PubMed] [Google Scholar]
- 6.Pocock B, Boon LM, Vikkula M. Molecular basis of vascular birthmarks. Sem Plast Surg. 2006;20:149. [Google Scholar]
- 7.Jinnin M, Medici D, Park L, et al. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008;14:1236. doi: 10.1038/nm.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bischoff J. Progenitor cells in infantile hemangioma. J Craniofac Surg. 2009;20 (Suppl 1):695. doi: 10.1097/SCS.0b013e318193d6ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boye E, Yu Y, Paranya G, et al. Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest. 2001;107:745. doi: 10.1172/JCI11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Y, Flint AF, Mulliken JB, et al. Endothelial progenitor cells in infantile hemangioma. Blood. 2004;103:1373. doi: 10.1182/blood-2003-08-2859. [DOI] [PubMed] [Google Scholar]
- 11.Yu Y, Fuhr J, Boye E, et al. Mesenchymal stem cells and adipogenesis in hemangioma involution. Stem Cells. 2006;24:1605. doi: 10.1634/stemcells.2005-0298. [DOI] [PubMed] [Google Scholar]
- 12.Khan ZA, Melero-Martin JM, Wu X, et al. Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood. 2006;108:915. doi: 10.1182/blood-2006-03-006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan ZA, Boscolo E, Picard A, et al. Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest. 2008;118:2592. doi: 10.1172/JCI33493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.North PE, Waner M, Mizeracki A, et al. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000;31:11. doi: 10.1016/s0046-8177(00)80192-6. [DOI] [PubMed] [Google Scholar]
- 15.Frieden IJ, Reese V, Cohen D. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132:307. doi: 10.1001/archderm.132.3.307. [DOI] [PubMed] [Google Scholar]
- 16.Limaye N, Boon LM, Vikkula M. From germline towards somatic mutations in the pathophysiology of vascular anomalies. Hum Mol Genet. 2009;18:R65. doi: 10.1093/hmg/ddp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boon LM, Mulliken JB, Enjolras O, et al. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140:971. doi: 10.1001/archderm.140.8.971. [DOI] [PubMed] [Google Scholar]
- 18.Dompmartin A, Acher A, Thibon P, et al. Association of localized intravascular coagulopathy with venous malformations. Arch Dermatol. 2008;144:873. doi: 10.1001/archderm.144.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dompmartin A, Bailleux F, Thibon P, et al. Elevated D-dimer level is diagnostic for venous malformations. Arch Dermatol. 2009;145 doi: 10.1001/archdermatol.2009.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wouters V, Limaye N, Uebelhoer M, et al. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur J Hum Genet. 2009 doi: 10.1038/ejhg.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boon LM, Mulliken JB, Vikkula M, et al. Assignment of a locus for dominantly inherited venous malformations to chromosome 9p. Hum Mol Genet. 1994;3:1583. doi: 10.1093/hmg/3.9.1583. [DOI] [PubMed] [Google Scholar]
- 22.Vikkula M, Boon LM, Carraway KL, 3rd, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 23.Calvert JT, Riney TJ, Kontos CD, et al. Allelic and locus heterogeneity in inherited venous malformations. Hum Mol Genet. 1999;8:1279. doi: 10.1093/hmg/8.7.1279. [DOI] [PubMed] [Google Scholar]
- 24.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mallory SB, Enjolras O, Boon LM, et al. Congenital plaque-type glomuvenous malformations presenting in childhood. Arch Dermatol. 2006;142:892. doi: 10.1001/archderm.142.7.892. [DOI] [PubMed] [Google Scholar]
- 26.Goodman TF, Abele DC. Multiple glomus tumors. A clinical and electron microscopic study. Arch Dermatol. 1971;103:11. doi: 10.1001/archderm.103.1.11. [DOI] [PubMed] [Google Scholar]
- 27.Brouillard P, Enjolras O, Boon LM, et al. Glomulin and glomuvenous malformation. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development. 2. New Jork: Oxford University Press; 2008. p. 1561. [Google Scholar]
- 28.Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”) Am J Hum Genet. 2002;70:866. doi: 10.1086/339492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boon LM, Brouillard P, Irrthum A, et al. A gene for inherited cutaneous venous anomalies (“glomangiomas”) localizes to chromosome 1p21-22. Am J Hum Genet. 1999;65:125. doi: 10.1086/302450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chambraud B, Radanyi C, Camonis JH, et al. FAP48, a new protein that forms specific complexes with both immunophilins FKBP59 and FKBP12. Prevention by the immunosuppressant drugs FK506 and rapamycin. J Biol Chem. 1996;271:32923. doi: 10.1074/jbc.271.51.32923. [DOI] [PubMed] [Google Scholar]
- 31.Grisendi S, Chambraud B, Gout I, et al. Ligand-regulated binding of FAP68 to the hepatocyte growth factor receptor. J Biol Chem. 2001;276:46632. doi: 10.1074/jbc.M104323200. [DOI] [PubMed] [Google Scholar]
- 32.Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218. [PubMed] [Google Scholar]
- 33.Mulliken JB, Young AE. Vascular birthmarks: hemangiomas and malformations. Philadelphia: WB Saunders; 1988. [Google Scholar]
- 34.Eerola I, Boon LM, Watanabe S, et al. Locus for susceptibility for familial capillary malformation (‘port-wine stain’) maps to 5q. Eur J Hum Genet. 2002;10:375. doi: 10.1038/sj.ejhg.5200817. [DOI] [PubMed] [Google Scholar]
- 35.Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240. doi: 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Revencu N, Boon LM, Mulliken JB, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29:959. doi: 10.1002/humu.20746. [DOI] [PubMed] [Google Scholar]
- 37.Boon LM, Mulliken JB, Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15:265. doi: 10.1016/j.gde.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Thiex R, Mulliken JB, Revencu N, et al. A novel association between RASA1 mutations and spinal arteriovenous anomalies. Am J Neuroradiol. doi: 10.3174/ajnr.A1907. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ballester R, Marchuk D, Boguski M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 40.Kulkarni SV, Gish G, van der Geer P, et al. Role of p120 Ras-GAP in directed cell movement. J Cell Biol. 2000;149:457. doi: 10.1083/jcb.149.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17:860. doi: 10.1038/ejhg.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 43.Shovlin CL, Hughes JM, Scott J, et al. Characterization of endoglin and identification of novel mutations in hereditary hemorrhagic telangiectasia. Am J Hum Genet. 1997;61:68. doi: 10.1086/513906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cymerman U, Vera S, Karabegovic A, et al. Characterization of 17 novel endoglin mutations associated with hereditary hemorrhagic telangiectasia. Hum Mutat. 2003;21:482. doi: 10.1002/humu.10203. [DOI] [PubMed] [Google Scholar]
- 45.Johnson DW, Berg JN, Gallione CJ, et al. A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res. 1995;5:21. doi: 10.1101/gr.5.1.21. [DOI] [PubMed] [Google Scholar]
- 46.Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 47.Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 48.Cole SG, Begbie ME, Wallace GM, et al. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42:577. doi: 10.1136/jmg.2004.028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vikkula M. Vascular Pathologies: Angiogenomics: towards a genetic nosology and nderstanding of vascular anomalies. Eur J Hum Genet. 2007 doi: 10.1038/sj.ejhg.5201856. [DOI] [PubMed] [Google Scholar]
- 50.Blinkenberg EO, Brendehaug A, Sandvik AK, et al. Angioma serpiginosum with oesophageal papillomatosis is an X-linked dominant condition that maps to Xp11.3-Xq12. Eur J Hum Genet. 2007;15:543. doi: 10.1038/sj.ejhg.5201800. [DOI] [PubMed] [Google Scholar]
- 51.Katta R, Wagner A. Angioma serpiginosum with extensive cutaneous involvement. J Am Acad Dermatol. 2000;42:384. doi: 10.1016/s0190-9622(00)90119-1. [DOI] [PubMed] [Google Scholar]
- 52.Houge G, Oeffner F, Grzeschik KH. An Xp11.23 deletion containing PORCN may also cause angioma serpiginosum, a cosmetic skin disease associated with extreme skewing of X-inactivation. Eur J Hum Genet. 2008;16:1027. doi: 10.1038/ejhg.2008.87. [DOI] [PubMed] [Google Scholar]
- 53.Grzeschik KH, Bornholdt D, Oeffner F, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet. 2007;39:833. doi: 10.1038/ng2052. [DOI] [PubMed] [Google Scholar]
- 54.Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med. 1988;319:343. doi: 10.1056/NEJM198808113190605. [DOI] [PubMed] [Google Scholar]
- 55.Revencu N, Vikkula M. Cerebral cavernous malformation: new molecular and clinical insights. J Med Genet. 2006;43:716. doi: 10.1136/jmg.2006.041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laberge-le Couteulx S, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- 57.Sahoo T, Johnson EW, Thomas JW, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999;8:2325. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- 58.Serebriiskii I, Estojak J, Sonoda G, et al. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene. 1997;15:1043. doi: 10.1038/sj.onc.1201268. [DOI] [PubMed] [Google Scholar]
- 59.Denier C, Goutagny S, Labauge P, et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;74:326. doi: 10.1086/381718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liquori CL, Berg MJ, Siegel AM, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergametti F, Denier C, Labauge P, et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liquori CL, Berg MJ, Squitieri F, et al. Low frequency of PDCD10 mutations in a panel of CCM3 probands: potential for a fourth CCM locus. Hum Mutat. 2006;27:118. doi: 10.1002/humu.9389. [DOI] [PubMed] [Google Scholar]
- 63.Riant F, Bergametti F, Ayrignac X, et al. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. Febs J. doi: 10.1111/j.1742-4658.2009.07535.x. [DOI] [PubMed] [Google Scholar]
- 64.Labauge P, Enjolras O, Bonerandi JJ, et al. An association between autosomal dominant cerebral cavernomas and a distinctive hyperkeratotic cutaneous vascular malformation in 4 families. Ann Neurol. 1999;45:250. doi: 10.1002/1531-8249(199902)45:2<250::aid-ana17>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 65.Eerola I, Plate KH, Spiegel R, et al. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. 2000;9:1351. doi: 10.1093/hmg/9.9.1351. [DOI] [PubMed] [Google Scholar]
- 66.Toll A, Parera E, Gimenez-Arnau AM, et al. Cutaneous venous malformations in familial cerebral cavernomatosis caused by KRIT1 gene mutations. Dermatology. 2009;218:307. doi: 10.1159/000199461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sirvente J, Enjolras O, Wassef M, et al. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol. 2009;23:1066. doi: 10.1111/j.1468-3083.2009.03263.x. [DOI] [PubMed] [Google Scholar]
- 68.Gault J, Shenkar R, Recksiek P, et al. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke. 2005;36:872. doi: 10.1161/01.STR.0000157586.20479.fd. [DOI] [PubMed] [Google Scholar]
- 69.Akers AL, Johnson E, Steinberg GK, et al. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet. 2009;18:919. doi: 10.1093/hmg/ddn430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pagenstecher A, Stahl S, Sure U, et al. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet. 2009;18:911. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Denier C, Gasc JM, Chapon F, et al. Krit1/cerebral cavernous malformation 1 mRNA is preferentially expressed in neurons and epithelial cells in embryo and adult. Mech Dev. 2002;117:363. doi: 10.1016/s0925-4773(02)00209-5. [DOI] [PubMed] [Google Scholar]
- 72.Zawistowski JS, Stalheim L, Uhlik MT, et al. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005;14:2521. doi: 10.1093/hmg/ddi256. [DOI] [PubMed] [Google Scholar]
- 73.Boulday G, Blecon A, Petit N, et al. Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations. Dis Model Mech. 2009;2:168. doi: 10.1242/dmm.001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Plummer NW, Gallione CJ, Srinivasan S, et al. Loss of p53 sensitizes mice with a mutation in Ccm1 (KRIT1) to development of cerebral vascular malformations. Am J Pathol. 2004;165:1509. doi: 10.1016/S0002-9440(10)63409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Uhlik MT, Abell AN, Johnson NL, et al. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol. 2003;5:1104. doi: 10.1038/ncb1071. [DOI] [PubMed] [Google Scholar]
- 76.Hilder TL, Malone MH, Bencharit S, et al. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007;6:4343. doi: 10.1021/pr0704276. [DOI] [PubMed] [Google Scholar]
- 77.Whitehead KJ, Plummer NW, Adams JA, et al. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437. doi: 10.1242/dev.01036. [DOI] [PubMed] [Google Scholar]
- 78.Whitehead KJ, Chan AC, Navankasattusas S, et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 2009;15:177. doi: 10.1038/nm.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lazareth I. Classification of lymphedema. Rev Med Interne. 2002;23 (Suppl 3):375s. doi: 10.1016/s0248-8663(02)80378-2. [DOI] [PubMed] [Google Scholar]
- 80.Irrthum A, Karkkainen MJ, Devriendt K, et al. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet. 2000;67:295. doi: 10.1086/303019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Finegold DN, Kimak MA, Lawrence EC, et al. Truncating mutations in FOXC2 cause multiple lymphedema syndromes. Hum Mol Genet. 2001;10:1185. doi: 10.1093/hmg/10.11.1185. [DOI] [PubMed] [Google Scholar]
- 82.Doffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 83.Irrthum A, Devriendt K, Chitayat D, et al. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am J Hum Genet. 2003;72:1470. doi: 10.1086/375614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alders M, Hogan BM, Gjini E, et al. Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nat Genet. 2009 doi: 10.1038/ng.484. [DOI] [PubMed] [Google Scholar]
- 85.Brice G, Child AH, Evans A, et al. Milroy disease and the VEGFR-3 mutation phenotype. J Med Genet. 2005;42:98. doi: 10.1136/jmg.2004.024802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ghalamkarpour A, Morlot S, Raas-Rothschild A, et al. Hereditary lymphedema type I associated with VEGFR3 mutation: the first de novo case and atypical presentations. Clin Genet. 2006;70:330. doi: 10.1111/j.1399-0004.2006.00687.x. [DOI] [PubMed] [Google Scholar]
- 87.Ghalamkarpour A, Debauche C, Haan E, et al. Sporadic in utero generalized edema caused by mutations in the lymphangiogenic genes VEGFR3 and FOXC2. J Pediatr. 2009;155:90. doi: 10.1016/j.jpeds.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 88.Ghalamkarpour A, Holnthoner W, Saharinen P, et al. Recessive primary congenital lymphoedema caused by a VEGFR3 mutation. J Med Genet. 2009;46:399. doi: 10.1136/jmg.2008.064469. [DOI] [PubMed] [Google Scholar]
- 89.Dale RF. The inheritance of primary lymphoedema. J Med Genet. 1985;22:274. doi: 10.1136/jmg.22.4.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xue Y, Cao R, Nilsson D, et al. FOXC2 controls Ang-2 expression and modulates angiogenesis, vascular patterning, remodeling, and functions in adipose tissue. Proc Natl Acad Sci U S A. 2008;105:10167. doi: 10.1073/pnas.0802486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hayashi H, Sano H, Seo S, et al. The Foxc2 transcription factor regulates angiogenesis via induction of integrin beta3 expression. J Biol Chem. 2008;283:23791. doi: 10.1074/jbc.M800190200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hayashi H, Kume T. Foxc transcription factors directly regulate Dll4 and Hey2 expression by interacting with the VEGF-Notch signaling pathways in endothelial cells. PLoS One. 2008;3:e2401. doi: 10.1371/journal.pone.0002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Petrova TV, Karpanen T, Norrmen C, et al. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med. 2004;10:974. doi: 10.1038/nm1094. [DOI] [PubMed] [Google Scholar]
- 94.Hennekam RC, Geerdink RA, Hamel BC, et al. Autosomal recessive intestinal lymphangiectasia and lymphedema, with facial anomalies and mental retardation. Am J Med Genet. 1989;34:593. doi: 10.1002/ajmg.1320340429. [DOI] [PubMed] [Google Scholar]
- 95.Hogan BM, Bos FL, Bussmann J, et al. Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat Genet. 2009;41:396. doi: 10.1038/ng.321. [DOI] [PubMed] [Google Scholar]
- 96.Connell F, Kalidas K, Ostergaard P, et al. Linkage and sequence analysis indicate that CCBE1 is mutated in recessively inherited generalised lymphatic dysplasia. Hum Genet. 127:231. doi: 10.1007/s00439-009-0766-y. [DOI] [PubMed] [Google Scholar]
- 97.Malik S, Grzeschik KH. Congenital, low penetrance lymphedema of lower limbs maps to chromosome 6q16.2-q22.1 in an inbred Pakistani family. Hum Genet. 2008;123:197. doi: 10.1007/s00439-007-0458-4. [DOI] [PubMed] [Google Scholar]
- 98.Sigstad H, Aagenaes O, Bjorn-Hansen RW, et al. Primary lymphoedema combined with hereditary recurrent intrahepatic cholestasis. Acta Med Scand. 1970;188:213. doi: 10.1111/j.0954-6820.1970.tb08028.x. [DOI] [PubMed] [Google Scholar]
- 99.Bull LN, Roche E, Song EJ, et al. Mapping of the locus for cholestasis-lymphedema syndrome (Aagenaes syndrome) to a 6.6-cM interval on chromosome 15q. Am J Hum Genet. 2000;67:994. doi: 10.1086/303080. [DOI] [PMC free article] [PubMed] [Google Scholar]