

Abstract
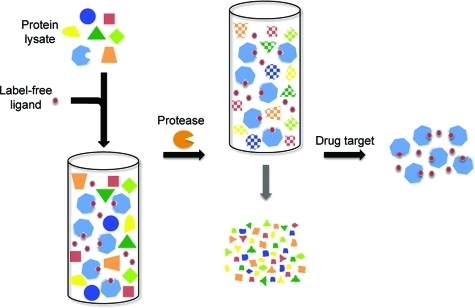
Small-molecule target identification is a vital and daunting task for the chemical biology community as well as for researchers interested in applying the power of chemical genetics to impact biology and medicine. To overcome this “target ID” bottleneck, new technologies are being developed that analyze protein–drug interactions, such as drug affinity responsive target stability (DARTS), which aims to discover the direct binding targets (and off targets) of small molecules on a proteome scale without requiring chemical modification of the compound. Here, we review the DARTS method, discuss why it works, and provide new perspectives for future development in this area.
Keywords: 2D-PAGE: A two-dimensional gel-based method that combines isoelectric focusing and SDS–PAGE to separate proteins by charge and molecular weight., Affinity chromatography: Traditional method of target identification that uses immobilized small molecules to pull down binding proteins from a complex protein mixture., Degradomics: The subfield of proteomics devoted to the study of proteases and their biological substrates., Drug affinity responsive target stability (DARTS): A method for target identification that relies on drug-induced protease resistance., MudPIT: A gel-free proteomics technique that uses online separation of tryptic peptides by strong cation exchange and reversed-phase HPLC to analyze very complex protein mixtures by mass spectrometry., SILAC: Stable isotope labeling of amino acids in cell culture., Target identification: Determination of which proteins a particular small molecule binds in the cell.
Humans have been using small molecule drugs for their medicinal, hallucinogenic, and toxic properties for at least thousands of years; the oldest materia medica might be traced back to the legendary Shen Nong (the Divine Farmer) of 2737 B.C. Until very recently, Nature was the exclusive source of all such drugs. There are literally millions of species of plants, fungi, bacteria, and animals on Earth that produce a plethora of compounds with equally diverse chemical structures and pharmacological properties, many of which are still unknown to man. Only in the last one and a half centuries, and mainly during the past few decades, have we begun to acquire the ability to create ourselves compounds with desired properties, such as binding to and modulating the activity (or expression) of specific proteins (1–15). That being said, even when not using nature as the direct source of new drugs, we still often rely on inspiration from natural product compounds in our search for new medicines (16–18). Studies on the molecular mechanism of action of natural small molecules has also long been a goldmine for identifying the function and importance of proteins from many classes, including membrane and intracellular receptors, signaling proteins, and proteins involved in regulating and carrying out the cell cycle, cell growth, mitosis, DNA replication and repair, transcription, and translation (reviewed in refs (19–21)).
A large percentage of compounds of current interest were derived from screens using cell culture or whole organisms and phenotypic or molecular readouts (22–30). While the opportunity for finding novel drugs or drug uses from such assays is high, their primary limitation is that they typically give no indication of the potential drug targets, and finding the direct targets is often the most challenging and time-consuming step of the project. Moreover, drugs screened for or designed to specifically modulate activity of a given protein of interest are still likely to bind multiple other proteins, many of which may not be predicted simply by sequence or structural homology.
Although mechanistic studies of drug action has a long history, identification of the direct targets of drugs has its roots in the pioneering work on affinity chromatography, biochemical fractionation, and radioactive ligand binding assays. Affinity purification of enzymes with small molecule inhibitors was first developed in the early 1950s by Leonard Lerman (31) and subsequently used for novel identifications by the McCormick and Anfinson laboratories during the 1960s (32,33). Around the same time others had begun to use radioisotope-labeled compounds and biochemical fractionation to enrich drug-binding proteins from crude tissue extracts and cell lysates (34–38). Starting with the proof of existence by purification of the lac repressor protein by Wally Gilbert, using radioligand binding measured by equilibrium dialysis as an assay (39), radioligand binding quickly became a successful method to identify receptors for hormones and neurotransmitters. Cuatrecasas isolated the insulin receptor, a very low abundance protein, using radioactive insulin binding and insulin covalently linked to agarose beads for affinity chromatography (reviewed in ref (40)). Sol Snyder and colleagues discovered the opiate receptors in brain using a radioligand binding assay (41), assisting the demonstration and identification of endogenous opioid peptides and their myriad biological functions. He followed this with rapid identification of the 10 most important neurotransmitter receptors, which hundreds of other workers had been attempting unsuccessfully for years. His success came from identification of suitable high affinity and high specificity ligands and the know-how to employ them properly. The first neurotransmitter receptor to be purified was the nicotinic acetylcholine receptor, using radioactive snake toxin as a binding assay and small molecule affinity chromatography (as well as a tissue source with high expression, electric rays and eels) (42). The first direct identification of amino acids involved in general anesthetic binding (etomidate) in its biotarget, GABA-A receptors in brain, has used photoaffinity labeling with a photoincorporatable radiolabeled aziridine analogue of etomidate that retained anesthetic activity and microsequencing (43). Many prominent natural product target identifications have made use of one of these two techniques (44–52). Today, affinity chromatography, also known as affinity purification, has become the most-used method (53), but radioaffinity and photoaffinity approaches occasionally find use (54).
Affinity Chromatography as a Classical Method for Target ID
Despite the large number of target identification techniques described to date, affinity chromatography remains the most widely used method (55,56). The typical project begins with structure–activity relationship (SAR) studies in which various functional groups of the small molecule of interest are modified or removed to determine which one(s) are dispensable for drug activity. These nonessential site(s) are then used as points of attachment to an affinity tag (e.g., biotin) or solid matrix (e.g., Affi-Gel agarose beads). Then, much like is performed during immunoprecipitation of specific proteins using an antibody and Protein A/G-conjugated beads, the drug-linked beads are incubated with protein extracts, followed by extensive washing to remove nonspecifically bound proteins. Finally the tightly binding proteins are eluted with excess free drug or under highly denaturing conditions. Most often the eluted proteins are subsequently analyzed by SDS–PAGE and protein bands are identified by mass spectrometry. Given that there are typically many more nonspecific binders than actual drug targets, negative control experiments using similarly tagged inactive analogues of the parental compound are performed simultaneously when possible. Once potential targets are identified, subsequent studies must confirm direct binding as well as its biological importance.
The primary limitation of affinity chromatography is the need to derivatize the small molecules of interest. SAR studies are time-consuming and require extensive medicinal chemistry expertise that is often lacking in the academic laboratories performing forward chemical genetics and phenotypic small molecule screens. Even when available, many small molecules cannot be modified without affecting bioactivity, and presumably binding, or cannot be easily obtained or synthesized in quantities large enough to permit SAR and subsequent studies. However, in comparison to other newly developed target identification methods that rely on various phenotypic or molecular signatures to narrow down potential targets, the advantage of affinity-based methods is that they rely solely on binding of the drug to its target protein, rather than specific cellular or biochemical readouts that are only useful for a fraction of compounds (reviewed in refs (57–62)).
A recent major advance in the affinity chromatography approach for target identification takes advantage of the quantitative capability of SILAC to drastically increase the sensitivity of this approach for target identification (63). This quantitative proteomic approach eliminates the need to visualize the target protein by gel staining, instead relying upon identification of the target by tandem mass spectrometry, which also allows the identification of multiple targets (polypharmacology). Furthermore, by comparison of the target pull-down with a well-designed control pull-down, true target proteins can be more easily distinguished from the many nonspecific binders due to their unique enrichment in the target pull-down sample. While this development is likely to enhance many target identification projects, it is still reliant upon SAR studies and limited to small molecules with derivatizable moieties. It is also contingent on the availability of a suitable inactive small molecule that can serve as the negative control, which is often not possible to obtain.
In summary, affinity chromatography of small molecules is severely limited due to the vast structural diversity and complexity of biologically active small molecules. Unlike affinity fusion (e.g., to glutathione S-transferase, or GST) and iodination of peptides, which are carried out routinely by the standard molecular biological (64,65) and chloramine-T (66,67) methods, respectively, no universal or simple solution has been obtained to immobilize or label organic compounds.
Drug Affinity Responsive Target Stability (DARTS)
Given the shortcomings of current target identification methodologies, a breakthrough will likely require thinking outside the “chemically modifying the small molecule” box. We imagined that a target identification strategy that only relied on drug–protein binding yet did not require modification of the small molecule would be ideal. Such an approach could potentially identify any protein targets of small molecules with no limitations posed by chemistry or mechanism of action. The idea that a small molecule drug would stabilize its target protein’s structure and result in protease resistance offered a possible solution to the problem of target identification, so long as the decreased proteolysis could be readily detected in complex samples. Stabilization of lysozyme structure upon substrate binding is a well-known phenomenon that has been demonstrated by increased resistance to denaturation by heat and chaotropic agents (68). This enhanced stability is postulated to result from a shift in the thermodynamic landscape of the protein to favor the ligand-bound state, which prevents much of the protein’s innate flexibility and movement (or protein “breathing”) (69–71) from being realized (1). One recent example that lends support for this model comes from the finding that FK506, which protects FKBP12 in DARTS (62), leads to stabilization of a high-energy backbone conformation that is otherwise minorly populated in the native conformational ensemble of free FKBP12 (72). Ligand-induced stabilization has been exploited by numerous techniques to detect and analyze specific protein–ligand interactions (73–77). It has also been instrumental for purification of natively folded recombinant proteins and subsequent structural determination by protein crystallization (78,79). Even ligand-induced resistance to proteolysis turns out to not be a completely new idea, as it was amply demonstrated for serum albumin (80,81), Staphylococcal nuclease and nuclease-T (82), firefly luciferase (83), etc. However, very few prior reports detailed small molecule-induced protease resistance, and there certainly had been no realization that it may be general enough to be used as a discovery approach for identifying unknown targets. Our work has shown just how widespread this phenomenon is and demonstrated the potential of utilizing it for target identification (62) (1).
Figure 1.
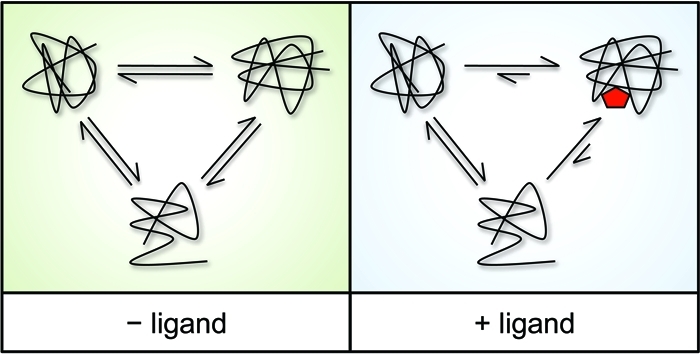
Theoretical model for why DARTS works. Under physiological conditions a protein is in dynamic equilibrium with multiple alternative conformations and may also exhibit some level of local, reversible unfolding (69,71,126,127). Upon saturation with a specific ligand, the equilibrium will shift to highly favor the conformation bound by the ligand as a result of the negative free energy change due to hydrophobic, hydrogen bonding, and/or electrostatic interactions that are formed between the protein and the drug ligand. This leads to a thermodynamically more stable state in which the target protein’s conformational fluctuations (“breathing”) and unfolding are dramatically decreased and resistance to denaturation and DARTS proteolysis is markedly increased. (The fate of the target protein in vivo, on the other hand, cannot be predicted; ligand binding can cause either increase, decrease, or no change in the stability and/or expression of its target depending on the biology (62).)
Table 1. Small molecule–protein interactions confirmed or identified by DARTSa.
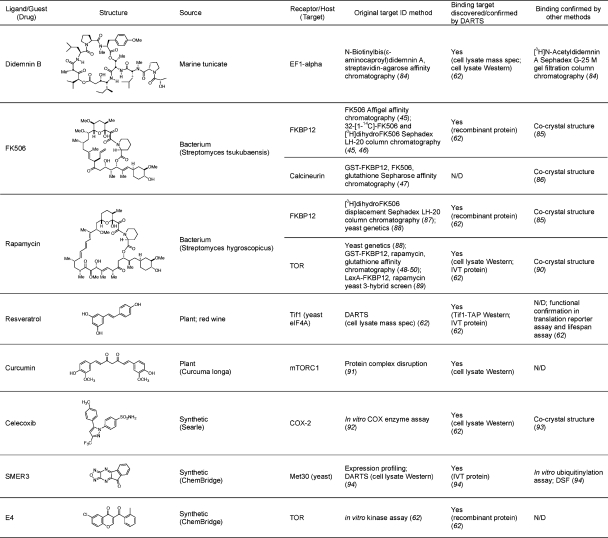 |
N/D: direct binding not determined. IVT: in vitro translated. DSF: differential scanning fluorimetry.
DARTS offers an unprecedented ability to identify new proteins targeted by small molecules. It is similar to affinity chromatography in that both are affinity-based methods that start with complex protein samples and selectively enrich the target protein(s) while depleting all nontarget proteins. However, whereas affinity chromatography utilizes positive enrichment by selectively pulling out the target proteins and leaving behind nontargets, DARTS uses negative enrichment by digesting away nontarget proteins while leaving behind the target proteins that are rendered protease-resistant (2). With several decades of affinity chromatography usage, its strengths and limitations have been well characterized. For example, a primary weakness is the often-high level of nonspecific binding of nontarget proteins to the matrix, which confounds isolation of the true targets. Although extensive washing of the matrix can help reduce the number of nonspecific binders, weaker true interactors will often be lost during the process. In contrast, DARTS does not require washing and can be used to analyze lower affinity interactions.
Figure 2.

Comparison of affinity chromatography and DARTS. (A) Using small molecule affinity chromatography, protein lysates are incubated with the immobilized small molecule to positively enrich target proteins. Unbound proteins are washed away, followed by elution of the target proteins with excess free drug. (B) With DARTS, protein lysates are incubated with native drug and then treated with proteases. The target proteins are negatively enriched due to their drug-induced protease resistance while nontarget proteins are digested away.
DARTS is also unique in that native small molecules are used without the need for chemical derivatization or even knowing the chemical identity or purity of the compound. As such, DARTS allows bioactive natural product extracts to be used for target ID prior to fractionation, thus enabling research of not only polypharmacology (95) but also polypharmacy (e.g., Chinese herbal medicine). This is a tremendous advantage over affinity chromatography and other prior methods.
Additional Protein-Based Target ID Platforms
Proteome Chips
The advent of proteome microarrays has revolutionized the global analysis of the biochemical activities of proteins, including the study of protein–small molecule interactions (96). In this method, small molecules are first labeled such that their physical presence and location can be followed upon binding to the proteome chip (24). Common labels used for affinity chromatography can also be used to probe proteome chips, including affinity tags (e.g., biotin), fluorescence tags, photochemical tags, and radioisotopes (97,98). A key advantage is the ability to analyze drug binding on a whole-proteome scale in a microarray format, revealing all binding targets, including “off-targets”. However, a current disadvantage shared with affinity chromatography is the need for small-molecule labeling and the requirement that the labeling moiety can be incorporated without abolishing the compound’s bioactivity. The power of this approach will be greatly enhanced by the development of label-free binding detection compatible with the proteome array format (99).
ABPP
A chemical proteomic approach that accesses protein targets via the use of reactive small molecule probes also constitutes a powerful means for identifying unknown targets. For example, activity-based protein profiling (ABPP) relies on small molecules containing reactive functional groups (mainly electrophiles) that can covalently attach to catalytic residues in an enzyme active site (100–103) (reviewed in ref (104)). Such activity-based probes allow the targeted enzymes to be labeled for purification and analysis. Many flavors of ABPs have been created, including photoactivatable, radioactive, and biotinylated (105,106), which greatly facilitates both discovering novel enzymes and inhibitor screening (reviewed in refs (54) and (107)). Covalent linkage between a reactive probe and its target can thus serve the purpose for target identification (52,108), although the requirement for derivatization of each small molecule still exists.
SPROX
In the realm of using label-free small molecules for binding target identification, another affinity-based label-free methodology was introduced in 2010 (109). Stability of proteins from rates of oxidation (SPROX) is similar to DARTS in that it detects small molecule-induced changes in the folding and thermodynamic stability of target proteins in complex samples (109). Instead of using differential proteolysis as a readout, SPROX measures ligand-induced changes in the rate of methionine oxidation for target proteins. This approach is similar in essence to hydrogen–deuterium exchange, which has been used extensively to analyze protein–ligand interactions and determine ligand binding domains within individual proteins, except that methionine oxidation levels are more readily measurable in complex protein mixtures (110,111). Although SPROX clearly has many of the same advantages as DARTS, it has many additional limitations. The major limiting factor of SPROX is that only the most abundant proteins in each sample can be identified and accurately quantified. Unlike affinity chromatography and DARTS, SPROX has no mechanism to enrich the target proteins from nontarget proteins. Therefore, obtaining proteome-wide coverage using SPROX would require extensive up-front fractionation of the sample. Additionally, only methionine-containing peptides are useful with SPROX analysis, and not all methionine residues exhibit differential oxidation rates informative for determining thermodynamic changes. This is in contrast to the DARTS and affinity chromatography which can utilize information from any identifiable peptides.
Methodology and Practice of DARTS
The simplest procedure for DARTS simply involves separation of compound-treated and control protein samples digested with varying amounts of protease by 1D SDS–PAGE, staining the gel with Coomassie Blue, Sypro Ruby, or silver stain, and analyzing the respective lanes of the gel for bands that are more intense in one sample over the other. Upon finding a band whose abundance differs between the compound-treated and control samples, each band can be cut out, digested with trypsin, and analyzed by liquid chromatography coupled to tandem mass spectrometry (LC−MS/MS). After annotating the peptides and proteins identified in each gel band, label-free quantitative analysis using spectral counting, LC/MS extracted ion currents (XIC), or MS/MS total ion current (TIC) can determine which identified protein has been enriched in your DARTS experiment (112–114). Despite its simplicity, we have shown the utility of the 1D gel approach for identifying previously known and novel drug targets with DARTS (62).
The initial success of DARTS with 1D gels is likely due to two factors: the targets of the chosen small molecules are both high abundance proteins, and neither protein is extremely sensitive or resistant to the proteases used (62). Given the need not only to visualize the target protein on the gel but also to determine that it is more abundant in one lane versus another, it is not hard to imagine that many potential drug targets would be missed by this nonsensitive analytical approach. The target protein could either not be sufficiently abundant in the cell to be visibly stained, or even if it is abundant enough to see, its enrichment in one sample over another could be masked because the protein comigrates with many other proteins of the same molecular weight on the gel. If just one of these comigrating proteins is much more abundant than your target protein, the fact that it is present at the same amount in both samples will hide the fact that the target protein is highly enriched in one sample. Moreover, the presence of several low or moderately abundant proteins could just as easily mask the differential abundance of the comigrating target protein. Given the limitations of this approach to DARTS, our lab and others are currently pursuing several different proteomics (mainly mass spectrometry) and nonproteomics (mainly cDNA based) platforms that offer vastly increased sensitivity and throughput; only the proteomics approaches will be discussed herein.
Perspective for Future Advances in DARTS
Proteomics
Perhaps the easiest way to more readily detect changes in protein levels between samples is to perform two-dimensional gel electrophoresis, also known as 2D-PAGE, in which proteins are first separated by charge in the first dimension, followed by SDS–PAGE in the second dimension (115). The two orthogonal separation techniques utilized by 2D-PAGE allows for more sensitive visualization of changes in protein abundance between samples because only proteins with the same isoelectric point and molecular weight will be present in the exact same spot on the gel. Whereas with 1D SDS–PAGE a single band on the gel will likely contain many, potentially dozens of different proteins, most spots on a 2D gel will consist of a single or very small number of proteins. Therefore, abundance differences in single proteins between two samples are much less likely to be masked by other comigrating proteins in 2D-PAGE. Additionally, the development of difference gel electrophoresis (DIGE) allows for even more sensitive analysis of protein abundance differences. With DIGE, two or three samples are analyzed simultaneously by labeling proteins in each sample with differently colored fluorescent tags, which are then combined and separated on a single 2D gel (116). Through automated quantitative analysis of fluorescent signals at each gel spot, similar to a microarray for gene expression analysis, this method eliminates the challenge of comparing all the spots between two or more separate gels, as well as any worries about reproducibility of protein migration patterns between multiple gels. Any spot that exhibits differential abundance can then be identified by MALDI–MS or LC–MS to identify the putative target protein.
An alternative proteomics approach that can potentially be paired with DARTS is multidimensional protein identification technology (MudPIT) (117). MudPIT is analogous to 2D-PAGE in the idea that two orthogonal separation techniques are used to provide enhanced coverage of proteins, yet it provides several fold higher sensitivity over 2D-PAGE. Rather than separate intact proteins, however, MudPIT is performed on the tryptic peptides derived from complex protein samples. The peptides are first separated by charge using strong cation exchange (SCX). As the peptides are eluted from the SCX phase, they directly enter the second separation phase consisting of C18 reversed-phase matrix, which then fractionates the peptides based on hydrophobicity just as in traditional LC–MS analysis. After elution from the reversed-phase matrix the peptides are directly sprayed into the mass spectrometer for analysis. This method eliminates the use of gels, as well as the need to visually analyze your samples to find differentially abundant proteins. Two or more samples can be analyzed by MudPIT separately using spectral counting or other label-free methods for quantitative analysis. Alternatively, multiple samples can be labeled using SILAC, ICAT, or iTRAQ and analyzed together in a single MudPIT run for even more sensitive quantitative comparison (118–120).
We have devised a strategy in which the protein samples after DARTS protease treatment are dialyzed to remove all the small peptides and amino acids resulting from digestion, enriching the samples with intact proteins or protein fragments above a given molecular weight (e.g., 10 kDa). After dialysis, the samples are trypsin digested in solution and subjected to MudPIT analysis (3). The dialysis step is necessary because the large number of small, nontryptic peptides generated during the DARTS protease treatment would compete with tryptic peptides during the MudPIT analysis both for binding to the SCX and reversed-phase matrices as well as for MS/MS analysis by the ion trap mass spectrometer. Their elimination from the sample greatly reduces the sample complexity and increases the likelihood of identifying target proteins of the small molecules under study. This approach is potentially much more sensitive than a gel-based approach and is compatible with all the mentioned label and label-free quantitative methods.
Figure 3.
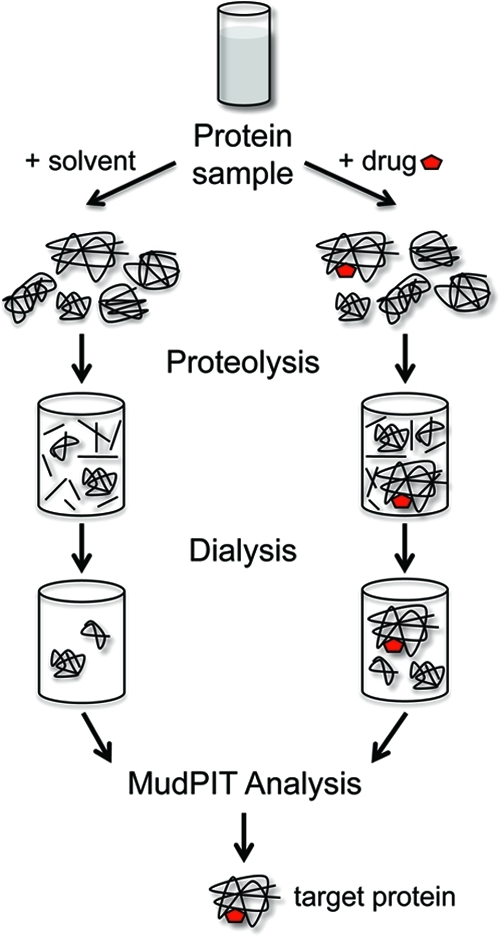
Combined DARTS-MudPIT approach. Protein lysates are split into two and incubated with the small molecule or solvent as a control. After proteolysis, each sample is dialyzed to remove digested peptides, followed by MudPIT analysis to look for enriched proteins in the drug-containing sample. Quantitation can be performed with any isotopic-labeling strategy or using label-free approaches (see text).
Degradomics
In addition to the arsenal of proteomics tools already described, new developments in a subfield of proteomics called degradomics may also be applicable in DARTS. The degradomics field is devoted to the study of proteases and the identification of all their biological substrates, aka degradomes, and many ingenious techniques have been developed and used to successfully identify hundreds of previously unknown protease substrates (reviewed in refs (121–123)). Whereas degradomics aims to identify those few proteins that are proteolyzed against a larger background of undigested proteins, DARTS seeks to find those few proteins that are either not digested or are digested to a lesser extent in one sample versus another, against a larger background of proteins that are equally digested in both samples. Despite these differences, we anticipate that current and future developments in degradomics could have an enormous impact on DARTS by facilitating identification of proteins that are differentially proteolyzed in the presence of small molecule drugs and ligands.
Experimental Conditions
In addition to using more sensitive proteomics techniques, the choice of protease, lysis buffer, and detergents may also influence the likelihood of successfully identifying drug targets with DARTS. The goal of current methods development is to maximize digestion of all nontarget background proteins without compromising the protection of the target protein, as well as to minimize the number of conditions that must be tried for each drug. An ideal DARTS condition would be one in which all proteins are digested at the same rate and extent so that the only intact proteins remaining after proteolysis would be drug targets. However, given the huge diversity of protein sequences, structures, conformational dynamics, complexes and interactions, it is no surprise that proteins vary dramatically in their sensitivity to proteolysis. Furthermore, given that different proteases target specific residues or short motifs for degradation sites as well as have a variety of preferences for folding or location of the recognition site within a polypeptide, proteome-wide susceptibility to proteolysis also varies depending on the protease used.
The initial use of thermolysin and a gentle, nondenaturing lysis buffer meant that only proteins that spontaneously unfold or have native unstructured regions could be digested, despite it being rather nonspecific with regards to substrate sequence. While the protection from thermolysin proteolysis in DARTS experiments was exceptionally robust, presumably because binding of the drugs prevented unfolding of the target proteins, a large fraction of background proteins always remained that were refractory to thermolysin digestion. This complicated proteomic analysis because the remaining samples were very complex, biasing all identifications toward high abundance proteins. Furthermore, if the drug target had been among those proteins not digestible by thermolysin, it would never be identified by DARTS. To overcome this, we first tried another single protease that can digest natively folded and denatured proteins. Using subtilisin we were able to protect target proteins, albeit to a lesser extent, under conditions in which almost all proteins were at least partially digested. Subsequently we reasoned that a mixture of many different proteases with various substrate specificities might work even better than any single protease. This seems to be generally true, as the commercially available Pronase, a mixture of endo- and exoproteases that digest native and unfolded proteins, can digest all proteins to a similar extent as subtilisin while allowing for a high level of protection similar to that seen with thermolysin. It is conceivable that other homemade protease mixtures could be optimized for DARTS that would not suffer from lot-to-lot variability as does a crude extract like Pronase (which, nevertheless, can be “calibrated” using homemade protein or lysate standards).
Another rather simple approach to maximize sensitivity is to perform subcellular fractionation prior to DARTS analysis. This could particularly be useful when the target of a given drug is believed or known to reside in a specific cellular compartment, such as the nucleus, mitochondria, ER, Golgi, or plasma membrane. Simple and robust methods already exist to purify each of these subcellular structures to near homogeneity from a variety of cell and tissue types. The proteins isolated by most subcellular fractionation techniques are natively folded and fully compatible with DARTS analysis. Indeed, such an approach to improve sensitivity of target identification has been used with other affinity-based approaches (124).
Drug Screening with DARTS
In addition to target identification, protease resistance in DARTS can also potentially be used as a screening readout. Although protein stability measurements made by differential scanning fluorimetry (DSF), differential static light scattering (DSLS), and isothermal denaturation (ITD) are readily performed in microscale and can be rapidly adapted to high-throughput screening of nearly any protein regardless of whether function has been demonstrated, these methods all require relatively large amounts of pure protein in order to screen large compound libraries (125). On the other hand, DARTS is unique in that it does not require using pure proteins. While DARTS can be performed with pure protein as well, its main advantage is that the level of protein purity is not a concern. Moreover, by using miniaturization techniques, high-throughput small-molecule DARTS screening could be performed against any protein in a complex protein mixture. Screening with DARTS, like DSF, DSLS, and ITD, has the potential of identifying compounds that bind to a large number of sites on the target protein. Such molecules may be activators or inhibitors of the protein that can individually or in combination serve as leads for specific probes useful for dissecting its molecular functions across cell types and species.
Conclusions
Drug target identification has been a decades-long quest that until recently was largely dominated by a single technique, affinity chromatography, with minimal improvements. However, with the introduction of many newer affinity-based and affinity-free approaches with diverse chemical and biological mechanisms for identifying drug targets, it may be difficult to choose which methods to use for a particular project. Methods that require extensive research resources or technical expertise have a high “activation barrier”, which often precludes them from being used regardless of their likelihood of success. Furthermore, a priori prediction of the methods most likely to work for a given drug is usually not possible.
Although in its infancy, DARTS has the potential to be a major player in drug target identification. The DARTS phenomenon holds over such a wide range of molecules (even for high-micromolar ligands) and conditions (buffers, detergents, proteases, etc.) that essentially no (or little) experimental optimization is required. Getting started with DARTS is thus straightforward and can be performed in any lab with basic molecular biology or biochemistry setup. It is also quick to carry out and can be performed with any small molecule and protein samples from any organism. Future improvements in DARTS methodology are expected to increase its sensitivity and decrease the number of proteolysis conditions that must be tested for each small molecule. The use of more sensitive proteomics techniques with DARTS should greatly enhance its capability to identify lower abundance targets, although which exact methods will prove most useful is to be determined. Furthermore, a nonproteomics approach using cDNA libraries provides an alternative means to perform DARTS against the proteins encoded by any genome of interest (62).
Last but not least, DARTS may also be used as a means of target validation. It provides a simple and fast method for verifying direct binding of small molecules to presumed target proteins that were identified by alternative approaches. This may be particularly useful when a list of potential target proteins is suspected by phenotypic analysis or obtained through omic screens or computational prediction (which may implicate proteins of a particular complex or pathway in the compound’s mechanism of action but are unable to show which protein if any is directly bound by the drug (94)). DARTS can also be used to identify the binding domain within the target protein because with more extensive protease treatment often the full-length polypeptide of the target will not survive but a smaller fragment corresponding to the binding domain will retain protease resistance. For both target validation and target domain mapping, the robustness of DARTS with proteins generated from DNA constructs (e.g., expressed in cell lysates or by in vitro transcription-translation) becomes extremely handy (62,94). With such versatility, DARTS is likely to contribute considerably to the identification and analysis of drug–protein interactions for a long time to come.
Acknowledgments
We thank M. Kirschner for insights and discussions on why DARTS works, and the National Institutes of Health (R01 CA124974 and R21 CA149774 to J.H. and R01 AA007680 to R.W.O.) and the American Cancer Society (RSG-07-035-01-CCG to J.H.) for funding support. B.L. is a trainee of the National Institutes of Health UCLA Chemistry-Biology Interface Predoctoral Training Program (T32 GM008496).
Funding Statement
National Institutes of Health, United States
References
- Fischer E. (1898) Bedeutung der Stereochemie für die Physiologie. Z. Physiol Chem. 26, 60–87. [Google Scholar]
- Corey E. J., and Cheng X.-m. (1989) The Logic of Chemical Synthesis, John Wiley, New York. [Google Scholar]
- Burke M. D.; Schreiber S. L. (2004) A planning strategy for diversity-oriented synthesis. Angew. Chem., Int. Ed. 43, 46–58. [DOI] [PubMed] [Google Scholar]
- Cane D. E.; Walsh C. T.; Khosla C. (1998) Harnessing the biosynthetic code: combinations, permutations, and mutations. Science 282, 63–68. [DOI] [PubMed] [Google Scholar]
- Li X.; Liu D. R. (2004) DNA-templated organic synthesis: nature’s strategy for controlling chemical reactivity applied to synthetic molecules. Angew. Chem., Int. Ed. 43, 4848–4870. [DOI] [PubMed] [Google Scholar]
- Wrenn S. J.; Harbury P. B. (2007) Chemical evolution as a tool for molecular discovery. Annu. Rev. Biochem. 76, 331–349. [DOI] [PubMed] [Google Scholar]
- Shuker S. B.; Hajduk P. J.; Meadows R. P.; Fesik S. W. (1996) Discovering high-affinity ligands for proteins: SAR by NMR. Science 274, 1531–1534. [DOI] [PubMed] [Google Scholar]
- Schumacher T. N.; Mayr L. M.; Minor D. L. Jr.; Milhollen M. A.; Burgess M. W.; Kim P. S. (1996) Identification of D-peptide ligands through mirror-image phage display. Science 271, 1854–1857. [DOI] [PubMed] [Google Scholar]
- Spencer D. M.; Wandless T. J.; Schreiber S. L.; Crabtree G. R. (1993) Controlling signal transduction with synthetic ligands. Science 262, 1019–1024. [DOI] [PubMed] [Google Scholar]
- Bishop A.; Buzko O.; Heyeck-Dumas S.; Jung I.; Kraybill B.; Liu Y.; Shah K.; Ulrich S.; Witucki L.; Yang F.; Zhang C.; Shokat K. M. (2000) Unnatural ligands for engineered proteins: new tools for chemical genetics. Annu. Rev. Biophys. Biomol. Struct. 29, 577–606. [DOI] [PubMed] [Google Scholar]
- Druker B. J.; Talpaz M.; Resta D. J.; Peng B.; Buchdunger E.; Ford J. M.; Lydon N. B.; Kantarjian H.; Capdeville R.; Ohno-Jones S.; Sawyers C. L. (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39. [DOI] [PubMed] [Google Scholar]
- Walensky L. D.; Kung A. L.; Escher I.; Malia T. J.; Barbuto S.; Wright R. D.; Wagner G.; Verdine G. L.; Korsmeyer S. J. (2004) Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A.; Xu S.; Montgomery M. K.; Kostas S. A.; Driver S. E.; Mello C. C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. [DOI] [PubMed] [Google Scholar]
- Dervan P. B. (2001) Molecular recognition of DNA by small molecules. Bioorg. Med. Chem. 9, 2215–2235. [DOI] [PubMed] [Google Scholar]
- Clardy J.; Walsh C. (2004) Lessons from natural molecules. Nature 432, 829–837. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. (2007) Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 70, 461–477. [DOI] [PubMed] [Google Scholar]
- Harvey A. L. (2008) Natural products in drug discovery. Drug Discovery Today 13, 894–901. [DOI] [PubMed] [Google Scholar]
- Yamamoto K. R. (1985) Steroid receptor regulated transcription of specific genes and gene networks. Annu. Rev. Genet. 19, 209–252. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf D. J.; Thummel C.; Beato M.; Herrlich P.; Schutz G.; Umesono K.; Blumberg B.; Kastner P.; Mark M.; Chambon P.; Evans R. M. (1995) The nuclear receptor superfamily: the second decade. Cell 83, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung D. T.; Jamison T. F.; Schreiber S. L. (1996) Understanding and controlling the cell cycle with natural products. Chem. Biol. 3, 623–639. [DOI] [PubMed] [Google Scholar]
- Haggarty S. J.; Mayer T. U.; Miyamoto D. T.; Fathi R.; King R. W.; Mitchison T. J.; Schreiber S. L. (2000) Dissecting cellular processes using small molecules: identification of colchicine-like, taxol-like and other small molecules that perturb mitosis. Chem. Biol. 7, 275–286. [DOI] [PubMed] [Google Scholar]
- Koeller K. M.; Haggarty S. J.; Perkins B. D.; Leykin I.; Wong J. C.; Kao M. C.; Schreiber S. L. (2003) Chemical genetic modifier screens: small molecule trichostatin suppressors as probes of intracellular histone and tubulin acetylation. Chem. Biol. 10, 397–410. [DOI] [PubMed] [Google Scholar]
- Huang J.; Zhu H.; Haggarty S. J.; Spring D. R.; Hwang H.; Jin F.; Snyder M.; Schreiber S. L. (2004) Finding new components of the target of rapamycin (TOR) signaling network through chemical genetics and proteome chips. Proc. Natl. Acad. Sci. U.S.A. 101, 16594–16599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert U. S.; Kiger A. A.; Richter C.; Perlman Z. E.; Perrimon N.; Mitchison T. J.; Field C. M. (2004) Parallel chemical genetic and genome-wide RNAi screens identify cytokinesis inhibitors and targets. PLoS Biol. 2, e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae C. A.; Peterson R. T. (2003) Zebrafish-based small molecule discovery. Chem. Biol. 10, 901–908. [DOI] [PubMed] [Google Scholar]
- Ding S.; Schultz P. G. (2004) A role for chemistry in stem cell biology. Nat. Biotechnol. 22, 833–840. [DOI] [PubMed] [Google Scholar]
- Inglese J.; Johnson R. L.; Simeonov A.; Xia M.; Zheng W.; Austin C. P.; Auld D. S. (2007) High-throughput screening assays for the identification of chemical probes. Nat. Chem. Biol. 3, 466–479. [DOI] [PubMed] [Google Scholar]
- Petrascheck M.; Ye X.; Buck L. B. (2007) An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature 450, 553–556. [DOI] [PubMed] [Google Scholar]
- Pieper A. A.; Xie S.; Capota E.; Estill S. J.; Zhong J.; Long J. M.; Becker G. L.; Huntington P.; Goldman S. E.; Shen C. H.; Capota M.; Britt J. K.; Kotti T.; Ure K.; Brat D. J.; Williams N. S.; MacMillan K. S.; Naidoo J.; Melito L.; Hsieh J.; De Brabander J.; Ready J. M.; McKnight S. L. (2010) Discovery of a proneurogenic, neuroprotective chemical. Cell 142, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman L. S. (1953) A biochemically specific method for enzyme isolation. Proc. Natl. Acad. Sci. U.S.A. 39, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenis C.; McCormick D. B. (1964) Purification of liver flavokinase by column chromatography on flavin-cellulose compounds. J. Biol. Chem. 239, 3093–3097. [PubMed] [Google Scholar]
- Cuatrecasas P.; Wilchek M.; Anfinsen C. B. (1968) Selective enzyme purification by affinity chromatography. Proc. Natl. Acad. Sci. U.S.A. 61, 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisy G. G.; Taylor E. W. (1967) The mechanism of action of colchicine. Binding of colchincine-3H to cellular protein. J. Cell Biol. 34, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisy G. G.; Taylor E. W. (1967) The mechanism of action of colchicine. Colchicine binding to sea urchin eggs and the mitotic apparatus. J. Cell Biol. 34, 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelanski M. L.; Taylor E. W. (1967) Isolation of a protein subunit from microtubules. J. Cell Biol. 34, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelanski M. L.; Taylor E. W. (1968) Properties of the protein subunit of central-pair and outer-doublet microtubules of sea urchin flagella. J. Cell Biol. 38, 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberg R. C.; Borisy G. G.; Taylor E. W. (1968) The colchicine-binding protein of mammalian brain and its relation to microtubules. Biochemistry 7, 4466–4479. [DOI] [PubMed] [Google Scholar]
- Gilbert W.; Muller-Hill B. (1966) Isolation of the lac repressor. Proc. Natl. Acad. Sci. U.S.A. 56, 1891–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuatrecasas P.; Hollenberg M. D. (1976) Membrane receptors and hormone action. Adv. Protein Chem. 30, 251–451. [DOI] [PubMed] [Google Scholar]
- Pert C. B.; Snyder S. H. (1973) Opiate receptor: demonstration in nervous tissue. Science 179, 1011–1014. [DOI] [PubMed] [Google Scholar]
- Meunier J. C.; Sealock R.; Olsen R.; Changeux J. P. (1974) Purification and properties of the cholinergic receptor protein from Electrophorus electricus electric tissue. Eur. J. Biochem. 45, 371–394. [DOI] [PubMed] [Google Scholar]
- Li G. D.; Chiara D. C.; Sawyer G. W.; Husain S. S.; Olsen R. W.; Cohen J. B. (2006) Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 26, 11599–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschumacher R. E.; Harding M. W.; Rice J.; Drugge R. J.; Speicher D. W. (1984) Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226, 544–547. [DOI] [PubMed] [Google Scholar]
- Harding M. W.; Galat A.; Uehling D. E.; Schreiber S. L. (1989) A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 341, 758–760. [DOI] [PubMed] [Google Scholar]
- Siekierka J. J.; Hung S. H.; Poe M.; Lin C. S.; Sigal N. H. (1989) A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature 341, 755–757. [DOI] [PubMed] [Google Scholar]
- Liu J.; Farmer J. D. Jr.; Lane W. S.; Friedman J.; Weissman I.; Schreiber S. L. (1991) Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66, 807–815. [DOI] [PubMed] [Google Scholar]
- Brown E. J.; Albers M. W.; Shin T. B.; Ichikawa K.; Keith C. T.; Lane W. S.; Schreiber S. L. (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369, 756–758. [DOI] [PubMed] [Google Scholar]
- Sabatini D. M.; Erdjument-Bromage H.; Lui M.; Tempst P.; Snyder S. H. (1994) RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78, 35–43. [DOI] [PubMed] [Google Scholar]
- Sabers C. J.; Martin M. M.; Brunn G. J.; Williams J. M.; Dumont F. J.; Wiederrecht G.; Abraham R. T. (1995) Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 270, 815–822. [DOI] [PubMed] [Google Scholar]
- Fenteany G.; Standaert R. F.; Lane W. S.; Choi S.; Corey E. J.; Schreiber S. L. (1995) Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 268, 726–731. [DOI] [PubMed] [Google Scholar]
- Taunton J.; Hassig C. A.; Schreiber S. L. (1996) A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272, 408–411. [DOI] [PubMed] [Google Scholar]
- Rix U.; Superti-Furga G. (2009) Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 5, 616–624. [DOI] [PubMed] [Google Scholar]
- Sadakane Y.; Hatanaka Y. (2006) Photochemical fishing approaches for identifying target proteins and elucidating the structure of a ligand-binding region using carbene-generating photoreactive probes. Anal. Sci. 22, 209–218. [DOI] [PubMed] [Google Scholar]
- Sleno L.; Emili A. (2008) Proteomic methods for drug target discovery. Curr. Opin. Chem. Biol. 12, 46–54. [DOI] [PubMed] [Google Scholar]
- Sato S.; Murata A.; Shirakawa T.; Uesugi M. (2010) Biochemical target isolation for novices: affinity-based strategies. Chem. Biol. 17, 616–623. [DOI] [PubMed] [Google Scholar]
- Lamb J.; Crawford E. D.; Peck D.; Modell J. W.; Blat I. C.; Wrobel M. J.; Lerner J.; Brunet J. P.; Subramanian A.; Ross K. N.; Reich M.; Hieronymus H.; Wei G.; Armstrong S. A.; Haggarty S. J.; Clemons P. A.; Wei R.; Carr S. A.; Lander E. S.; Golub T. R. (2006) The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Parsons A. B.; Lopez A.; Givoni I. E.; Williams D. E.; Gray C. A.; Porter J.; Chua G.; Sopko R.; Brost R. L.; Ho C. H.; Wang J.; Ketela T.; Brenner C.; Brill J. A.; Fernandez G. E.; Lorenz T. C.; Payne G. S.; Ishihara S.; Ohya Y.; Andrews B.; Hughes T. R.; Frey B. J.; Graham T. R.; Andersen R. J.; Boone C. (2006) Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126, 611–625. [DOI] [PubMed] [Google Scholar]
- Hoon S.; Smith A. M.; Wallace I. M.; Suresh S.; Miranda M.; Fung E.; Proctor M.; Shokat K. M.; Zhang C.; Davis R. W.; Giaever G.; St Onge R. P.; Nislow C. (2008) An integrated platform of genomic assays reveals small-molecule bioactivities. Nat. Chem. Biol. 4, 498–506. [DOI] [PubMed] [Google Scholar]
- Terstappen G. C.; Schlupen C.; Raggiaschi R.; Gaviraghi G. (2007) Target deconvolution strategies in drug discovery. Nat. Rev. Drug Discovery 6, 891–903. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Mitchison T. J.; Bender A.; Young D. W.; Tallarico J. A. (2009) Multi-parameter phenotypic profiling: using cellular effects to characterize small-molecule compounds. Nat. Rev. Drug Discovery 8, 567–578. [DOI] [PubMed] [Google Scholar]
- Lomenick B.; Hao R.; Jonai N.; Chin R. M.; Aghajan M.; Warburton S.; Wang J.; Wu R. P.; Gomez F.; Loo J. A.; Wohlschlegel J. A.; Vondriska T. M.; Pelletier J.; Herschman H. R.; Clardy J.; Clarke C. F.; Huang J. (2009) Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U.S.A. 106, 21984–21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S. E.; Schenone M.; Margolin A. A.; Li X.; Do K.; Doud M. K.; Mani D. R.; Kuai L.; Wang X.; Wood J. L.; Tolliday N. J.; Koehler A. N.; Marcaurelle L. A.; Golub T. R.; Gould R. J.; Schreiber S. L.; Carr S. A. (2009) Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc. Natl. Acad. Sci. U.S.A. 106, 4617–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. B.; Johnson K. S. (1988) Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31–40. [DOI] [PubMed] [Google Scholar]
- Einarson M. B., Orlinick J. R. (2002) Identification of Protein-Protein Interactions with Glutathione S-Transferase Fusion Proteins, in Protein-Protein Interactions: A Molecular Cloning Manual, pp 37–57, Cold Spring Harbor Laboratory Press, Woodbury, NY. [Google Scholar]
- Hunter W. M.; Greenwood F. C. (1962) Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature 194, 495–496. [DOI] [PubMed] [Google Scholar]
- Greenwood F. C.; Hunter W. M.; Glover J. S. (1963) The preparation of I-131-labelled human growth hormone of high specific radioactivity. Biochem. J. 89, 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace C. N.; McGrath T. (1980) Substrate stabilization of lysozyme to thermal and guanidine hydrochloride denaturation. J. Biol. Chem. 255, 3862–3865. [PubMed] [Google Scholar]
- Englander S. W.; Rolfe A. (1973) Hydrogen exchange studies of respiratory proteins. 3. Structural and free energy changes in hemoglobin by use of a difference method. J. Biol. Chem. 248, 4852–4861. [PubMed] [Google Scholar]
- Makowski L.; Rodi D. J.; Mandava S.; Minh D. D.; Gore D. B.; Fischetti R. F. (2008) Molecular crowding inhibits intramolecular breathing motions in proteins. J. Mol. Biol. 375, 529–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972. [DOI] [PubMed] [Google Scholar]
- Brath U.; Akke M. (2009) Differential responses of the backbone and side-chain conformational dynamics in FKBP12 upon binding the transition-state analog FK506: implications for transition-state stabilization and target protein recognition. J. Mol. Biol. 387, 233–244. [DOI] [PubMed] [Google Scholar]
- Pantoliano M. W.; Petrella E. C.; Kwasnoski J. D.; Lobanov V. S.; Myslik J.; Graf E.; Carver T.; Asel E.; Springer B. A.; Lane P.; Salemme F. R. (2001) High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screening 6, 429–440. [DOI] [PubMed] [Google Scholar]
- Senisterra G. A.; Markin E.; Yamazaki K.; Hui R.; Vedadi M.; Awrey D. E. (2006) Screening for ligands using a generic and high-throughput light-scattering-based assay. J. Biomol. Screening 11, 940–948. [DOI] [PubMed] [Google Scholar]
- Senisterra G. A.; Soo Hong B.; Park H. W.; Vedadi M. (2008) Application of high-throughput isothermal denaturation to assess protein stability and screen for ligands. J. Biomol. Screening 13, 337–342. [DOI] [PubMed] [Google Scholar]
- Leavitt S.; Freire E. (2001) Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 11, 560–566. [DOI] [PubMed] [Google Scholar]
- Park C.; Marqusee S. (2005) Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2, 207–212. [DOI] [PubMed] [Google Scholar]
- Elleby B.; Svensson S.; Wu X.; Stefansson K.; Nilsson J.; Hallen D.; Oppermann U.; Abrahmsen L. (2004) High-level production and optimization of monodispersity of 11beta-hydroxysteroid dehydrogenase type 1. Biochim. Biophys. Acta 1700, 199–207. [DOI] [PubMed] [Google Scholar]
- Vedadi M.; Niesen F. H.; Allali-Hassani A.; Fedorov O. Y.; Finerty P. J. Jr.; Wasney G. A.; Yeung R.; Arrowsmith C.; Ball L. J.; Berglund H.; Hui R.; Marsden B. D.; Nordlund P.; Sundstrom M.; Weigelt J.; Edwards A. M. (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U.S.A. 103, 15835–15840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus G. (1965) Protein substrate conformation and proteolysis. Proc. Natl. Acad. Sci. U.S.A. 54, 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus G.; McClintock D. K.; Castellani B. A. (1967) Ligand-stabilized conformations in serum albumin. J. Biol. Chem. 242, 4402–4408. [PubMed] [Google Scholar]
- Taniuchi H.; Moravek L.; Anfinsen C. B. (1969) Ligand-induced resistance of staphylococcal nuclease and nuclease-T to proteolysis by subtilisin, alpha-chymotrypsin, and thermolysin. J. Biol. Chem. 244, 4600–4606. [PubMed] [Google Scholar]
- Thompson J. F.; Hayes L. S.; Lloyd D. B. (1991) Modulation of firefly luciferase stability and impact on studies of gene regulation. Gene 103, 171–177. [DOI] [PubMed] [Google Scholar]
- Crews C. M.; Collins J. L.; Lane W. S.; Snapper M. L.; Schreiber S. L. (1994) GTP-dependent binding of the antiproliferative agent didemnin to elongation factor 1 alpha. J. Biol. Chem. 269, 15411–15414. [PubMed] [Google Scholar]
- Van Duyne G. D.; Standaert R. F.; Karplus P. A.; Schreiber S. L.; Clardy J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124. [DOI] [PubMed] [Google Scholar]
- Griffith J. P.; Kim J. L.; Kim E. E.; Sintchak M. D.; Thomson J. A.; Fitzgibbon M. J.; Fleming M. A.; Caron P. R.; Hsiao K.; Navia M. A. (1995) X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 82, 507–522. [DOI] [PubMed] [Google Scholar]
- Bierer B. E.; Mattila P. S.; Standaert R. F.; Herzenberg L. A.; Burakoff S. J.; Crabtree G.; Schreiber S. L. (1990) Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. U.S.A. 87, 9231–9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J.; Movva N. R.; Hall M. N. (1991) Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253, 905–909. [DOI] [PubMed] [Google Scholar]
- Chiu M. I.; Katz H.; Berlin V. (1994) RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc. Natl. Acad. Sci. U.S.A. 91, 12574–12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.; Chen J.; Schreiber S. L.; Clardy J. (1996) Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239–242. [DOI] [PubMed] [Google Scholar]
- Beevers C. S.; Chen L.; Liu L.; Luo Y.; Webster N. J.; Huang S. (2009) Curcumin disrupts the mammalian target of rapamycin-raptor complex. Cancer Res. 69, 1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning T. D.; Talley J. J.; Bertenshaw S. R.; Carter J. S.; Collins P. W.; Docter S.; Graneto M. J.; Lee L. F.; Malecha J. W.; Miyashiro J. M.; Rogers R. S.; Rogier D. J.; Yu S. S.; Anderson G. D.; Burton E. G.; Cogburn J. N.; Gregory S. A.; Koboldt C. M.; Perkins W. E.; Seibert K.; Veenhuizen A. W.; Zhang Y. Y.; Isakson P. C. (1997) Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib). J. Med. Chem. 40, 1347–1365. [DOI] [PubMed] [Google Scholar]
- Kurumbail R. G.; Stevens A. M.; Gierse J. K.; McDonald J. J.; Stegeman R. A.; Pak J. Y.; Gildehaus D.; Miyashiro J. M.; Penning T. D.; Seibert K.; Isakson P. C.; Stallings W. C. (1996) Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 384, 644–648. [DOI] [PubMed] [Google Scholar]
- Aghajan M.; Jonai N.; Flick K.; Fu F.; Luo M.; Cai X.; Ouni I.; Pierce N.; Tang X.; Lomenick B.; Damoiseaux R.; Hao R.; Del Moral P. M.; Verma R.; Li Y.; Li C.; Houk K. N.; Jung M. E.; Zheng N.; Huang L.; Deshaies R. J.; Kaiser P.; Huang J. (2010) Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat. Biotechnol. 28, 738–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L. (2007) Network pharmacology. Nat. Biotechnol. 25, 1110–1111. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Bilgin M.; Bangham R.; Hall D.; Casamayor A.; Bertone P.; Lan N.; Jansen R.; Bidlingmaier S.; Houfek T.; Mitchell T.; Miller P.; Dean R. A.; Gerstein M.; Snyder M. (2001) Global analysis of protein activities using proteome chips. Science 293, 2101–2105. [DOI] [PubMed] [Google Scholar]
- Colca J. R.; Harrigan G. G. (2004) Photo-affinity labeling strategies in identifying the protein ligands of bioactive small molecules: examples of targeted synthesis of drug analog photoprobes. Comb. Chem. High Throughput Screening 7, 699–704. [DOI] [PubMed] [Google Scholar]
- Mitsopoulos G.; Walsh D. P.; Chang Y. T. (2004) Tagged library approach to chemical genomics and proteomics. Curr. Opin. Chem. Biol. 8, 26–32. [DOI] [PubMed] [Google Scholar]
- Lu H., Wen J. A., Wang X., Yuan K., Li W., Lu H. B., Zhou Y. L., Jin K. J., Ruan K. C., and Yang G. Z. (2010) Detection of the specific binding on protein microarrays by oblique-incidence reflectivity difference method, J. Opt. 12, published online September 6, 2010, DOI: 10.1088/2040-8978/12/9/095301. [Google Scholar]
- Kato D.; Boatright K. M.; Berger A. B.; Nazif T.; Blum G.; Ryan C.; Chehade K. A.; Salvesen G. S.; Bogyo M. (2005) Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 1, 33–38. [DOI] [PubMed] [Google Scholar]
- Greenbaum D. C.; Arnold W. D.; Lu F.; Hayrapetian L.; Baruch A.; Krumrine J.; Toba S.; Chehade K.; Bromme D.; Kuntz I. D.; Bogyo M. (2002) Small molecule affinity fingerprinting. A tool for enzyme family subclassification, target identification, and inhibitor design. Chem. Biol. 9, 1085–1094. [DOI] [PubMed] [Google Scholar]
- Speers A. E.; Cravatt B. F. (2005) A tandem orthogonal proteolysis strategy for high-content chemical proteomics. J. Am. Chem. Soc. 127, 10018–10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M. J.; Saghatelian A.; Sorensen E. J.; Cravatt B. F. (2005) Target discovery in small-molecule cell-based screens by in situ proteome reactivity profiling. Nat. Biotechnol. 23, 1303–1307. [DOI] [PubMed] [Google Scholar]
- Bottcher T.; Pitscheider M.; Sieber S. A. (2010) Natural products and their biological targets: proteomic and metabolomic labeling strategies. Angew. Chem., Int. Ed. 49, 2680–2698. [DOI] [PubMed] [Google Scholar]
- Bogyo M.; Verhelst S.; Bellingard-Dubouchaud V.; Toba S.; Greenbaum D. (2000) Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chem. Biol. 7, 27–38. [DOI] [PubMed] [Google Scholar]
- Dalhoff C.; Huben M.; Lenz T.; Poot P.; Nordhoff E.; Koster H.; Weinhold E. (2010) Synthesis of S-adenosyl-l-homocysteine capture compounds for selective photoinduced isolation of methyltransferases. ChemBioChem 11, 256–265. [DOI] [PubMed] [Google Scholar]
- Evans M. J.; Cravatt B. F. (2006) Mechanism-based profiling of enzyme families. Chem. Rev. 106, 3279–3301. [DOI] [PubMed] [Google Scholar]
- Drahl C.; Cravatt B. F.; Sorensen E. J. (2005) Protein-reactive natural products. Angew. Chem., Int. Ed. 44, 5788–5809. [DOI] [PubMed] [Google Scholar]
- West G. M.; Tucker C. L.; Xu T.; Park S. K.; Han X.; Yates J. R. 3rd; Fitzgerald M. C. (2010) Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements. Proc. Natl. Acad. Sci. U.S.A. 107, 9078–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderegg R. J.; Wagner D. S. (1995) Mass-spectrometric characterization of a protein ligand interaction. J. Am. Chem. Soc. 117, 1374–1377. [Google Scholar]
- Wang F.; Blanchard J. S.; Tang X. J. (1997) Hydrogen exchange/electrospray ionization mass spectrometry studies of substrate and inhibitor binding and conformational changes of Escherichia coli dihydrodipicolinate reductase. Biochemistry 36, 3755–3759. [DOI] [PubMed] [Google Scholar]
- Liu H.; Sadygov R. G.; Yates J. R. 3rd. (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201. [DOI] [PubMed] [Google Scholar]
- Chelius D.; Bondarenko P. V. (2002) Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J. Proteome Res. 1, 317–323. [DOI] [PubMed] [Google Scholar]
- Asara J. M.; Christofk H. R.; Freimark L. M.; Cantley L. C. (2008) A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics 8, 994–999. [DOI] [PubMed] [Google Scholar]
- O’Farrell P. H. (1975) High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 250, 4007–4021. [PMC free article] [PubMed] [Google Scholar]
- Unlu M.; Morgan M. E.; Minden J. S. (1997) Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis 18, 2071–2077. [DOI] [PubMed] [Google Scholar]
- Washburn M. P.; Wolters D.; Yates J. R. 3rd. (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247. [DOI] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Gygi S. P.; Rist B.; Gerber S. A.; Turecek F.; Gelb M. H.; Aebersold R. (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999. [DOI] [PubMed] [Google Scholar]
- DeSouza L.; Diehl G.; Rodrigues M. J.; Guo J.; Romaschin A. D.; Colgan T. J.; Siu K. W. (2005) Search for cancer markers from endometrial tissues using differentially labeled tags iTRAQ and cICAT with multidimensional liquid chromatography and tandem mass spectrometry. J. Proteome Res. 4, 377–386. [DOI] [PubMed] [Google Scholar]
- Impens F.; Colaert N.; Helsens K.; Plasman K.; Van Damme P.; Vandekerckhove J.; Gevaert K. (2010) MS-driven protease substrate degradomics. Proteomics 10, 1284–1296. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Wells J. A. (2009) Methods for the proteomic identification of protease substrates. Curr. Opin. Chem. Biol. 13, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet A.; Butler G. S.; Rodriguez D.; Prudova A.; Overall C. M. (2008) Metadegradomics: toward in vivo quantitative degradomics of proteolytic post-translational modifications of the cancer proteome. Mol. Cell. Proteomics 7, 1925–1951. [DOI] [PubMed] [Google Scholar]
- Kotake Y.; Sagane K.; Owa T.; Mimori-Kiyosue Y.; Shimizu H.; Uesugi M.; Ishihama Y.; Iwata M.; Mizui Y. (2007) Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 3, 570–575. [DOI] [PubMed] [Google Scholar]
- Senisterra G. A.; Finerty P. J. Jr. (2009) High throughput methods of assessing protein stability and aggregation. Mol. Biosyst. 5, 217–223. [DOI] [PubMed] [Google Scholar]
- Freire E. (1999) The propagation of binding interactions to remote sites in proteins: analysis of the binding of the monoclonal antibody D1.3 to lysozyme. Proc. Natl. Acad. Sci. U.S.A. 96, 10118–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D. E.; Maragakis P.; Lindorff-Larsen K.; Piana S.; Dror R. O.; Eastwood M. P.; Bank J. A.; Jumper J. M.; Salmon J. K.; Shan Y.; Wriggers W. (2010) Atomic-level characterization of the structural dynamics of proteins. Science 330, 341–346. [DOI] [PubMed] [Google Scholar]